
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial polyketides from Trichoderma koningiopsis QA-3, an endophytic fungus obtained from the medicinal plant Artemisia argyi†
Xiao-Shan Shiabc,
Dun-Jia Wangb,
Xiao-Ming Lia,
Hong-Lei Lia,
Ling-Hong Menga,
Xin Lia,
Yan Pib,
Xing-Wang Zhou*b and
Bin-Gui Wang *a
*a
aKey Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Laboratory of Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Nanhai Road 7, Qingdao 266071, P. R. China. E-mail: wangbg@ms.qdio.ac.cn
bCollege of Chemistry and Chemical Engineering, Hubei Normal University, Cihu Road 11, Huangshi 435002, P. R. China. E-mail: 228689100@qq.com
cUniversity of Chinese Academy of Sciences, Yuquan Road 19A, Beijing 100049, P. R. China
First published on 3rd November 2017
Abstract
Artemisia argyi is broadly cultivated as a medicinal plant in Qichun of Hubei province in central China. Five new fungal polyketides (1–5) and two known analogues (6 and 7) were isolated and identified from the culture extract of Trichoderma koningiopsis QA-3, an endophytic fungus obtained from the inner tissue of Artemisia argyi that was collected from Qichun. Their structures were elucidated by detailed interpretation of the spectroscopic data and the structures and absolute configurations of compounds 1–4 were confirmed by X-ray crystallographic analysis. Compounds 1–3 are tricyclic polyketides possessing octahydrochromene framework and having ketal unit in their structures, while compounds 4/7 and 5/6 are related bicyclic and tricyclic analogues, respectively. The antibacterial activities against human pathogen E. coli and seven marine-derived aquatic pathogens as well as against eight agro-pathogenic fungi for each of the isolated compounds were evaluated. Compounds 1–7 showed activity against human pathogen Escherichia coli (each with MIC 64 μg mL−1), while 1 and 7 inhibited most of the tested aquatic bacteria and agro-pathogenic fungi (MICs ranging from 4 to 64 μg mL−1), respectively.
Introduction
Artemisia argyi, a typical medicinal and industrial plant that is widely cultivated in Qichun in the Hubei Province in central China,1 is frequently used for the treatment of diseases such as eczema,2 hemorrhaging,3 and inflammation.4 Multiple active metabolites including sesquiterpenes,5,6 triterpenes,6–8 flavonoids,9–11 and caffeoylquinic derivatives12,13 as well as essential oils14–16 were described from A. argyi. Endophytic fungi has been an essential source of novel and bioactivity natural products,17–19 but the chemical constituents of endophytic fungi from A. argyi have not yet been reported. As part of our continuous research for new bioactive natural products from endophytic fungi,20 a fungal strain Trichoderma koningiopsis QA-3, which was obtained from the inner tissue of A. argyi, attracted our attention. The organic extract of the fungal culture exhibited antimicrobial activity against several marine-derived pathogens in our primary screening. Chemical investigations on the fungal culture resulted in the isolation and identification of five new polyketides (1–5) and two known analogues (6 and 7) (Fig. 1). The structures of these compounds were determined by detailed analysis of the spectroscopic data and the structures and absolute configurations of compounds 1–4 were confirmed by single-crystal X-ray diffraction analysis. This paper describes the isolation, structure determination, stereochemical assignment, and antimicrobial activities of compounds 1–7.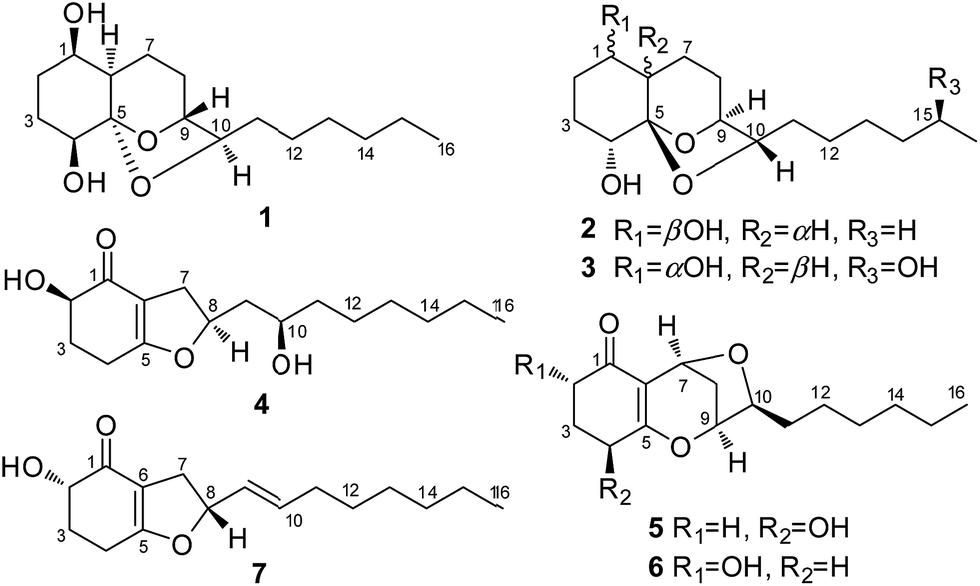 | ||
| Fig. 1 Chemical structures of compounds 1–7. | ||
Results and discussion
The culture of T. koningiopsis QA-3 was exhaustively extracted with EtOAc to afford an organic extract, which was further purified by a combination of column chromatography on Sephadex LH-20, Si gel, and Lobar Li Chroprep RP-18 as well as by preparative TLC to yield compounds 1–7.Compound 1 was isolated as colorless crystals and its molecular formula was determined as C16H28O4 on the basis of HRESIMS data, implying three degrees of unsaturation. The 1H and 13C NMR data of 1 indicated the presence of one methyl, nine methylenes, five methines (with four oxygenated), and one oxygenated non-protonated carbons as well as two exchangeable protons at δH 3.70 and 3.68 (Tables 1 and 2). Detailed analysis of the 1H and 13C NMR spectroscopic data as well as COSY and HMBC correlations (Fig. 2) indicated that the planar structure of 1 was same as that of koninginin A, a tricyclic fungal polyketide identified from the fungal strain T. koningii (ATCC no. 46314),21 whose absolute configuration was determined to be 1S, 4R, 5S, 6S, 9S, and 10S by total synthesis22 and by a single-crystal X-ray diffraction experiment.23
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 3.70, br s | 3.47, m | 3.68, m (overlap) | ||
| 2α | 1.39, m | 1.73, m (overlap) | 1.40, m | 3.93, dd (11.1, 4.7) | 2.21, m |
| 2β | 1.45, m (overlap) | 1.40, m (overlap) | 1.57, m | 2.34, ddd (12.9, 7.7, 4.5) | |
| 3α | 1.57, m | 1.45, m | 1.75, q (12.6) | 2.10, m | 2.01, m |
| 3β | 1.74, m | 1.87, m | 1.60, m (overlap) | 1.76, dd (12.4, 5.3) | 1.77, m |
| 4α | 3.34, dd (11.8, 4.4) | 3.38, brs | 3.36, br s | 2.51, m | 4.22, dd (12.6, 5.1) |
| 4β | 2.39, m | ||||
| 6 | 1.60, m | 1.90, m (overlap) | 1.46, m (overlap) | ||
| 7α | 1.95, m | 1.39, m (overlap) | 1.49, m (overlap) | 2.78, m | 4.78, d (4.6) |
| 7β | 1.49, m (overlap) | 1.96, m | 1.94, m | 2.39, m | |
| 8α | 1.37, m (overlap) | 1.76, m (overlap) | 2.07, m | 4.93, m | 2.13, m |
| 8β | 2.07, m | 1.56, dd (13.3, 5.7) | 1.39, m | 1.98, m | |
| 9α | 4.15, br s | 4.10, br s | 4.16, br s | 1.82, ddd (11.6, 9.7, 4.7) | 4.98, br s |
| 9β | 1.63, m | ||||
| 10 | 3.87, t (6.5) | 4.03, t (6.3) | 3.88, t (6.3) | 3.50, m | 3.96, t (7.0) |
| 11a | 1.46, m | 1.36, m (overlap) | 1.44, m | 1.35, m | 1.46, m |
| 11b | 1.36, m (overlap) | 1.34, m (overlap) | 1.36, m | 1.23, m (overlap) | |
| 12 | 1.24, m (overlap) | 1.28, m (overlap) | 1.24, m (overlap) | 1.23, m (overlap) | 1.29, m |
| 13 | 1.24, m (overlap) | 1.25, m (overlap) | 1.26, m (overlap) | 1.23, m (overlap) | 1.26, m (overlap) |
| 14 | 1.24, m (overlap) | 1.25, m (overlap) | 1.32, m (overlap) | 1.23, m (overlap) | 1.26, m (overlap) |
| 15 | 1.24, m (overlap) | 1.25, m (overlap) | 3.55, br s | 1.23, m (overlap) | 1.26, m (overlap) |
| 16 | 0.85, t (6.5) | 0.86, t (6.8) | 1.02, d (6.0) | 0.84, t (6.7) | 0.85, t (6.7) |
| 1-OH | 3.70, s | 3.46, s | 3.69, s | ||
| 2-OH | 5.04, br s | ||||
| 4-OH | 3.68, s | 4.88, s | 4.04, d (5.4) | 5.54, d (5.6) | |
| 10-OH | 4.54, s | ||||
| 15-OH | 4.30, s |
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 71.0, CH | 67.8, CH | 71.0, CH | 194.6, C | 193.5, C |
| 2 | 31.0, CH2 | 24.6, CH2 | 31.1, CH2 | 70.6, CH | 32.7, CH2 |
| 3 | 26.6, CH2 | 26.6, CH2 | 26.7, CH2 | 31.1, CH2 | 29.8, CH2 |
| 4 | 69.0, CH | 69.3, CH | 69.1, CH | 22.0, CH2 | 63.7, CH |
| 5 | 108.6, C | 108.4, C | 108.6, C | 176.4, C | 172.3, C |
| 6 | 41.5, CH | 38.2, CH | 41.5, CH | 110.5, C | 117.4, C |
| 7 | 20.5, CH2 | 18.2, CH2 | 20.5, CH2 | 37.2, CH2 | 65.0, CH |
| 8 | 26.6, CH2 | 27.8, CH2 | 26.6, CH2 | 84.4, CH | 31.7, CH2 |
| 9 | 77.9, CH | 77.3, CH | 77.9, CH | 43.2, CH2 | 78.4, CH |
| 10 | 77.6, CH | 79.1, CH | 77.7, CH | 66.4, CH | 84.9 CH |
| 11 | 34.5, CH2 | 35.0, CH2 | 34.6, CH2 | 30.1, CH2 | 31.1, CH2 |
| 12 | 25.0, CH2 | 24.7, CH2 | 25.4, CH2 | 25.0, CH2 | 25.8, CH2 |
| 13 | 28.7, CH2 | 28.6, CH2 | 25.3, CH2 | 28.8, CH2 | 28.6, CH2 |
| 14 | 31.3, CH2 | 31.1, CH2 | 39.0, CH2 | 31.3, CH2 | 31.2, CH2 |
| 15 | 22.0, CH2 | 21.9, CH2 | 65.8, CH | 21.9, CH2 | 21.9, CH2 |
| 16 | 13.9, CH3 | 13.8, CH3 | 23.6, CH3 | 13.9, CH3 | 13.9, CH3 |
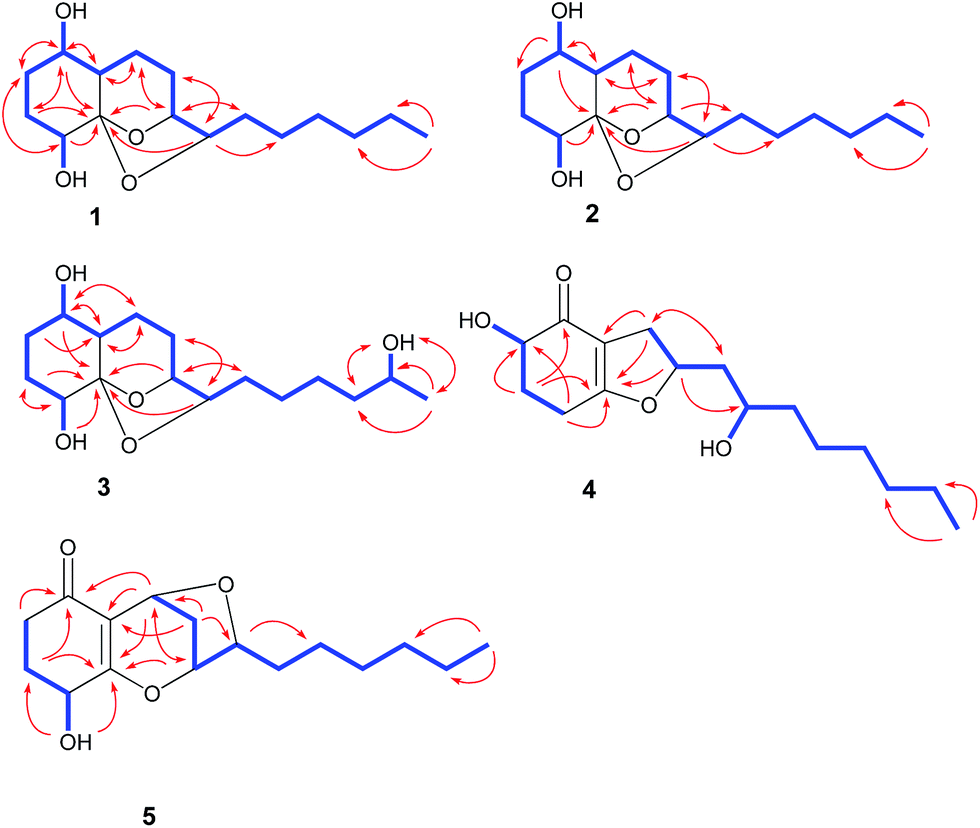 | ||
| Fig. 2 Key HMBC (arrows) and COSY (bold lines) correlations for compounds 1–5. | ||
The relative configuration of compound 1 was determined by NOESY data. NOE correlations from H-8β to OH-1 and H-9 and from H-3β to OH-1 and OH-4 indicated the cofacial orientation of these groups (Fig. 3), while correlations from H-4 to H-6 and H-8α to H-10 suggested that these protons were on the other side of the molecule. However, the relative configuration at the chiral center C-5 could not be determined by NOESY spectrum since no diagnostic NOE could be observed. The relative and absolute configuration of 1 was unambiguously established by a single-crystal X-ray diffraction experiment using Cu Kα radiation (Fig. 4). The Flack parameter 0.0(5) allowed for the establishment of the absolute configuration of 1 as 1R, 4S, 5R, 6R, 9R, and 10R, which indicated the enantiomeric nature as that of koninginin A. On the basis of the above data, the structure of 1 was determined and it was named as ent-koninginin A.
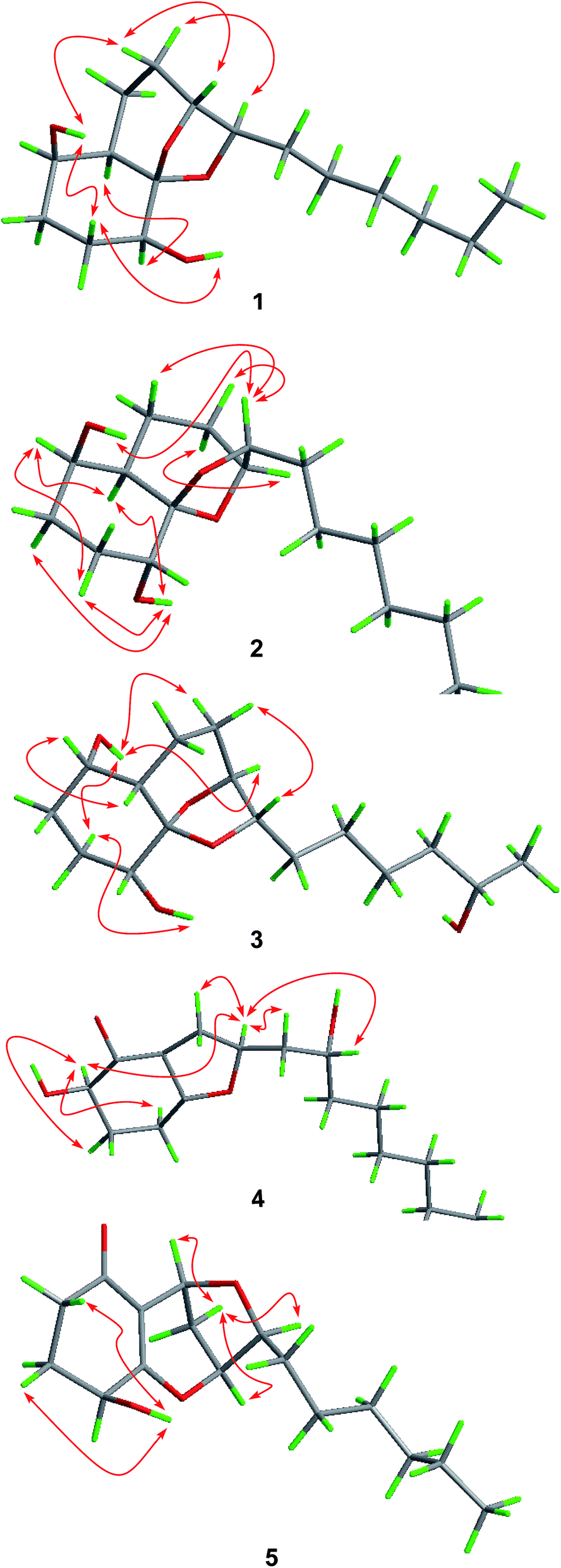 | ||
| Fig. 3 Key NOESY correlations of 1–5. | ||
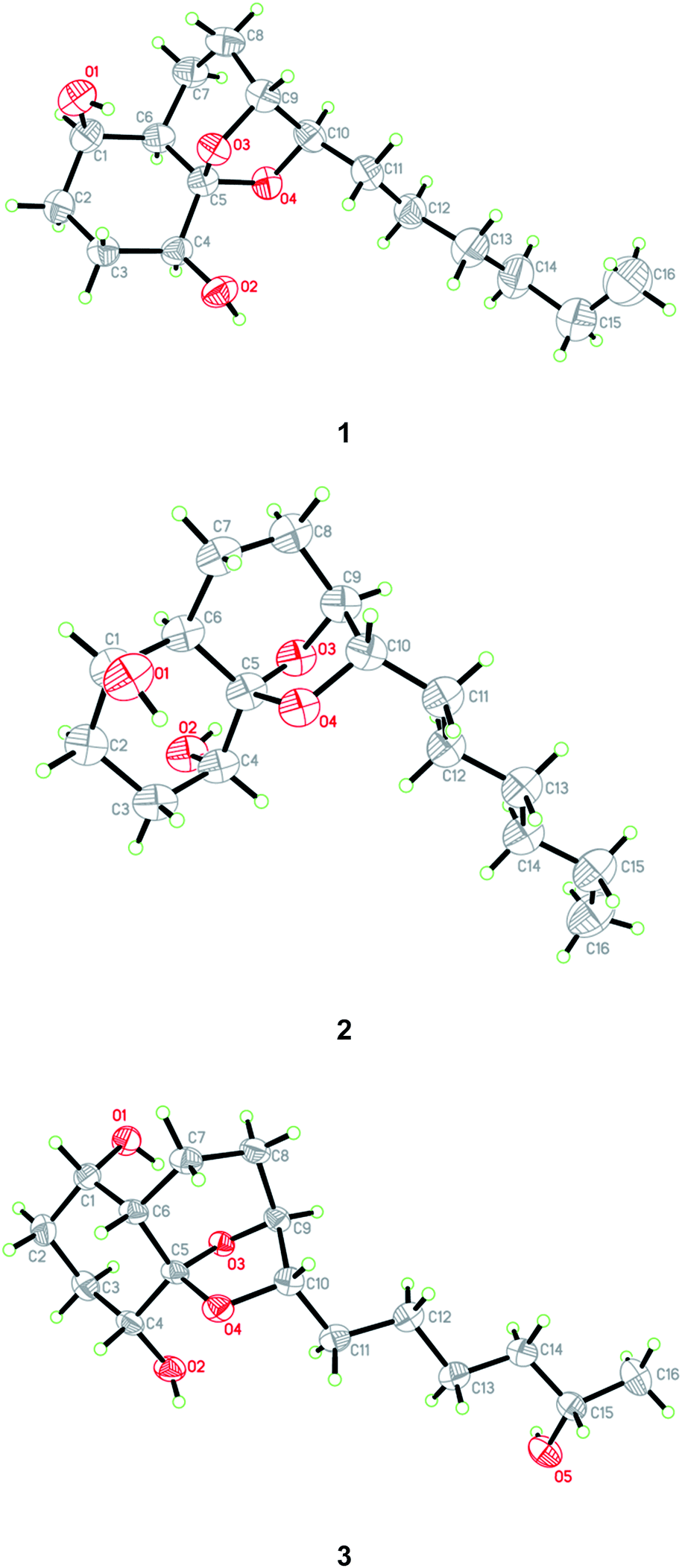 | ||
| Fig. 4 X-ray crystallographic structures of compounds 1–3. | ||
Compound 2 was originally obtained as an amorphous powder. The molecular formula was assigned on the basis of HRESIMS as C16H28O4, indicating three degrees of unsaturation, same as that of 1. The 1H and 13C NMR spectroscopic data of 2 (Tables 1 and 2) were very similar to those of 1, except for minor differences in the chemical shifts for CH-1, CH2-2, CH-6, and CH-10. Inspection of the COSY and HMBC correlations suggested that compound 2 contains same carbon and proton connectivity as that of 1 (Fig. 2). Upon slow evaporation of the solvent (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 10:1) by storing in a refrigerator, quality single crystals of compound 2 were obtained, making feasible an X-ray diffraction analysis that unequivocally confirmed its relative and absolute configuration (Fig. 4). The final refinement of the Cu Kα data resulted in a 0.0(7) Flack parameter, allowing for the unambiguous assignment of the absolute configuration as 1R, 4R, 5S, 6R, 9S, and 10S. Thus, compound 2 was determined as 1,6-di-epi-koninginin A.
:H2O = 10:1) by storing in a refrigerator, quality single crystals of compound 2 were obtained, making feasible an X-ray diffraction analysis that unequivocally confirmed its relative and absolute configuration (Fig. 4). The final refinement of the Cu Kα data resulted in a 0.0(7) Flack parameter, allowing for the unambiguous assignment of the absolute configuration as 1R, 4R, 5S, 6R, 9S, and 10S. Thus, compound 2 was determined as 1,6-di-epi-koninginin A.
15-Hydroxykoninginin A (3) was initially obtained as an amorphous powder and its molecular formula was determined as C16H28O5 (three degrees of unsaturation), with one oxygen atom more than 2, on the basis of HRESIMS data. The 1H and 13C NMR spectroscopic data of 3 (Tables 1 and 2) was very similar to those of 2, suggesting that 3 was an analogue of 2. However, resonances for a methylene at δH 1.25 (2H, H-15) and δC 21.9 (C-15) in the NMR spectra of 2 disappeared in those of 3. Instead, resonances for an oxygenated methine resonating at δH 3.55 (H-15) and δC 65.8 (C-15) were observed in the NMR spectra of 3. This revealed that the methylene group (CH2-15) in 2 was hydroxylated in 3, which was confirmed by the COSY and HMBC correlations (Fig. 2).
In the NOESY experiments, NOEs from OH-1 to H-8α and H-9 and from H-3α to OH-1 and OH-4 indicated the cofacial orientation of these groups (Fig. 3), while correlations from H-1 to H-6 and from H-8β to H-10 suggested that these protons were on the other side of this molecule. However, the relative configuration of the chiral centers at C-5 and C-15 could not be determined by NOE spectrum. Using the same method, quality single crystals of 3 were obtained, making feasible an X-ray diffraction analysis (Cu Kα radiation) that unambiguously confirmed its relative and absolute configurations (Fig. 4). The Flack parameter 0.0(3) allowed for the establishment of the absolute configuration of 3 as 1S, 4R, 5S, 6S, 9S, 10S, and 15S.
HRESIMS analysis of 4 revealed two pseudo-molecular ion peaks at m/z 283.1907 [M + H]+ and 305.1722 [M + Na]+, consistent with the molecular formula C16H26O4, and indicating the presence of four degrees of unsaturation. The skeleton of compound 4 was determined to be the same as that of koningiopisin D, a bicyclic polyketide isolated from the fungus T. koningiopsis YIM PH 30002,24 by detailed analysis of the 1H and 13C NMR spectroscopic data (Tables 1 and 2). However, the OAc group connected to C-10 was replaced by OH group in compound 4, as determined by the NMR data as well as supported by the HRESIMS data. The presence of OH group at C-10 was also determined by the COSY correlation from H-9 to H-10 and by the HMBC correlations from H-8 to C-10 as well as by the chemical shifts of CH-10 (δH 3.50 and δC 66.4).
The relative and absolute configuration of 4 were unambiguously established by a single-crystal X-ray diffraction experiment using Cu Kα radiation (Fig. 5). The Flack parameter 0.0(9) allowed for the establishment of the absolute configuration of 4 as 2R, 8S, and 10R. On the basis of the above data, the structure of 4 was determined, and was name as 10-deacetylkoningiopisin D.
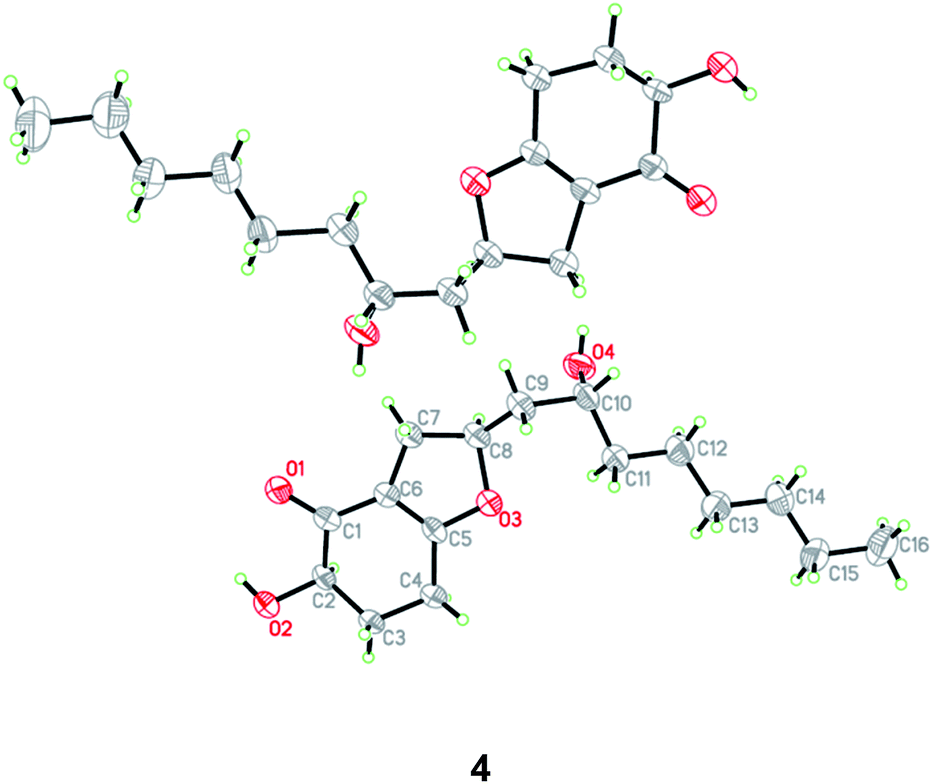 | ||
| Fig. 5 X-ray crystallographic structures of compound 4. | ||
Compound 5 was isolated as an amorphous powder and its molecular formula was assigned as C16H24O4 on the basis of HRESIMS. Analysis of the NMR data (Tables 1 and 2) showed that 5 possessed a structural similarity with koninginin L (6), which was isolated from the fungus T. koningii 8662.25 However, COSY correlations from H-4 to H-3 and the proton of 4-OH as well as HMBC correlations from the proton of 4-OH to C-3, C-4, and C-5 confirmed the presence of an OH group at C-4 in 5. NOE correlations of H-8α with H-7, H-9, and H-10 revealed that these protons were on the same face of the molecule, same as those of koninginin L (6). Based on the octant rule for the cyclohexenones,26–28 the positive Cotton effect (CE) at 306 nm in the ECD spectrum of 5 determined the absolute configuration of C-4 as S. The similar ECD curves of 5 and 6, which showed negative CE around 265 nm and positive CE around 222 nm, assigned the absolute configuration as 7S, 9S, and 10S. Thus, the structure of 5 was determined and the trivial name koninginin T was assigned to this compound.
In addition to compounds 1–5, two known compounds including koninginin L (6)25 and trichoketide A (7)29 were also isolated and identified from the fungal strain T. koningiopsis QA-3. Their structures were elucidated by comparing their NMR data with those reported in the literature.
Compounds 1–7 were assayed for their antimicrobial activities against human pathogen Escherichia coli, marine-derived aquatic bacteria (Edwardsiella tarda, Micrococcus luteus, Pseudomonas aeruginosa, Vibrio alginolyticus, V. anguillarum, V. parahemolyticus, and V. vulnificus), and agro-pathogenic fungi (Bipolaris sorokiniana, Ceratobasidium cornigerum, Colletottichum gloeosporioides Penz, Fusarium graminearum, F. oxysporum, Penicillium digitatum, Physalospora piricola Nose, and Valsa mali). As presented in Table 3, compounds 1 and 7 showed activity against human pathogen E. coli and aquatic bacteria E. tarda, V. anguillarum, and V. parahemolyticus, with MICs ranging from 8 to 64 μg mL−1. Compound 1 further showed activity against aquatic bacteria M. luteus and P. aeruginosa and agro-pathogen. In the antibacterial assays (Table 3), compounds 1–7 showed activity against human pathogen E. coli (each with MIC value 64 μg mL−1) and activity against marine-derived aquatic bacteria E. tarda, V. alginolyticus, and V. anguillarum (with MICs ranging from 8 to 64 μg mL−1) while compounds 1 and 7 further exhibited activity against M luteus, V. parahemolyticus, and V. vulnificus (with MIC values ranging from 4 to 64 μg mL−1), and the activity of compound 1 against V. vulnificus was stronger than that of the positive control chloramphenicol (MIC, 4 μg mL−1 vs. 8 μg mL−1). In the antifungal assays (Table 4), compound 7 showed a broad-spectrum activity against all of the eight tested agro-pathogenic fungi, and had potent activity against C. cornigerum and P. digitatum, both with MIC value 4.0 μg mL−1 (for the positive control amphotericin B, MIC = 1 and 0.5 μg mL−1, respectively), while compounds 2–6 showed weak or moderate activity against C. cornigerum, F. oxysporum, and/or P. digitatum, with MICs ranging from 8 to 64 μg mL−1.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Chloramphenicol | |
|---|---|---|---|---|---|---|---|---|
| a EC: E. coli. ET: E. tarda. ML: M. luteus. PA: P. aeruginosa. VAl: V. alginolyticus. VAn: V. anguillarum. VP: V. parahemolyticus. VV: V. vulnificus. —: no activity. | ||||||||
| EC | 64 | 64 | 64 | 64 | 64 | 64 | 64 | 2 |
| ET | 64 | 64 | 64 | 64 | 64 | 64 | 64 | 0.25 |
| ML | 64 | 64 | — | — | — | — | 64 | 2 |
| PA | 64 | — | — | — | — | — | — | 1 |
| VAl | 64 | 64 | 64 | 64 | 32 | 64 | 64 | 0.25 |
| VAn | 8 | 32 | 16 | 32 | 32 | 32 | 32 | 1 |
| VP | 64 | — | — | — | — | — | 64 | 2 |
| VV | 4 | — | — | — | — | — | 16 | 8 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Amphotericin B | |
|---|---|---|---|---|---|---|---|---|
| a BS: B. sorokiniana. CC: C. cornigerum. CG: C. gloeosporioides. FG: F. graminearum. FO: F. oxysporum. PD: P. digitatum. PP: P. piricola Nose. VM: V. mali. —: no activity. | ||||||||
| BS | 64 | — | — | — | — | — | 8 | 1 |
| CC | 8 | 64 | 64 | 32 | — | 16 | 4 | 1 |
| CG | — | — | — | 64 | — | — | 8 | 1 |
| FG | — | — | — | — | — | — | 64 | 0.25 |
| FO | 64 | 64 | — | 64 | — | — | 8 | 1 |
| PD | 32 | 64 | 64 | 8 | 16 | — | 4 | 0.5 |
| PP | 16 | — | — | — | — | — | 8 | 1 |
| VM | — | — | — | — | — | — | 64 | 1 |
Conclusions
In summary, we isolated and characterized five new fungal polyketides including ent-koninginin A (1), 1,6-di-epi-koninginin A (2), 15-hydroxykoninginin A (3), 10-deacetylkoningiopisin D (4), and koninginin T (5), together with two known analogues, koninginin L (6) and trichoketide A (7) from the culture extract of endophytic fungus T. koningiopsis QA-3. Their structures were elucidated by detailed interpretation of the spectroscopic data and the structures and absolute configurations of compounds 1–4 were confirmed by X-ray crystallographic analysis. Compounds 1–3 are tricyclic polyketides possessing octahydrochromene framework and having ketal unit in their structures, while compounds 5/6 and 4/7 are related tricyclic and bicyclic analogues, respectively. The antimicrobial activities of compounds 1–7 were evaluated and compounds 1 and 7 inhibited human pathogen E. coli and all of the seven marine-derived aquatic bacteria with MIC values ranging from 4 to 64 μg mL−1. In addition, compound 7 showed a broad-spectrum activity against all of the eight tested agro-pathogenic fungi.Experimental section
General experimental procedures
Melting points were determined on an SGW X-4 micro-melting-point apparatus. Optical rotations were measured with an Optical Activity AA-55 polarimeter. The ECD spectra were recorded on a Chirascan spectropolarimeter, using CH3OH as a solvent. UV spectra were recorded on a PuXi TU-1810 UV-visible spectrophotometer. 1D and 2D NMR spectra were recorded at 500 for 1H and 125 MHz and 13C in DMSO-d6, respectively, on a Bruker Avance 500 spectrometer with TMS as internal standard. ESIMS and HRESIMS data were obtained on a Waters Micromass Q-TOF Premier and a Thermo Fisher Scientific LTQ Orbitrap XL spectrometers, respectively. Analytical HPLC were performed using a Dionex HPLC system equipped with P680 pump, ASI-100 automated sample injector, and UVD340U multiple wavelength detector controlled by Chromeleon software (version 6.80). Open column chromatography was performed with commercially available Si gel (200–300 mesh, Qingdao Haiyang Chemical Co.), Lobar LiChroprep RP-18 (40–63 μm, Merck), and Sephadex LH-20 (Pharmacia). TLC was carried out using silica gel GF254 (Qingdao Haiyang Chemical Factory) plates. All solvents used were distilled prior to use.Fungal material
The endophytic fungus T. koningiopsis QA-3 was obtained from the inner tissue of the cultivated medicinal plant Artemisia argyi collected at Qichun of the Hubei Province in the central China, in July 2014, using a protocol as described in our previous report.30 Fungal identification was performed by analysis of its ITS region of the rDNA as described previously.30 The resulting sequence was same (100%) to that of T. koningiopsis DMC 795b (compared with EU 718083.1), and has been deposited in GenBank (with accession no MF616361). The strain is preserved at the Key Laboratory of Experimental Marine Biology, Institute of Oceanology of the Chinese Academy of Sciences (IOCAS).Cultivation
For chemical investigations, the fresh mycelia of T. koningiopsis QA-3 were grown on PDA medium at 28 °C for 7 days and were then inoculated for 30 days at room temperature in 90 × 1 L conical flasks with solid rice medium (each flask contained 70 g rice, 0.1 g corn flour, 0.3 g peptone, and 100 mL distilled water).Extraction and isolation
The whole fermented cultures were extracted three times with EtOAc, which was evaporated under reduced pressure to afford an extract (76.7 g). The extract was fractionated by Si gel vacuum liquid chromatography (VLC) using different solvents of increasing polarity from petroleum ether (PE) to MeOH to yield ten fractions (Frs. 1–10) based on TLC and HPLC analysis. Purification of Fr. 5 (4.3 g) by reversed-phase column chromatography (CC) over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 10:90 to 100:0) yielded eight subfractions (Frs. 5.1–5.8). Fr. 5.6 (728.1 mg) was purified by CC on Sephadex LH-20 (MeOH) and then by recrystallization to obtain compound 1 (57.4 mg), and the residue was further purified by preparative TLC (plate: 20 × 20 cm, developing solvents: PE–EtOAc, 1:1) to obtain compound 2 (5.0 mg). Fr. 6(7.3 g) was fractioned by CC over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 10:90 to 100:0) and then purified by CC on Sephadex LH-20 (MeOH) and Si gel (PE–EtOAc, from 10:1 to 2:1) to yield compounds 6 (12.5 mg) and 7 (27.1 mg). Purification of Fr. 7 (10.2 g) by reversed-phase column chromatography (CC) over Lobar LiChroprep RP-18 with a MeOH–H2O gradient (from 10:90 to 100:0) yielded fifteen subfractions (Frs. 7.1–7.15). Fr. 7.4 (1.9 g) was further purified on Si gel eluting with a CH2Cl2–MeOH gradient (from 100:1 to 10:1) and then by preparative TLC (plate: 20 × 20 cm, developing solvents: CH2Cl2–acetone, 5:1) to obtain compound 3 (61.2 mg). Fr. 7.5 (2.8 g) was purified by CC over Si gel eluting with a CH2Cl2–EtOAc gradient (from 20:1 to 2:1) and then by preparative TLC (plate: 20 × 20 cm, developing solvent: CH2Cl2/MeOH, 20:1) and Sephadex LH-20 (MeOH) to afford compounds 4 (12.1 mg) and 5 (14.0 mg).
ε) 271 (3.51) nm; ECD (3.39 mM, MeOH) λmax (Δε) 264 (+34.25) nm; 1H and 13C NMR data: see Tables 1 and 2; ESIMS m/z 283.19 [M + H]+, 305.17 [M + Na]+; HRESIMS m/z 283.1907 [M + H]+ (calcd for C16H27O4, 283.1904) and m/z 305.1722 [M + Na]+ (calcd for C16H26O4Na, 305.1729).ε) 267 (3. 69) nm; ECD (3.26 mM, MeOH) λmax (Δε) 222 (+49.21), 267 (−87.27), 306 (+35.99) nm; 1H and 13C NMR data: see Tables 1 and 2; ESIMS m/z 281.18 [M + H]+; HRESIMS m/z 281.1750 [M + H]+ (calcd for C16H25O4, 281.1753).X-ray crystallographic analysis of compounds 1–4 (ref. 31)
All crystallographic data were collected on an Agilent Xcalibur Eos Gemini CCD plate diffractometer equipped with graphite-monochromatic Cu-Kα radiation (λ = 1.54178 Å) at 293(2) K. The data were corrected for absorption by using the program SADABS.32 The structures were solved by direct methods with the SHELXTL software package.33 All non-hydrogen atoms were refined anisotropically. The H atoms were located by geometrical calculations, and their positions and thermal parameters were fixed during the structures refinement. The structures were refined by full-matrix least-squares techniques.34Antimicrobial assays
Antimicrobial evaluation against human pathogen Escherichia coli IOCAS-1, marine-derived aquatic bacteria (E. tarda QDIO-2, M. luteus QDIO-3, P. aeruginosa QDIO-4, V. alginolyticus QDIO-5, V. anguillarum QDIO-6, V. parahemolyticus QDIO-8, and V. vulnificus QDIO-9) and plant-pathogenic fungi (B. sorokiniana QDAU-7, C. cornigerum QDAU-8, C. gloeosporioides Penz QDAU-9, F. graminearum QDAU-10, F. oxysporum QDAU-5, P. digitatum QDAU-11, P. piricola Nose QDAU-12, and V. mali QDAU-13) was carried out by the microplate assay.35 The aquatic pathogens were obtained from the Institute of Oceanology, Chinese Academy of Sciences, while the plant pathogens were provided by the Qingdao Agricultural University. Chloramphenicol and amphotericin B were used as the positive control against bacteria and fungi, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Scientific and Technological Innovation Project of Qingdao National Laboratory for Marine Science and Technology (No. 2015ASKJ02). B.-G. Wang acknowledges the support of Taishan Scholar Project from Shandong Province.Notes and references
- L. J. Wan, J. Q. Lu and S. N. Guo, Med. Plant, 2016, 7, 1 Search PubMed.
- Q. F. Huang, H. G. Wu, J. Liu and J. Hong, J. Acupunct. Tuina Sci., 2012, 10, 342 CrossRef.
- Q. C. Zhao, H. Kiyohara and H. Yamada, Phytochemistry, 1994, 35, 73 CrossRef CAS PubMed.
- N. R. Shin, H. W. Ryu, J. W. Ko, S. H. Park, H. J. Yuk, H. J. Kim, J. C. Kim, S. H. Jeong and I. S. J. Shin, J. Ethnopharmacol., 2017, 209, 108 CrossRef PubMed.
- S. Wang, J. Li, J. Sun, K. W. Zeng, J. R. Cui, Y. Jiang and P. F. Tu, Fitoterapia, 2013, 85, 169 CrossRef CAS PubMed.
- M. Yoshikawa, H. Shimada, H. Matsuda, J. Yamahara and N. Murakami, Chem. Pharm. Bull., 1996, 44, 1656 CrossRef CAS PubMed.
- M. Adams, T. Efferth and R. Bauer, Planta Med., 2006, 72, 862 CAS.
- N. Li, Y. Mao, C. H. Deng and X. M. Zhang, J. Chromatogr. Sci., 2008, 46, 401 CAS.
- J. L. Lv, Z. Z. Li and L. B. Zhang, Nat. Prod. Res., 2017, 15, 1 CrossRef PubMed.
- J. M. Seo, H. M. Kang, K. H. Son, J. H. Kim, C. W. Lee, H. M. Kim, S. I. Chang and B. M. Kwon, Planta Med., 2003, 69, 218 CrossRef CAS PubMed.
- L. B. Zhang, J. L. Lv, H. L. Chen, X. Q. Yan and J. A. Duan, Biochem. Syst. Ecol., 2013, 50, 455 CrossRef CAS.
- J. C. Lim, S. Y. Park, Y. Nam, T. T. Nguyen and U. D. Sohn, Korean J. Physiol. Pharmacol., 2012, 16, 313 CrossRef CAS PubMed.
- J. L. Lv, J. A. Duan, B. Shen and Y. Y. Yin, Chem. Nat. Compd., 2013, 49, 8 CrossRef CAS.
- L. L. Chen, H. J. Zhang, J. Chao and J. F. Liu, J. Ethnopharmacol., 2017, 204, 107 CrossRef CAS PubMed.
- Y. B. Ge, Z. G. Wang, Y. Xiong, X. J. Huang, Z. N. Mei and Z. G. Hong, J. Nat. Med., 2016, 70, 531 CrossRef CAS PubMed.
- H. C. Huang, H. F. Wang, K. H. Yih, L. Z. Chang and T. M. Chang, Int. J. Mol. Sci., 2012, 13, 14679 CrossRef CAS PubMed.
- P. F. Zhang, B. L. Shi, J. L. Su, Y. X. Yue, Z. X. Cao, W. B. Chu, K. Li and S. M. Yan, J. Anim. Physiol. Anim. Nutr., 2017, 101, 251 CrossRef CAS PubMed.
- G. A. Strobel, R. N. Kharwar and V. C. Verma, Nat. Prod. Commun., 2009, 4, 1511 Search PubMed.
- V. M. Chapla, C. R. Biasetto and A. R. Araujo, Rev. Virtual Quim., 2013, 5, 421 CrossRef CAS.
- P. Zhang, L. H. Meng, A. Mándi, X. M. Li, T. Kurtán and B. G. Wang, RSC Adv., 2015, 5, 39870 RSC.
- H. G. Cutler, D. S. Himmelsbach, R. F. Arrendale, P. D. Cole and R. H. Cox, Agric. Biol. Chem., 1989, 53, 2605 CAS.
- X. X. Xu and Y. H. Zhu, Tetrahedron Lett., 1995, 36, 9173 CrossRef CAS.
- K. Mori, M. Bando and K. Abe, Biosci., Biotechnol., Biochem., 2002, 66, 1779 CrossRef CAS PubMed.
- K. Liu, Y. B. Yang, C. P. Miao, Y. K. Zheng, J. L. Chen, Y. W. Chen, L. H. Xu, H. L. Guang, Z. T. Ding and L. X. Zhao, Planta Med., 2016, 82, 371 CAS.
- B. Y. Lang, J. Li, X. X. Zhou, Y. H. Chen, Y. H. Yang, X. N. Li, Y. Zeng and P. Zhao, J. Phytochem. Lett., 2015, 11, 1 CrossRef CAS.
- X. L. Ye, Stereochemistry, Beijing University Press, Beijing, 1999, p. 242 Search PubMed.
- Y. Sun, L. Tian, J. Huang, H. Y. Ma, Z. Zheng, A. L. Lv, K. Yasukawa and Y. H. Pei, Org. Lett., 2008, 10, 393 CrossRef CAS PubMed.
- F. H. Song, H. Q. Dai, Y. J. Tong, B. Ren, C. X. Chen, N. Sun, X. Y. Liu, J. Bian, M. Liu, H. Gao, H. W. Liu, X. P. Chen and L. X. Zhang, J. Nat. Prod., 2010, 73, 806 CrossRef CAS PubMed.
- H. Yamazaki, R. Saito, O. Takahashi, R. Kirikoshi, K. Toraiwa, K. Iwasaki, Y. Izumikawa, W. Nakayama and M. Namikoshi, J. Antibiot., 2015, 68, 628 CrossRef CAS PubMed.
- S. Wang, X. M. Li, F. Teuscher, D. L. Li, A. Diesel, R. Ebel, P. Proksch and B. G. Wang, J. Nat. Prod., 2006, 69, 1622 CrossRef CAS PubMed.
- Crystallographic data of compounds 1–4 have been deposited in the Cambridge Crystallographic Data Centre as CCDC 1566573 (for 1), CCDC 1566574 (for 2), CCDC 1566575 (for 3), and CCDC 1566583 (for 4).
- G. M. Sheldrick, SADABS, Software for Empirical Absorption Correction, University of Gottingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL, Structure Determination Software Programs, Bruker Analytical X-ray System Inc., Madison, WI, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement, University of Gottingen, Germany, 1997 Search PubMed.
- C. G. Pierce, P. Uppuluri, A. R. Tristan, F. L. Wormley Jr, E. Mowat, G. Ramage and J. L. Lopez-Ribot, Nat. Protoc., 2008, 3, 1494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS spectra of compounds 1–5. Selected 1D and 2D NMR (1–5) and ECD (3–5) spectra (PDF). X-ray crystallographic files of 1–4 (CIF). CCDC 1566573–1566575, and 1566583 for compounds 1–4 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11122c |
| This journal is © The Royal Society of Chemistry 2017 |