
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of an archaeal photoreceptor/transducer complex from Natronomonas pharaonis assembled within styrene–maleic acid lipid particles
N. Voskoboynikova†
 a,
W. Mosslehy†a,
A. Colbasevicia,
T. T. Ismagulovab,
D. V. Bagrovb,
A. A. Akovantsevad,
P. S. Timashevcd,
A. Y. Mulkidjaniana,
V. N. Bagratashvilid,
K. V. Shaitanb,
M. P. Kirpichnikovb and
H.-J. Steinhoff*a
a,
W. Mosslehy†a,
A. Colbasevicia,
T. T. Ismagulovab,
D. V. Bagrovb,
A. A. Akovantsevad,
P. S. Timashevcd,
A. Y. Mulkidjaniana,
V. N. Bagratashvilid,
K. V. Shaitanb,
M. P. Kirpichnikovb and
H.-J. Steinhoff*a
aDepartment of Physics, University of Osnabrück, 49069, Osnabrück, Germany. E-mail: hsteinho@uos.de
bDepartment of Bioengineering, Faculty of Biology, Lomonosov Moscow State University, 1-12 Leninskiye Gory, 119234, Moscow, Russia
cInstitute for Regenerative Medicine of I. M. Sechenov First Moscow State Medical University, 8-2 Trubetskaya St., Moscow, 119991, Russia
dInstitute of Photonic Technologies of Research Center “Crystallography and Photonics” of RAS, 2 Pionerskaya St., Troitsk, Moscow, 142190, Russia
First published on 3rd November 2017
Abstract
The styrene–maleic acid (SMA) copolymers enable a detergent-free extraction of membrane proteins from lipid bilayers yielding stable water-soluble nanocontainers with lipid-encased proteins. Here, we demonstrate the ability of the SMA copolymer to extract a complex of interacting membrane proteins from proteoliposomes, namely the archaeal sensory rhodopsin II from Natronomonas pharaonis, NpSRII, in complex with its transducer, NpHtrII. The NpSRII/NpHtrII complexes retained their structural integrity within SMA lipid particles as revealed by diverse biophysical techniques. Pulse electron paramagnetic resonance data showed the SMA-encased NpSRII/NpHtrII complexes as native-like 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dimers with a distance between NpSRII sites 159 according to the “V”-shaped conformation found in a crystal structure.
:2 dimers with a distance between NpSRII sites 159 according to the “V”-shaped conformation found in a crystal structure.
Introduction
The membrane-embedded protein photoreceptor sensory rhodopsin II (NpSRII) from the halophilic archaeon Natronomonas pharaonis mediates the photophobic response to harmful blue light.1–3 NpSRII is structurally and functionally related to the light driven ion pumps bacteriorhodopsin (BR), halorhodopsin (HR), and channelrhodopsin.4 In addition, NpSRII is distantly related to G-protein coupled receptors (GPCRs) including visual rhodopsins.5 Similar to other rhodopsins, NpSRII comprises seven transmembrane helices (A–G) with a retinal chromophore covalently bound to a conserved lysine residue of helix G via a protonated Schiff base. In membranes, NpSRII interacts with its cognate transducer (NpHtrII), which consists of a membrane-embedded N-terminal domain with two transmembrane α-helices, TM1 and TM2, and an elongated α-helical coiled-coil cytoplasmic signalling domain that protrudes into the cytoplasm by ∼26 nm.6 In membrane lipids, the NpSRII/NpHtrII complexes are present in a 2:2 molecular stoichiometry.7,8 The structure of the transmembrane domains of the NpSRII/NpHtrII complex, with two receptor molecules flanking a transducer dimer, as initially proposed from electron paramagnetic resonance (EPR) data,9 was resolved by X-ray crystallography.1
Owing to the availability of the crystal structure,1 the possibility to trigger the reaction cycle of NpSRII by a short laser flash, and the homology between NpHtrII and other prokaryotic sensory transducers,10 the NpSRII/NpHtrII complex serves as a paradigmal system for studying sensory transduction.11 Isomerization of the retinal chromophore in response to light activation of NpSRII leads to an outward movement of helix F,7,9,12,13 which induces a rotary/screw-like motion of the transducer helix TM2 that may be accompanied by a displacement,9,14,15 presumably analogous to the piston-like motion in chemoreceptors.16–18 These transient changes in the transmembrane part of NpHtrII lead to subsequent signal propagating along the cytoplasmic part of NpHtrII to the intracellular signalling pathway,19 which modulates the cell motility similarly to the bacterial two-component chemotaxis systems (reviewed e.g. in ref. 6, 10 and 20–22). In cell membranes, the 2:2 NpSRII/NpHtrII complexes are likely further organized in trimers,19,23,24 similarly to the E. coli chemoreceptors,25 which hinders investigation of protein interactions within separate NpSRII/NpHtrII complexes. Still, the inter-protein interaction within the 2:2 photoreceptor/transducer complexes are of considerable interest for elucidation of the signalling pathway.
To facilitate studying of separate 2:2 photoreceptor/transducer complexes in a lipid environment, we have recently established reconstitution of the detergent-solubilized NpSRII/NpHtrII complex into nanodiscs, self-assembling soluble monodisperse lipid discoidal particles of controlled nanometer-scale size, where a fragment of lipid bilayer is surrounded and stabilized by an amphiphilic helical protein belt.26 We used single-molecule (SM) fluorescence imaging, fluorescence cross-correlation spectroscopy (FCCS) analysis, and site-directed spin labeling (SDSL) electron paramagnetic resonance (EPR) spectroscopy to confirm the reconstitution of the 2:2 NpSRII/NpHtrII complex into nanodiscs. Photocycle measurements confirmed functional activity of the reconstituted NpSRII.26
Recently, lipid discoidal particles stabilized by amphipathic styrene–maleic acid (SMA) copolymers were suggested as alternative nanocontainers for membrane proteins. While preparation of nanodiscs implies application of detergents, the treatment by SMA copolymers enables a direct, detergent-free extraction of membrane proteins both from artificial and natural lipid bilayers providing stable membrane patches with incorporated protein(s) (reviewed, e.g., in ref. 27–29). SMA-encased nanolipoprotein particles are comprised of lipid/protein cores surrounded by a polymer belt and have, depending on the preparation routine, diameters of 10–30 nm. The SMA copolymer with a 3:1 styrene-to-maleic acid molar ratio (3:1 SMA) was shown to extract integral membrane proteins, both α-helical bundle proteins such as bacteriorhodopsin30,31 and β-barrel proteins, such as lipid A palmitoyltransferase PagP30 from 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) liposomes. Moreover, the SMA copolymer is able to solubilize, in a detergent-free manner, hetero- and homo-oligomeric membrane protein complexes.27,28,32–34 The resulting lipoprotein-SMA particles were 11–12 nm in diameter and maintained the protein integrity and activity.30,31 The 3:1 SMA polymer-mediated lipid solubilisation was also shown to be non-selective in regard to the lipid type.35,36 The properties of SMA-solubilized membrane proteins were studied using different biophysical methods. The integral membrane protein KCNE1, e.g., extracted by 3:1 SMA from 1-palmitoyl-2-oleoyl-phosphatidylcholine/1-palmitoyl-2-oleoyl-phosphatidylglycerol (POPC/POPG) liposomes could be characterized by electron paramagnetic resonance (EPR) spectroscopy with significantly improved resolution when compared to micelles.37,38
Here, we have tested whether SMA copolymer can solubilize an archaeal photoreceptor–transducer complex in the absence of detergents. We have shown that the 3:1 SMA copolymer is able to extract, in a detergent-free manner, the NpSRII/NpHtrII complex from protein-containing liposomes (proteoliposomes) made of E. coli polar lipids. The obtained NpSRII/NpHtrII-SMA lipid particles (SMALPs) were characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM), atomic force microscopy (AFM), resonance Raman spectroscopy, as well as by continuous wave (cw) and pulse EPR spectroscopy. NpSRII/NpHtrII was found to be present in SMALPs as structurally integer separate complexes with a photoreceptor: transducer stoichiometry of 2:2.
Materials and methods
The polar lipid extract of E. coli membranes in chloroform was purchased from Avanti Polar Lipids, Inc. (Alabaster; AL; USA). N-Dodecyl-β-D-maltoside (DDM) was bought from Anatrace (Affymetrix, Cleveland, Ohio, US). Isopropyl-β-D-thiogalactopyranosid (IPTG) was acquired from Carl Roth (Karlsruhe, Germany). EDTA-free complete protease inhibitor cocktail was procured from Roche Life Science (Mannheim, Germany). Ni-NTA superflow agarose was purchased from Qiagen (Hilden, Germany). The styrene maleic acid (SMA) copolymer with styrene-to-maleic acid molar ratio of 3:1 (MW 9500 Da, supplied as an aqueous sodium salt solution SMA 3000 HNa) was kindly provided as a gift by Cray Valley (Exton; PA; USA). All other reagents were of analytical grade.
Protein concentrations were determined using an UV-vis spectrophotometer (UV-2450, Shimadzu Corporation, Kyoto, Japan). The concentration of NpSRII was determined by using the known extinction coefficient of 50000 M−1 cm−1 at λ = 280 nm. The concentration of NpHtrII157 was determined according to ref. 39. The concentration of MSP1E3D1 was determined by using the known extinction coefficient of 29400 M−1 cm−1 at λ = 280 nm.
Protein expression and purification
For purification purposes, all proteins had a C-terminal 6xHis-tag. NpSRII-His and its respective cysteine mutant (NpSRII-L159C) and NpHtrII157-His (transducer construct that was truncated at position 157) were expressed in E. coli BL21 (DE3) cells and purified according to ref. 23, 40 and 41 with minor modifications. Briefly, transformed cells were grown in Luria–Bertani (LB) medium containing 50 mg ml−1 kanamycin at 37 °C to an optical density OD580 of 1.0. The overexpression of the protein was induced by addition of IPTG to a final concentration of 0.5 mM. In the case of NpSRII, 10 μM all-trans retinal (Sigma) was also added. After an induction period of 3 h at 37 °C, cells were harvested (4200 × g; 15 min; 4 °C), washed and resuspended (1/100 culture volume) in 150 mM NaCl, 25 mM NaPi (pH 8.0), 2 mM EDTA buffer containing a protease inhibitor mix. Cells were disrupted by sonication (Branson Sonifier II W-250, Heinemann, Germany). Membranes were isolated by centrifugation (50000 × g; 1 h; 4 °C), and solubilized in buffer A (300 mM NaCl, 50 mM NaPi (pH 8.0), 2% (w/v) DDM) overnight at 4 °C. Solubilized membrane proteins were isolated by centrifugation (50000 × g; 1 h; 4 °C) followed by chromatography using Ni-NTA superflow material which was pre-equilibrated with buffer B (300 mM NaCl, 50 mM NaPi (pH 8.0), 0.05% (w/v) DDM). Non-specifically bound proteins were removed by washing extensively with buffer B containing 30 mM imidazole. His-tagged proteins were eluted with buffer B containing 200 mM imidazole. Fractions containing the desired protein were pooled and dialyzed against buffer C (500 mM NaCl, 10 mM Tris (pH 8.0), 0.05% (w/v) DDM) to remove imidazole. If not used directly for spin labeling and reconstitution, protein samples were flash frozen and stored at −80 °C.
MSP1E3D1 was expressed in E. coli BL21 (DE3) cells and was purified according to ref. 42 with minor modifications. Briefly, transformed cells were grown in LB medium containing 50 mg ml−1 kanamycin at 37 °C to an optical density OD580 of 1.0. The overexpression of the protein was induced by addition of IPTG to a final concentration of 0.5 mM. After an induction period of 4 h at 37 °C, cells were harvested (4200 × g; 15 min; 4 °C) and resuspended (1/100 culture volume) in buffer D (300 mM NaCl, 40 mM Tris (pH 8.0)) containing 1% Triton. Cells were disrupted by sonication (Branson Sonifier II W-250). The cell suspension was cleared by centrifugation (50000 × g; 45 min; 4 °C). The cleared supernatant was subsequently loaded on Ni-NTA superflow material which was pre-equilibrated with buffer D. The column was washed with 4 bed volumes of each of the following: (1) buffer D containing 1% Triton; (2) buffer D containing 50 mM cholate; (3) buffer D; (4) buffer D containing 50 mM imidazole. MSP1E3D1 was eluted with buffer D containing 300 mM imidazole. Fractions containing MSP1E3D1 were pooled and dialyzed against 100 mM NaCl, 10 mM Tris (pH 8.0) buffer to remove imidazole. If not used directly for reconstitution, protein samples were flash frozen and stored at −80 °C.
Preparation of proteoliposomes
E. coli polar lipids were transferred to a glass flask, and chloroform was removed by drying under nitrogen flow. After evaporation of chloroform, the lipid film was dried under vacuum for at least 1 hour, and then hydrated in buffer E (150 mM NaCl, 10 mM Tris (pH 8.0)) followed by 5× freeze–thaw cycles and, if not used directly, stored as aliquots at −80 °C. Before reconstitution, the suspension of liposomes was passed at least 11× through polycarbonate unipore membranes (Whatman) with a pore size of 400 nm or 200 nm using a Mini-Extruder Set (Avanti Polar Lipids, Alabaster, AL, USA).For reconstitution of the NpSRII/NpHtrII157 complex in liposomes, NpSRII and NpHtrII157 were mixed at a 1:1 molar ratio and then incubated for 20 min on ice followed by mixing with liposomes at a protein-to-lipid molar ratio of 1:172. The assembly mixture was subsequently incubated with Bio-Beads SM-2 (Bio-Rad Laboratories; Munich; Germany) (50 mg of beads/mg of detergent) pre-washed extensively with methanol and water. Finally, proteoliposomes were pelleted by centrifugation (15800 × g; 15 min; 4 °C) and resuspended in buffer E at a final lipid concentration of 20 mg ml−1.
Preparation of SMA lipid particles
SMA/lipid particles (SMALPs) containing the NpSRII/NpHtrII157 complex (in the following abbreviated as NpSRII/NpHtrII157-SMALPs) were assembled in vitro according to a protocol adapted from ref. 31. In the case of extrusion through the membrane with 400 nm pore size, the suspension of proteoliposomes was sonicated additionally for 30 min in a bath sonicator at room temperature. 5% (w/v) solution of SMA copolymer in buffer E was added dropwise over 2–3 min to the proteoliposome suspension to get a final lipid-to-SMA weight ratio of 1:2.5. The assembly mixture was allowed to equilibrate for 1 hour at room temperature and then for 16 h at 4 °C. The resulting samples were centrifuged (126000 × g; 30 min; 4 °C) to remove non-solubilized material. Subsequently, SMALP preparations were fractionated on a Superdex 200 10/200 GL column (GE Healthcare) using an ÄKTA Purifier FPLC system (GE Healthcare) with a flow rate of 0.5 ml min−1 at 4 °C.
Preparation of nanodiscs
Nanodiscs containing the NpSRII/NpHtrII157 complex were assembled in vitro according to ref. 26 using MSP1D1E3 as a scaffold protein. The assembly was performed in buffer E containing 5 mM DDM. NpSRII and NpHtrII157 were mixed at a 1:1 molar ratio and then incubated for 20 min on ice. Polar lipids from E. coli membranes and MSP1D1E3 were then added to the NpSRII/NpHtrII157 complex to give final concentrations of 0.76 mM lipids, 14 μM MSP1D1E3 and 7 μM NpSRII/NpHtrII157. After incubation for 1 h at room temperature, the assembly mixture was incubated with Bio-Beads SM-2 overnight at 4 °C. Non-solubilized proteins and any aggregated material were removed by centrifugation at 126000 × g for 15 min at 4 °C. Samples were stored at 4 °C until further use. Subsequently, preparations of nanodiscs were fractionated on a Superdex 200 10/200 GL column using an ÄKTA Purifier FPLC system, with a flow rate of 0.5 ml min−1 at 4 °C.
Site directed spin labeling
The spin label (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)methanethiosulfonate (MTSSL; Enzo life sciences, NY) was covalently bound to the cysteine residue of NpSRII-L159C as described previously.26 Excess label was extensively washed out by using a 10K Amicon Ultracel centrifugal filter device (Millipore, Schwalbach, Germany). In the following, the spin label side chain is abbreviated as R1.Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) at 550 nm and 25 °C. Data represent the average of three sets of 14 runs of 10 s each. The particle size distribution was obtained by using the ZETASIZER software package ver. 7.02 under the assumption that SMALPs were spherically shaped.Transmission electron microscopy
The transmission electron microscopy (TEM) images were obtained on a JEM-2100 (Jeol, Japan) transmission electron microscope operated at 200 kV. For TEM imaging, a solution of the SMALPs in 10 mM Tris (pH 8.0), 150 mM NaCl was deposited on Ted Pella grids treated with a glow discharge device Emitech K100X. A drop of solution was incubated for 1–2 minutes, then treated with 1% uranyl acetate for 1 minute and dried. The size of SMALPs was estimated using the ImageJ software.43Atomic force microscopy
The atomic force microscopy (AFM) images were obtained on a Solver PRO microscope (NT-MDT, Russian Federation) operating in semicontact mode in buffer and using MSCT-AUHW cantilevers. For AFM imaging, a solution of SMALPs was diluted to final NpSRII concentration of 15 μg ml−1 by 10 mM Tris (pH 8.0), 150 mM NaCl, 10 mM MgCl2 and deposited on freshly cleaved mica. The SMALP sample was incubated for 5 minutes, then mica was washed with 1 ml of the buffer and transferred to the AFM liquid cell and scanned. The obtained AFM images were processed with the FemtoScan Online software.Resonance Raman spectroscopy
Resonance Raman (RR) spectra were measured on a Nicolet Almega XR dispersive Raman spectrometer (Thermo Fisher Scientific, USA). Scattering was induced by excitation from the 532 nm second harmonic of a Nd-YAG laser. The laser beam was directed by a microscope (Olympus BX-51) objective (100×) to the sample. The diameter of the focused laser spot on the sample was about 0.6 μm. The laser power at the sample did not exceed 15 mW. Spectra were recorded in the range of 500–2000 cm−1 with a spectral resolution of 2 cm−1. Each spectrum was obtained by averaging 20 exposures for 2 seconds each. Samples were prepared by drying approximately 2 μl of the reconstituted NpSRII/NpHtrII157 complex (0.8 mg ml−1 NpSRII) in 10 mM Tris (pH 8.0), 150 mM NaCl on a SERS (Surface Enhanced Raman Scattering) substrate consisting of carbon nanowalls (CNWs) covered with a layer of gold. CNWs were prepared by plasma-enhanced chemical vapour deposition (PECVD) synthesis according to ref. 44.Electron paramagnetic resonance spectroscopy
Continuous wave (cw) electron paramagnetic resonance (EPR) measurements were performed as described previously.45 Briefly, cw EPR spectra were recorded at room temperature with a home-made EPR spectrometer equipped with a Bruker dielectric resonator (Bruker Biospin; Germany). The microwave power was set to 1.0 mW, the B-field modulation amplitude was adjusted to 0.15 mT, and the sample temperature was stabilized by a gas stream through the resonator to 297 ± 1 K. Samples were loaded into EPR glass capillaries (0.9 mm inner diameter) at final protein concentrations of 15–20 μM (sample volume of 15 μl).For pulsed EPR experiments, samples were concentrated to ∼100 μM, loaded into EPR quartz capillaries (3 mm outer diameter; sample volume of 40 μl) and frozen with 20% glycerol. The spin labeling efficiency determined by cw EPR and absorption spectroscopy was about 75%. Double electron–electron resonance (DEER) experiments were accomplished at X-band frequencies (9.3–9.4 GHz) with a Bruker Elexsys 580 spectrometer equipped with a Bruker Flexline split-ring resonator ER 4118X-MS3 and a continuous flow helium cryostat (CF935; Oxford Instruments) controlled by an Oxford Intelligent Temperature Controller ITC 503S. Measurements were performed using the four-pulse DEER sequence.46,47
| π/2(νobs) − τ1 − π(νobs) − τ′ − π(νpump) − (τ1 + τ2 − τ′) − π(νobs) − τ2 − echo. |
A two-step phase cycling (+〈x〉, −〈x〉) was performed on π/2(νobs). Time τ′ is varied, whereas τ1 and τ2 are kept constant. The dipolar evolution time is given by t = τ′ − τ1. Data were analysed only for t > 0. The resonator was overcoupled and the pump frequency νpump was set to the center of the resonator dip and coincided with the maximum of the nitroxide EPR spectrum, whereas the observer frequency νobs was ∼67 MHz higher (coinciding with the low field local maximum of the spectrum). All measurements were performed at a temperature of 50 K with observer pulse lengths of 16 ns for π/2 and 32 ns for π pulses and a pump pulse length of 12 ns. Proton modulation was averaged by adding traces at eight different τ1 values, starting at τ1,0 = 200 ns and incrementing by Δτ1 = 8 ns. Data points were collected in 8 ns time steps. The total measurement time for each sample was ∼24 h. Analysis of the data was performed with DeerAnalysis 2013.38,48
Rotamer analysis
Spin label conformations were calculated utilizing the software package MtsslWizard version 1.3,49 which is a plugin for the PyMOL molecular graphics system. The “tether-in-a-cone” approach50–52 was used which means that the program scans for all possible spin label rotamer conformations that do not clash with the given protein structure or with a label itself within a variable tolerance. The following settings were used: label – MTSSL; speed – painstaking search; vdW restraints – loose. In the case of the crystal structure published by V. Gordeliy et al.1 rotamer analysis was performed based on the relaxed structure19 of a dimer of dimers with the transducer truncated at position 157. In process of equilibration, the structure becomes unsymmetrical particularly due to asymmetry of the HAMP-domain (residues ∼ 131–157), which leads to differences in the sets of rotamers from each NpSRII molecule in the complex.Results
We utilized the SMA copolymer to isolate a heterogeneous complex of functionally and structurally interacting membrane proteins, NpSRII/NpHtrII, from proteoliposomes without the use of detergent. The resulting nanosized lipoprotein-containing SMA particles were subsequently characterized by a set of biophysical methods.We first reconstituted the purified detergent-solubilized NpSRII/NpHtrII157 complex in preformed liposomes of E. coli membrane polar lipids. Addition of the 3:1 SMA polymer at a lipid-to-SMA weight ratio of 1:2.5 to the suspension of the proteoliposomes obtained was followed by incubation for 1 hour at room temperature and then for 16 h at 4 °C. The concentration of 2.5% (w/v) SMA used was previously reported to be efficient in solubilization of DMPC liposomes.53 The resulting SMALP preparations were fractionated by size-exclusion chromatography (SEC). An exemplary SEC purification profile of assembled SMALP particles (Fig. 1A) shows a major peak with the presence of a minor peak in the void volume corresponding to residual aggregated species. The major peak has an absorption maximum at ∼500 nm, characteristic for the NpSRII bound retinal chromophore, which coincides with the absorption maximum of the detergent-solubilized NpSRII (Fig. 1B). Fractions from the major peak were pooled and further analysed by dynamic light scattering (DLS) at 550 nm and 25 °C to assess the average size of assembled SMALP particles (Fig. 1C) (see Materials and methods). The intensity-weighted particle diameter was estimated to be in the 10 nm range consistent with previously reported data.28,30,31,54 The DLS data indicate the monodisperse character of the SMALP preparations. Thus, incubation of NpSRII/NpHtrII157-liposomes with 3:1 SMA polymer resulted in formation of protein-containing SMA particles.
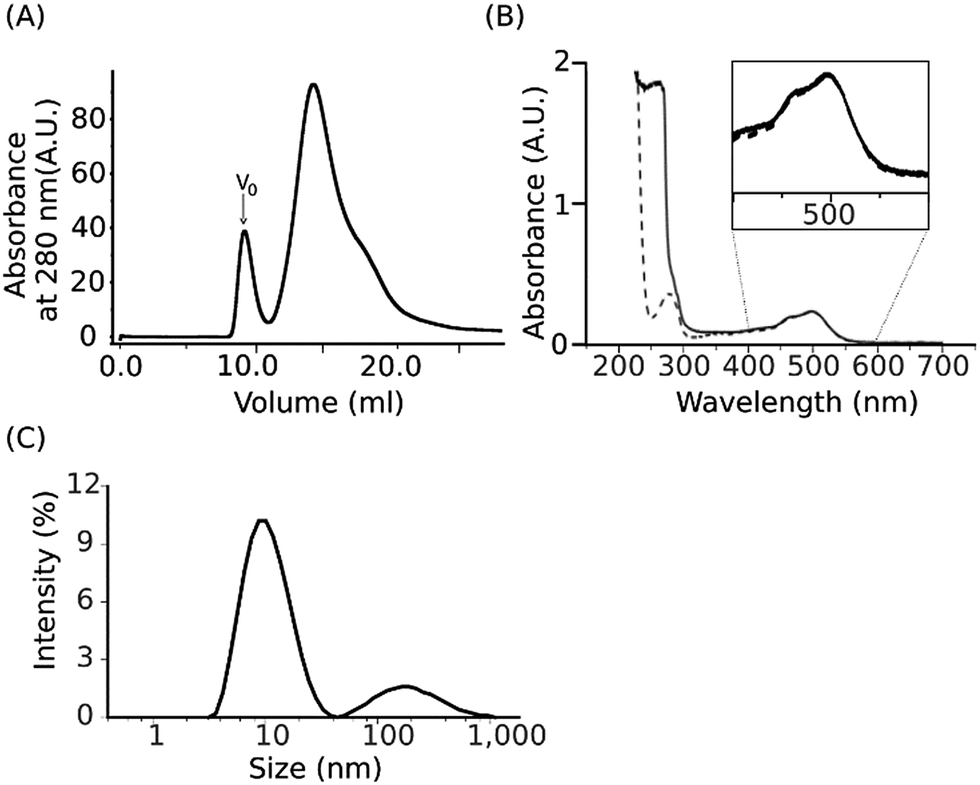 | ||
| Fig. 1 Physical characterization of the NpSRII/NpHtrII157 complex upon solubilization with the 3:1 SMA copolymer. (A) Size-exclusion chromatography purification profile of SMALP preparations. V0 indicates the void volume. (B) Absorption spectrum for the major SEC peak (solid line) and the detergent-solubilized NpSRII/NpHtrII157 complex (dashed line), respectively. (C) Size distribution based on DLS data obtained for the major SEC peak. | ||
Transmission electron microscopy imaging
Visualization of individual SMALPs deposited on the solid support was performed by transmission electron microscopy (TEM) imaging of negatively stained SMALPs. TEM images of SMALP preparations revealed monodisperse nanosized particles with well-defined edges (Fig. 2A). Measurements of 200 individual SMALPs (each particle measured twice) shows that their majority is 12–14 nm in diameter (Fig. 2B) in agreement with results obtained by other authors.31,55 These dimensions are larger than those detected by DLS (Fig. 1C) which may be due to the negative staining for TEM imaging.30,56 In addition, different physical characteristics were measured by DLS and TEM, namely the intensity-weighted diameter in solution versus the size of the projected area of the surface-adsorbed SMALPs.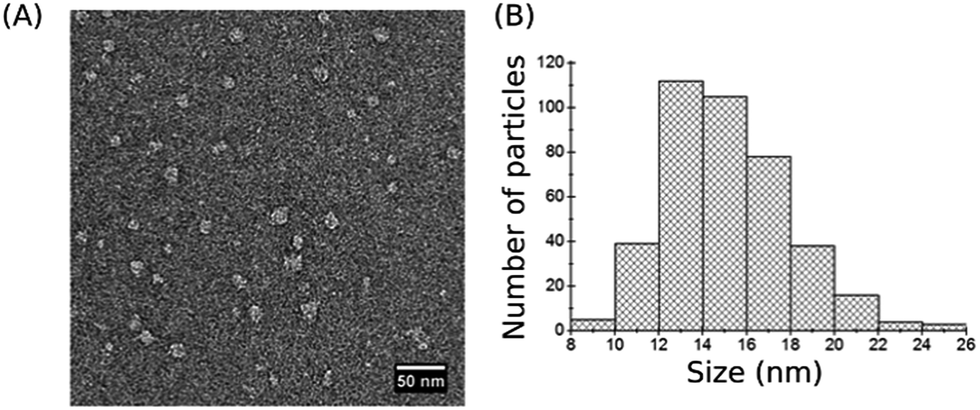 | ||
| Fig. 2 TEM imaging. (A) Negative-stain TEM image of assembled SMALP particles. (B) Size distribution based on TEM data. | ||
Atomic force microscopy imaging
We used atomic force microscopy (AFM) to estimate directly the thickness of assembled SMALPs under conditions similar to those that were shown to be suitable for nanodiscs.42,57,58 The AFM images of SMALPs were obtained in semicontact mode; the results are shown in Fig. 3. The mean height of SMALPs is 4.0 nm which is slightly less than the expected lipid bilayer thickness of about 4.0–5.0 nm. Seemingly, SMALPs either were flattened at the surface and/or compressed by the tip–sample interaction force. Both effects would decrease the measured height. The lateral radius of the SMALPs measured by AFM exceeded 23 nm, which is due to the well-known tip broadening effect of the silicon nitride cantilevers used with a curvature radius of up to 20 nm. The tip broadening effect could be decreased in future measurements by using tips with 1–3 nm radius.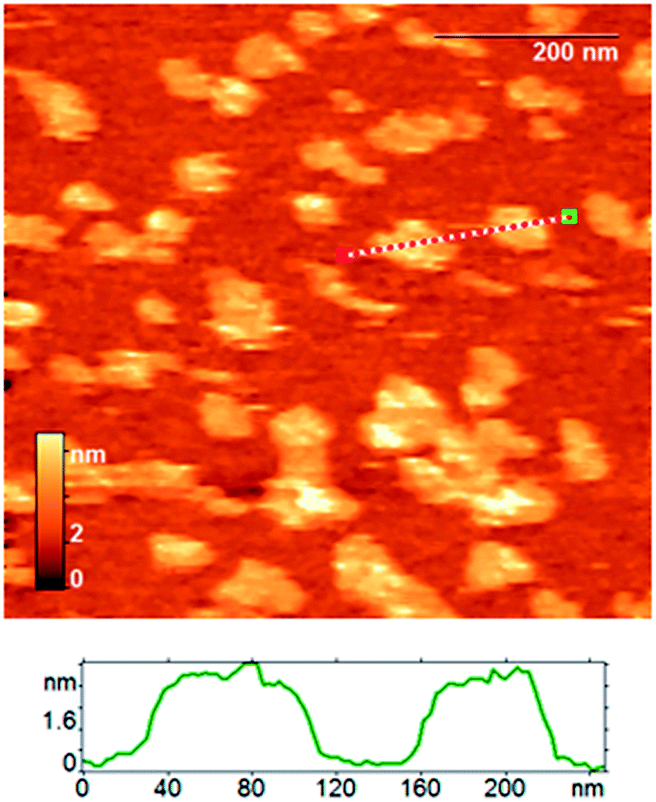 | ||
| Fig. 3 The AFM height image of assembled SMALP particles; an exemplary section profile is shown below. | ||
Resonance Raman spectroscopy
Resonance Raman (RR) spectroscopy is a suitable technique to study the configuration of the retinal chromophore in the protein. RR scattering enables to selectively probe the retinal vibrations because the retinal bands are selectively enhanced due to the resonance effect while the vibrational contributions of the apoprotein are negligible. Fig. 4 shows the RR spectrum of NpSRII/NpHtrII157-SMALPs (blue line). To define the possible contributions of empty SMALPs, their RR spectrum (red line) recorded under identical conditions is also shown, being very similar to the known spectra of SMA films.59 Vibrational bands of the RR spectrum of empty SMALPs clearly contribute to the RR spectrum of NpSRII/NpHtrII157-SMALPs, but still enable analysis of crucial regions of the retinal spectrum. The most prominent peak at 1541 cm−1 corresponds to the ethylenic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching vibration, νCC, of retinal. The high frequency of this band correlates with the absorption maximum, λmax, of 497 nm in the visible range based on the well-established correlation of νCC and λmax.60 The fingerprint region between 1100 and 1300 cm−1 with the predominant absorption of the C–C single bond stretches is known to display the conformation of the retinal polyene chain depending on the isomeric state of the retinal. The corresponding band pattern of NpSRII/NpHtrII157-SMALPs is very similar to that of detergent-solubilized NpSRII61 and lipid-reconstituted SRII from the halophilic archaeon Halobacterium salinarum62 (see Table 1). The characteristic peaks at 1273 cm−1, 1200 cm−1 and 1170 cm−1 are similar to those of parent states of bacteriorhodopsin and halorhodopsin, which include an all-trans retinal protonated Schiff base63 (see Table 1). The pronounced band at 1200 cm−1 characteristic for C14–C15 stretch vibrations and the band at 1170 cm−1 originating mainly from the C10–C11 stretch vibrations64 confirm the predominance of the all-trans retinal conformation. The occurrence of the weak peak at 1182 cm−1 indicates the presence of low amounts of 13-cis retinal.65 Thus, the present RR data confirm that reconstituted NpSRII retains its light-sensitive retinal chromophore, which functional configuration is not affected upon the SMA-mediated extraction of the NpSRII/NpHtrII157 complex.
C) stretching vibration, νCC, of retinal. The high frequency of this band correlates with the absorption maximum, λmax, of 497 nm in the visible range based on the well-established correlation of νCC and λmax.60 The fingerprint region between 1100 and 1300 cm−1 with the predominant absorption of the C–C single bond stretches is known to display the conformation of the retinal polyene chain depending on the isomeric state of the retinal. The corresponding band pattern of NpSRII/NpHtrII157-SMALPs is very similar to that of detergent-solubilized NpSRII61 and lipid-reconstituted SRII from the halophilic archaeon Halobacterium salinarum62 (see Table 1). The characteristic peaks at 1273 cm−1, 1200 cm−1 and 1170 cm−1 are similar to those of parent states of bacteriorhodopsin and halorhodopsin, which include an all-trans retinal protonated Schiff base63 (see Table 1). The pronounced band at 1200 cm−1 characteristic for C14–C15 stretch vibrations and the band at 1170 cm−1 originating mainly from the C10–C11 stretch vibrations64 confirm the predominance of the all-trans retinal conformation. The occurrence of the weak peak at 1182 cm−1 indicates the presence of low amounts of 13-cis retinal.65 Thus, the present RR data confirm that reconstituted NpSRII retains its light-sensitive retinal chromophore, which functional configuration is not affected upon the SMA-mediated extraction of the NpSRII/NpHtrII157 complex.
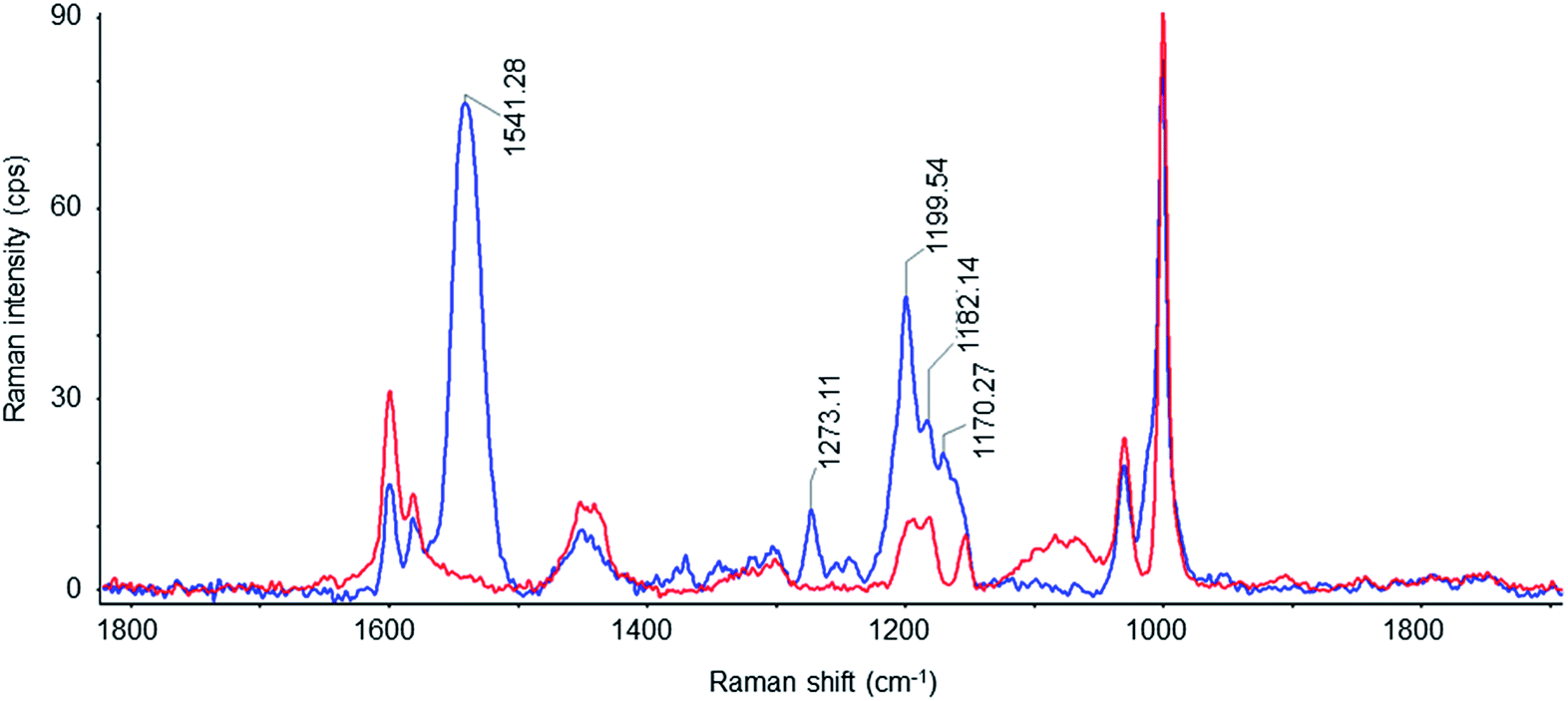 | ||
| Fig. 4 Resonance Raman spectra from empty SMALPs (red) and SMALPs loaded with the NpSRII/NpHtrII157 complex (blue) measured with 532 nm excitation. | ||
| Rhodopsin | CC stretch, cm−1 |
C14–C15 stretch, cm−1 | C10–C11 stretch, cm−1 | CH rocking, cm−1 | In-plane C–CH3 rocking, cm−1 |
|---|---|---|---|---|---|
| a In complex with NpHtrII in SMALPs.b From Natronomonas pharaonis, detergent-solubilized.c From Halobacterium salinarum (Hs), lipid-reconstituted.d From Hs, lipid-reconstituted.e From Hs, detergent-solubilized. | |||||
| NpSRIIa (here) | 1541 | 1200 | 1170 | 1273 | ∼1000 |
| SRIIb,61 | 1548 | 1202 | 1274 | 1012 | |
| SRIIc,62 | 1550 | 1200 | 1162 | 1273 | 1009 |
| BR570d,63 | 1529 | 1200 | 1169 | 1273 | 1008 |
| HR578e,63 | 1522 | 1200 | 1170 | 1272 | 1005 |
Electron paramagnetic resonance spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy is a powerful method to study structural details and conformational transitions of membrane proteins in lipid environments.66,67 Continuous wave (cw) EPR spectroscopy allows to probe the spin label side chain dynamics and can provide detailed information on the conformation and conformational dynamics of spin labeled membrane proteins embedded in lipid bilayers.13 Pulse double electron–electron resonance (DEER) enables measuring interspin distances in the range of 1.5–8 nm in site-directed spin labeled proteins, that provide valuable information to characterize the protein structure and structural changes.68,69Continuous wave electron paramagnetic resonance spectroscopy
The mutant of NpSRII, NpSRII-L159R1 (Fig. 5A), carrying the commonly used nitroxide spin label (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)methanethiosulfonate (MTSSL, side chain R1) at position 159, was chosen for EPR measurements. The spin label side chain of NpSRII-L159R1 resides at the cytoplasmic end of helix F outside of the membrane. NpSRII-L159R1 has been shown to provide a sensitive probe for EPR analyses of conformational changes involved in the light induced signal transfer from NpSRII to NpHtrII.13 Due to this property, NpSRII-L159R1 is utilized here to trace the possible impact of the nanoparticle environment on the NpSRII/NpHtrII complex conformation and dynamics. To monitor the spin-label dynamics by room temperature (RT) cw EPR measurements, NpSRII-L159R1 was reconstituted alone or in complex with its cognate transducer NpHtrII157, in SMALPs and, for comparison, in liposomes and nanodiscs. The spectra of the L159R1 side chains in the different lipid environments (Fig. 5B) are almost identical revealing a powder-like spectral pattern with a resolved hyperfine line in the high field region of the spectra. Moreover, these spectra are very similar to that of the NpSRII-L159R1/NpHtrII157 complex reconstituted in native purple membrane lipids (PML).13 The presence of the transducer NpHtrII157 does not influence the behaviour of L159R1 (not shown). The spectral shape reveals restriction of the side chain residual motion due to strong sterical interaction between the nitroxide and neighbouring residues of the protein in agreement with the inward facing orientation of the spin label binding site (see Fig. 5A and B). Hence, the nitroxide dynamics is not affected significantly by changes of the lipid environment.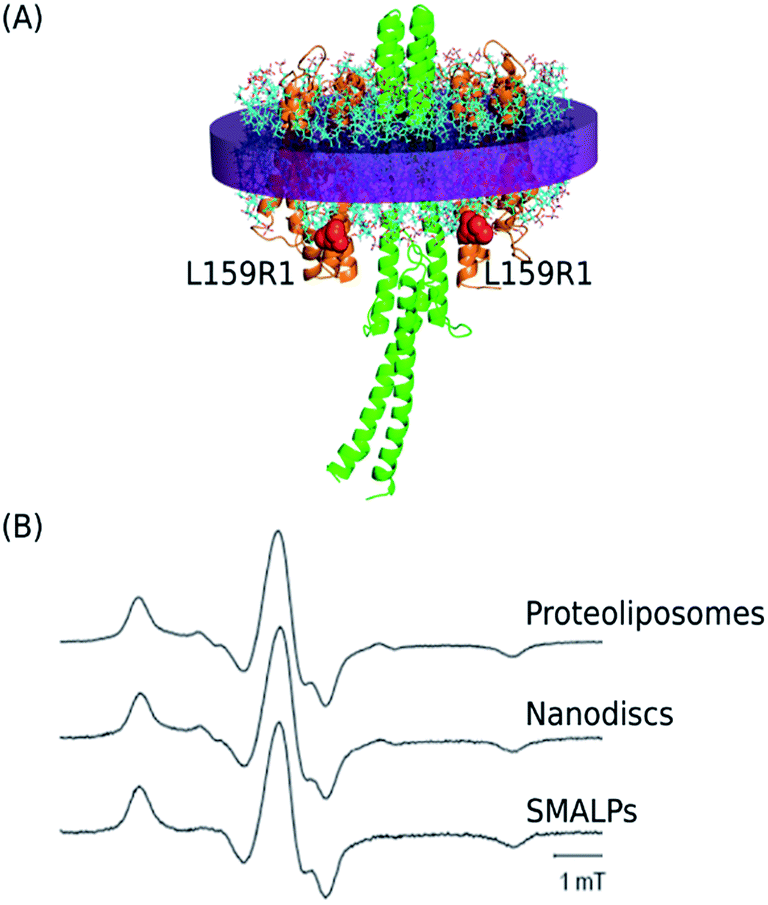 | ||
| Fig. 5 (A) Side view of the 2:2 NpSRII/NpHtrII157 complex (based on the structural model19 where the intermembrane part corresponds to the crystal structure (PDB: 1H2S)1). The spin labeled residue L159C is depicted in red. (B) cw EPR spectra revealing the spin label mobility for NpSRII-L159R1/NpHtrII157 after reconstitution in proteoliposomes, nanodiscs, and SMALPs. All spectra are normalized to maximum amplitude. | ||
Pulse electron paramagnetic resonance spectroscopy
Four-pulse double electron–electron resonance (DEER) measurements were performed on NpSRII-L159R1 in complex with NpHtrII157 to determine the distribution of distances between the nitroxides in the complex. The DEER data for the complex reconstituted in SMALPs (depicted in blue) or nanodiscs (depicted in red) are shown in Fig. 6. For both SMALP and nanodisc samples, the interspin distance distribution range accessible in the present experiment is between ∼1.5 and 5 nm. The DEER interspin distance distributions for NpSRII-L159R1/NpHtrII157 reconstituted in SMALPs are characterized by three maxima located at ∼2.3, 3.7, and 4.8 nm. These results are similar to the DEER data for the complex in nanodiscs, with maxima at ∼2.1, 3.6, and 4.8 nm, indicating occupation of multiple spin label side chain orientations in both cases (Fig. 6E). In nanodiscs these maxima are superimposed to a broad population density ranging from 3.6 nm down to below 2 nm. Since the minimal interspin distance in the NpSRII-L159R1/NpHtrII157 complex cannot fall below 2 nm due to sterical reasons (see below), we have to conclude that these contributions originate from a fraction of nanodiscs containing only NpSRII-L159R1 dimers.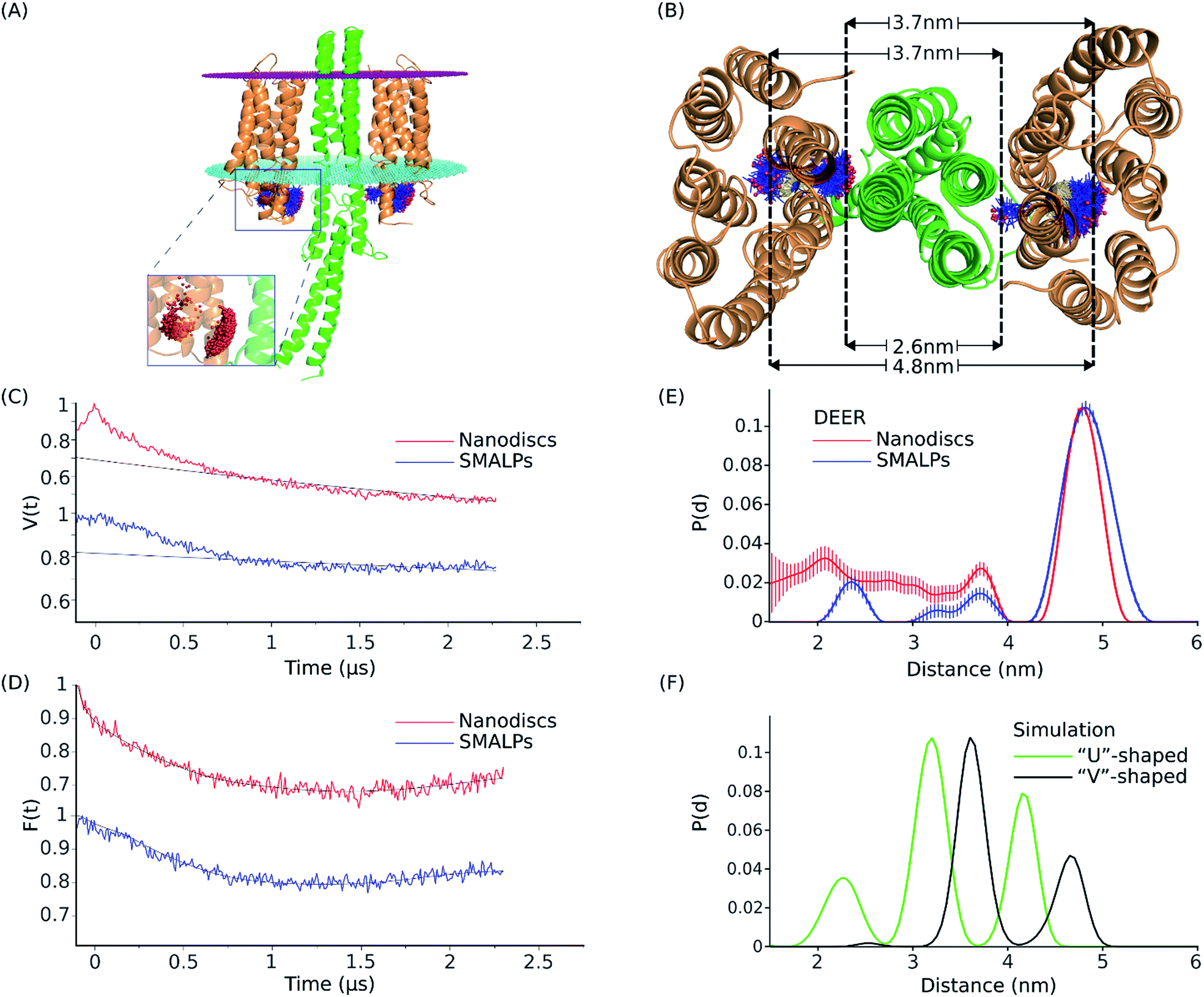 | ||
| Fig. 6 Structural model, DEER data and distance distribution for the NpSRII-L159R1/NpHtrII157 complex. (A) Equilibrated model of the NpSRII-L159R1/NpHtrII157 complex in “V”-shape conformation based on the crystal structure PDB: 1H2S.1,19 Side view of the protein complex with the membrane borders shown as magenta (extracellular side) and cyan (cytoplasmic side) dotted plates. Spin label conformations resulting from MtsslWizard simulation at the position L159 of NpSRII (orange) are shown as blue sticks with red spheres as nitroxide spin centres. Spin centres are shown more detailed in the zoomed part of the figure. (B) View from the cytoplasm of the model described in (A), where a set of three distance values for spin label rotamers, as predicted by in silico modeling for a single 2:2 NpSRII/NpHtrII157 dimer, is shown. Dashed lines show the approximate position of the maximum density of the spin centres and help to visualize their possible occurrence in the distance distribution data. (C) DEER data for the NpSRII-L159R1/NpHtrII157 complex reconstituted into SMALPs (blue) and nanodiscs (red). Experimental time domain data, V(t), superimposed with the corresponding background fits (black). (D) Background-corrected and normalized form factors, F(t), overlaid with the corresponding fits (black) calculated by Tikhonov regularization using DeerAnalysis.48. (E) Interspin distance distributions, normalized to unity, obtained by Tikhonov regularization using DeerAnalysis48 of the corresponding data shown in (D). (F) Normalized distance distributions obtained by MtsslWizard simulations for the computed model19 based on the “V”-shaped conformation (PDB: 1H2S)1 (black), and for the “U”-shaped conformation (PDB: 5JJE)66 (green). | ||
Rotamer analysis
To compare experimentally determined interspin distances with currently available quaternary structures,1,70 we performed in silico spin-labeling of two available crystal structures of NpSRII in complex with the membrane domain of NpHtrII followed by simulation of the expected interspin distances for the single 2:2 NpSRII-L159R1/NpHtrII dimer (see Materials and methods). Crystal structures of NpSRII in complex with truncated transducers in the ground state have been determined in the space groups P21212 and I212121.1,70 In both cases X-ray diffraction data showed electron density only for the transmembrane domain of the transducer but not for its adjacent HAMP domain. Although the structures of the rhodopsin and transducer monomers taken individually are nearly the same, the relative orientations of symmetrical parts of the two NpSRII proteins have been found to be parallel, “U”-shaped, in the space group I212121 (PDB: 5JJE), and gusset-like, “V”-shaped, in the space group P21212 (PDB: 1H2S). It has been suggested that the transition between “V”- and “U”-shaped conformations could be involved in signal transduction from the receptor to the signalling domain of NpHtrII.70 Here, we simulated interspin distances between spin labeled sites L159R1 for models based on both of these crystal structures. For the “V”-shaped conformation the transducer was complemented by homology modeling of the first HAMP domain and its attachment to the resolved membrane domain of NpHtrII.19 For the “U”-shaped conformation such data were not available so that the interspin calculations were performed in the absence of the HAMP domain. The results are shown in Fig. 6A, B and F, in comparison to the experimental interspin distance distributions presented in Fig. 6E. The calculated intradimer distance distribution reveals three maxima at ∼2.6, 3.7 and 4.8 nm for the “V”-shaped conformation (Fig. 6E). According to the structural model shown in Fig. 6B, the minimum distance value of ∼2.6 corresponds to the distance between spin label rotamers pointing to the center of the complex. The intermediate value of ∼3.7 is a combination of the distances between the “inward” and the “outward” rotamers (or vice versa) of the two NpSRII molecules in the complex and the maximum value of ∼4.8 marks the distance between rotamers oriented away from the center of the complex. For the “U”-shaped structure (PDB: 5JJE), the three respective maxima are found to be at ∼2.3, 3.2 and 4.2 nm. Interestingly, all three corresponding distances values predicted for the “U”-shaped structure are shifted to lower values compared to the respective values for the “V”-shaped conformation revealing the more compact structure of the complex and facilitating clear discrimination between these two conformations.
The set of three distance peak values as predicted by in silico modeling for the “V”-shaped conformation agrees well with the experimental interspin distance distribution peaks obtained for both SMALPs and nanodiscs. However, the peak amplitudes resulting from the experiment and the simulation are different. The DEER experiment suggests the outward orientation of the spin label side chains to be dominantly populated, whereas in the model the probability for inward orientation of at least one of the spin labels is high leading to the large peak at 3.7 nm.
All these data indicate that the NpSRII/NpHtrII complex in SMALPs forms the two-dimer-of-dimers structure similar to that found in nanodiscs26 with the complex in the “V”-shaped conformation. Hence, the investigated SMALPs (see Fig. 6E) provide enough space for reconstitution of a single 2:2 NpSRII/NpHtrII dimer.
Discussion
A variety of processes in cell membranes is triggered by interactions of membrane proteins. To investigate such interactions in detail, the complexes of interacting proteins need to be isolated in their entirety and studied in a model membrane system. Detergents usually used for the solubilization and isolation of membrane proteins could negatively influence their stability and functionality. SMA copolymers have been shown to enable a direct detergent-free extraction of membrane proteins or their complexes together with patches of lipid bilayer yielding stable nanocontainers, in which lipid-encased membrane proteins could be studied.27,30–34,71The archaeal photoreceptor/transducer complex, NpSRII/NpHtrII, provides a unique model system to study structural interactions between different integral membrane proteins and their conformational changes during the transmembrane signal transfer.11,72,73 Our study demonstrated the ability of the SMA polymer to extract the NpSRII/NpHtrII157 complex from proteoliposomes in a detergent-free manner as has been shown for other membrane protein complexes.27,33,34 The EPR spectra and DEER measurements revealed that the structural integrity of the NpSRII/NpHtrII157 complex reconstituted in SMALPs was preserved. SEC confirmed the stability of SMALP preparations. DLS measurements indicated the average particle size of assembled SMALPs to be 10 nm, which corresponds to reported DLS values.30,53,55,74,75 TEM images showed the individual SMALPs as monodisperse particles with well-defined shape. The lateral size was in the range of 12–14 nm, which is consistent with reported data of 10–15 nm obtained by TEM.27,30,32,76,77 The mean height, as measured by AFM is slightly less than the value of 4.6 nm obtained by small-angle neutron scattering experiments on SMALPs derived from DMPC vesicles.55 Such slight discrepancy can be attributed to the technical limitations of our AFM experiments.
Further, we applied spectroscopic methods for a more detailed structural characterization of SMALPs containing NpSRII/NpHtrII157. RR spectroscopy enabled to characterize the conformation of the retinal chromophore in NpSRII/NpHtrII157-SMALPs due to the resonance enhancement of characteristic retinal chromophore bands that provide molecular fingerprinting. The RR spectroscopic data clearly indicated that all-trans retinal is the predominant isomer in reconstituted NpSRII. Thus, reconstituted NpSRII retains its light-sensitive retinal prosthetic group with its functional conformation not affected upon the SMA-mediated extraction of the NpSRII/NpHtrII157 complex.
We used cw EPR spectroscopy to analyse the protein dynamics through tracking the residual motion of spin labeled side chains. cw EPR spectra of NpSRII-L159R1/NpHtrII157-SMALP samples revealed that SMALPs generally maintained the dynamic features of the reconstituted NpSRII/NpHtrII157 complex where the behaviour of the NpSRII-L159R1 side chain, either in complex with NpHtrII157 or without the transducer, was similar compared to that reconstituted in liposomes or nanodiscs. Hence, the conformation and dynamics of the NpSRII molecule in the vicinity of the functionally important helix F was not notably affected by the SMA polymer.
The DEER measurements showed that the interspin distance distribution in the SMALP-reconstituted NpSRII-L159R1/NpHtrII complexes was similar to that measured with nanodiscs, which contained 2:2 NpSRII/NpHtrII dimers.26 The positions of the interspin distance peaks agree with results of in silico modeling of a single 2:2 NpSRII/NpHtrII dimer for the “V”-shaped conformation.1 The difference in the peak amplitudes between simulation and experiment may indicate a more densely packed conformation of the NpSRII/NpHtrII-SMALP complex at the receptor-transducer interface, which renders inward orientations of the spin label side chain less probable. Hence, the DEER data indicate that NpSRII/NpHtrII complexes incorporate into SMALPs as 2:2 dimers with the distances between L159R1 sites according to the “V”-shaped conformation.1
Conclusions
We have demonstrated that the NpSRII/NpHtrII157 complex can be extracted from proteoliposomes in a detergent-free manner using the 3:1 SMA copolymer. The presented data indicate the integrity of this complex upon its reconstitution in SMA/lipid particles. Therefore, SMALPs that contain the NpSRII/NpHtrII complex can provide a valuable model system for studying the molecular events underlying the consecutive steps in receptor–transducer signalling. Time resolved analyses of light induced conformational changes of this system are currently performed.
Author contribution
N. V., A. Y. M., K. V. S. and H. J. S. designed the study. N. V. expressed and purified proteins for assays, developed a protocol for the preparation of NpSRII/NpHtrII157-nanodiscs, prepared samples of NpSRII/NpHtrII157-SMALPs, contributed to the TEM, AFM and RR spectroscopy experiments, and drafted the manuscript. W. M. developed a protocol for the preparation of NpSRII/NpHtrII157-SMALPs, prepared samples of NpSRII/NpHtrII157-containing liposomes, performed the cw and pulsed EPR experiments and wrote the respective sections of the manuscript. A. C. performed structural modeling, carried out pulsed EPR experiments and wrote the respective sections of the manuscript. D. B., T. I., M. P. K. and K. V. S. performed the TEM and AFM experiments, and wrote the respective sections of the manuscript. A. A., V. B. and P. T. performed the resonance Raman spectroscopy experiments and wrote the respective sections of the manuscript. H. J. S. conceived, supervised and directed the project, analysed data, and wrote the final version of the manuscript. All authors read, edited and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by German Research Foundation (DFG, STE640/14), the Federal Ministry of Education and Research of Germany (BMBF, 05K14MPA), the German Academic Exchange Service (DAAD) and the grants from the Russian Science Foundation (14-50-00029 (K. S.)) and the Russian Foundation for Basic Research (RFBR) (Project No. 15-54-12385 (K. S.), 16-02-00248 (P. T.)). We thank J. P. Klare for helpful discussions.Notes and references
- V. I. Gordeliy, J. Labahn, R. Moukhametzianov, R. Efremov, J. Granzin, R. Schlesinger, G. Buldt, T. Savopol, A. J. Scheidig, J. P. Klare and M. Engelhard, Nature, 2002, 419, 484–487 CrossRef CAS PubMed.
- A. Royant, P. Nollert, K. Edman, R. Neutze, E. M. Landau, E. Pebay-Peyroula and J. Navarro, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10131–10136 CrossRef CAS PubMed.
- H. Luecke, B. Schobert, J. K. Lanyi, E. N. Spudich and J. L. Spudich, Science, 2001, 293, 1499–1503 CrossRef CAS PubMed.
- M. Grote, M. Engelhard and P. Hegemann, Biochim. Biophys. Acta, 2014, 1837, 533–545 CrossRef CAS PubMed.
- D. N. Shalaeva, M. Y. Galperin and A. Y. Mulkidjanian, Biol. Direct, 2015, 10, 63 CrossRef PubMed.
- J. P. Klare, V. I. Gordeliy, J. Labahn, G. Buldt, H. J. Steinhoff and M. Engelhard, FEBS Lett., 2004, 564, 219–224 CrossRef CAS PubMed.
- A. A. Wegener, I. Chizhov, M. Engelhard and H. J. Steinhoff, J. Mol. Biol., 2000, 301, 881–891 CrossRef CAS PubMed.
- C. S. Yang and J. L. Spudich, Biochemistry, 2001, 40, 14207–14214 CrossRef CAS PubMed.
- A. A. Wegener, J. P. Klare, M. Engelhard and H. J. Steinhoff, EMBO J., 2001, 20, 5312–5319 CrossRef CAS PubMed.
- K. Wuichet and I. B. Zhulin, Sci. Signaling, 2010, 3, ra50 CrossRef PubMed.
- J. P. Klare, E. Bordignon, M. Engelhard and H. J. Steinhoff, Eur. J. Cell Biol., 2011, 90, 731–739 CrossRef CAS PubMed.
- J. P. Klare, E. Bordignon, M. Engelhard and H. J. Steinhoff, Photochem. Photobiol. Sci., 2004, 3, 543–547 CAS.
- E. Bordignon, J. P. Klare, J. Holterhues, S. Martell, A. Krasnaberski, M. Engelhard and H. J. Steinhoff, Photochem. Photobiol., 2007, 83, 263–272 CrossRef CAS PubMed.
- R. Moukhametzianov, J. P. Klare, R. Efremov, C. Baeken, A. Goppner, J. Labahn, M. Engelhard, G. Buldt and V. I. Gordeliy, Nature, 2006, 440, 115–119 CrossRef CAS PubMed.
- A. Ishchenko, E. Round, V. Borshchevskiy, S. Grudinin, I. Gushchin, J. P. Klare, T. Balandin, A. Remeeva, M. Engelhard, G. Buldt and V. Gordeliy, J. Photochem. Photobiol., B, 2013, 123, 55–58 CrossRef CAS PubMed.
- J. J. Falke and G. L. Hazelbauer, Trends Biochem. Sci., 2001, 26, 257–265 CrossRef CAS PubMed.
- K. M. Ottemann, W. Z. Xiao, Y. K. Shin and D. E. Koshland, Science, 1999, 285, 1751–1754 CrossRef CAS PubMed.
- D. Q. Yu, X. M. Ma, Y. H. Tu and L. H. Lai, Sci. Rep., 2015, 5, 8640 CrossRef CAS PubMed.
- P. S. Orekhov, D. Klose, A. Y. Mulkidjanian, K. V. Shaitan, M. Engelhard, J. P. Klare and H. J. Steinhoff, PLoS Comput. Biol., 2015, 11, e1004561 Search PubMed.
- W. D. Hoff, K. H. Jung and J. L. Spudich, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 223–258 CrossRef CAS PubMed.
- J. L. Spudich, C. S. Yang, K. H. Jung and E. N. Spudich, Annu. Rev. Cell Dev. Biol., 2000, 16, 365–392 CrossRef CAS PubMed.
- J. P. Klare, I. Chizhov and M. Engelhard, Results Probl. Cell Differ., 2008, 45, 73–122 CAS.
- I. P. Hohenfeld, A. A. Wegener and M. Engelhard, FEBS Lett., 1999, 442, 198–202 CrossRef CAS PubMed.
- P. Orekhov, A. Bothe, H. J. Steinhoff, K. V. Shaitan, S. Raunser, D. Fotiadis, R. Schlesinger, J. P. Klare and M. Engelhard, Photochem. Photobiol., 2017, 93, 796–804 CrossRef CAS PubMed.
- J. J. Falke and K. N. Piasta, Curr. Opin. Struct. Biol., 2014, 29, 85–94 CrossRef CAS PubMed.
- I. Orban-Glass, N. Voskoboynikova, K. B. Busch, D. Klose, C. Rickert, W. Mosslehy, F. Roder, V. Wilkens, J. Piehler, M. Engelhard, H. J. Steinhoff and J. P. Klare, Biochemistry, 2015, 54, 349–362 CrossRef CAS PubMed.
- A. R. Long, C. C. O'Brien, K. Malhotra, C. T. Schwall, A. D. Albert, A. Watts and N. N. Alder, BMC Biotechnol., 2013, 13, 41 CrossRef CAS PubMed.
- J. M. Dorr, S. Scheidelaar, M. C. Koorengevel, J. J. Dominguez, M. Schafer, C. A. van Walree and J. A. Killian, Eur. Biophys. J., 2016, 45, 3–21 CrossRef PubMed.
- S. C. Lee and N. L. Pollock, Biochem. Soc. Trans., 2016, 44, 1011–1018 CrossRef CAS PubMed.
- T. J. Knowles, R. Finka, C. Smith, Y. P. Lin, T. Dafforn and M. Overduin, J. Am. Chem. Soc., 2009, 131, 7484–7485 CrossRef CAS PubMed.
- M. Orwick-Rydmark, J. E. Lovett, A. Graziadei, L. Lindholm, M. R. Hicks and A. Watts, Nano Lett., 2012, 12, 4687–4692 CrossRef CAS PubMed.
- D. J. Swainsbury, S. Scheidelaar, R. van Grondelle, J. A. Killian and M. R. Jones, Angew. Chem., Int. Ed. Engl., 2014, 53, 11803–11807 CrossRef CAS PubMed.
- J. M. Dorr, M. C. Koorengevel, M. Schafer, A. V. Prokofyev, S. Scheidelaar, E. A. van der Cruijsen, T. R. Dafforn, M. Baldus and J. A. Killian, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18607–18612 CrossRef PubMed.
- A. J. Bell, L. K. Frankel and T. M. Bricker, J. Biol. Chem., 2015, 290, 18429–18437 CrossRef CAS PubMed.
- R. C. Arenas, J. Klingler, C. Vargas and S. Keller, Nanoscale, 2016, 8, 15016–15026 RSC.
- J. J. Dominguez Pardo, J. M. Dorr, A. Iyer, R. C. Cox, S. Scheidelaar, M. C. Koorengevel, V. Subramaniam and J. A. Killian, Eur. Biophys. J., 2017, 46, 91–101 CrossRef CAS PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- I. D. Sahu, R. M. McCarrick, K. R. Troxel, R. F. Zhang, H. J. Smith, M. M. Dunagan, M. S. Swartz, P. V. Rajan, B. M. Kroncke, C. R. Sanders and G. A. Lorigan, Biochemistry, 2013, 52, 6627–6632 CrossRef CAS PubMed.
- B. Ehresmann, P. Imbault and J. H. Weil, Anal. Biochem., 1973, 54, 454–463 CrossRef CAS PubMed.
- K. Shimono, M. Iwamoto, M. Sumi and N. Kamo, FEBS Lett., 1997, 420, 54–56 CrossRef CAS PubMed.
- N. Mennes, J. P. Klare, I. Chizhov, R. Seidel, R. Schlesinger and M. Engelhard, FEBS Lett., 2007, 581, 1487–1494 CrossRef CAS PubMed.
- T. H. Bayburt and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6725–6730 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- N. V. Suetin, S. A. Evlashin, A. V. Egorov, K. V. Mironovich, S. A. Dagesyan, L. V. Yashina, E. A. Goodilin and V. A. Krivchenko, Phys. Chem. Chem. Phys., 2016, 18, 12344–12349 RSC.
- D. Klose, N. Voskoboynikova, I. Orban-Glass, C. Rickert, M. Engelhard, J. P. Klare and H. J. Steinhoff, FEBS Lett., 2014, 588, 3970–3976 CrossRef CAS PubMed.
- R. E. Martin, M. Pannier, F. Diederich, V. Gramlich, M. Hubrich and H. W. Spiess, Angew. Chem., Int. Ed., 1998, 37, 2834–2837 CAS.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- G. Hagelueken, R. Ward, J. H. Naismith and O. Schiemann, Appl. Magn. Reson., 2012, 42, 377–391 CrossRef CAS PubMed.
- S. J. Hirst, N. Alexander, H. S. McHaourab and J. Meiler, J. Struct. Biol., 2011, 173, 506–514 CrossRef CAS PubMed.
- E. J. Hustedt, R. A. Stein, L. Sethaphong, S. Brandon, Z. Zhou and S. C. Desensi, Biophys. J., 2006, 90, 340–356 CrossRef CAS.
- Z. Zhou, S. C. DeSensi, R. A. Stein, S. Brandon, M. Dixit, E. J. McArdle, E. M. Warren, H. K. Kroh, L. Song, C. E. Cobb, E. J. Hustedt and A. H. Beth, Biochemistry, 2005, 44, 15115–15128 CrossRef CAS PubMed.
- S. Scheidelaar, M. C. Koorengevel, J. D. Pardo, J. D. Meeldijk, E. Breukink and J. A. Killian, Biophys. J., 2015, 108, 279–290 CrossRef CAS PubMed.
- D. V. Bagrov, N. Voskoboynikova, G. A. Armeev, W. Mosslehy, G. S. Gluhov, T. T. Ismagulova, A. Y. Mulkidjanian, M. P. Kirpichnikov, H.-J. Steinhoff and K. V. Shaitan, Biofizika, 2016, 61, 942–949 CAS.
- M. Jamshad, V. Grimard, I. Idini, T. J. Knowles, M. R. Dowle, N. Schofield, P. Sridhar, Y. P. Lin, R. Finka, M. Wheatley, O. R. T. Thomas, R. E. Palmer, M. Overduin, C. Govaerts, J. M. Ruysschaert, K. J. Edler and T. R. Dafforn, Nano Res., 2015, 8, 774–789 CrossRef CAS.
- T. H. Bayburt and S. G. Sligar, Protein Sci., 2003, 12, 2476–2481 CrossRef CAS PubMed.
- T. H. Bayburt, J. W. Carlson and S. G. Sligar, Langmuir, 2000, 16, 5993–5997 CrossRef CAS.
- C. D. Blanchette, J. A. Cappuccio, E. A. Kuhn, B. W. Segelke, W. H. Benner, B. A. Chromy, M. A. Coleman, G. Bench, P. D. Hoeprich and T. A. Sulchek, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 724–731 CrossRef CAS PubMed.
- P. Samyn and G. Schoukens, Polymers, 2014, 6, 820–845 CrossRef.
- B. Aton, A. G. Doukas, R. H. Callender, B. Becher and T. G. Ebrey, Biochemistry, 1977, 16, 2995–2999 CrossRef CAS PubMed.
- C. Gellini, B. Luttenberg, J. Sydor, M. Engelhard and P. Hildebrandt, FEBS Lett., 2000, 472, 263–266 CrossRef CAS PubMed.
- O. S. Mironova, R. G. Efremov, B. Person, J. Heberle, I. L. Budyak, G. Buldt and R. Schlesinger, FEBS Lett., 2005, 579, 3147–3151 CrossRef CAS PubMed.
- T. Alshuth, M. Stockburger, P. Hegemann and D. Oesterhelt, FEBS Lett., 1985, 179, 55–59 CrossRef CAS.
- S. O. Smith, A. B. Myers, R. A. Mathies, J. A. Pardoen, C. Winkel, E. M. van den Berg and J. Lugtenburg, Biophys. J., 1985, 47, 653–664 CrossRef CAS PubMed.
- S. O. Smith, J. A. Pardoen, J. Lugtenburg and R. A. Mathies, J. Phys. Chem., 1987, 91, 804–819 CrossRef CAS.
- E. Bordignon and Y. Polyhach, Methods Mol. Biol., 2013, 974, 329–355 CAS.
- H. J. Steinhoff, Biol. Chem., 2004, 385, 913–920 CrossRef CAS PubMed.
- G. W. Reginsson and O. Schiemann, Biochem. J., 2011, 434, 353–363 CrossRef CAS PubMed.
- A. Ishchenko, E. Round, V. Borshchevskiy, S. Grudinin, I. Gushchin, J. P. Klare, A. Remeeva, V. Polovinkin, P. Utrobin, T. Balandin, M. Engelhard, G. Buldt and V. Gordeliy, Sci. Rep., 2017, 7, 41811 CrossRef CAS PubMed.
- A. Ishchenko, E. Round, V. Borshchevskiy, S. Grudinin, I. Gushchin, J. P. Klare, A. Remeeva, V. Polovinkin, P. Utrobin, T. Balandin, M. Engelhard, G. Büldt and V. Gordeliy, Sci. Rep., 2017, 7, 41811 CrossRef CAS PubMed.
- C. Logez, M. Damian, C. Legros, C. Dupre, M. Guery, S. Mary, R. Wagner, C. M'Kadmi, O. Nosjean, B. Fould, J. Marie, J. A. Fehrentz, J. Martinez, G. Ferry, J. A. Boutin and J. L. Baneres, Biochemistry, 2016, 55, 38–48 CrossRef CAS PubMed.
- J. P. Klare, E. Bordignon, M. Doebber, J. Fitter, J. Kriegsmann, I. Chizhov, H. J. Steinhoff and M. Engelhard, J. Mol. Biol., 2006, 356, 1207–1221 CrossRef CAS PubMed.
- J. Holterhues, E. Bordignon, D. Klose, C. Rickert, J. P. Klare, S. Martell, L. Li, M. Engelhard and H. J. Steinhoff, Biophys. J., 2011, 100, 2275–2282 CrossRef CAS PubMed.
- M. C. Orwick, P. J. Judge, J. Procek, L. Lindholm, A. Graziadei, A. Engel, G. Grobner and A. Watts, Angew. Chem., Int. Ed. Engl., 2012, 51, 4653–4657 CrossRef CAS PubMed.
- R. Zhang, I. D. Sahu, L. Liu, A. Osatuke, R. G. Comer, C. Dabney-Smith and G. A. Lorigan, Biochim. Biophys. Acta, 2015, 1848, 329–333 CrossRef CAS PubMed.
- V. Postis, S. Rawson, J. K. Mitchell, S. C. Lee, R. A. Parslow, T. R. Dafforn, S. A. Baldwin and S. P. Muench, Biochim. Biophys. Acta, 2015, 1848, 496–501 CrossRef CAS PubMed.
- S. Gulati, M. Jamshad, T. J. Knowles, K. A. Morrison, R. Downing, N. Cant, R. Collins, J. B. Koenderink, R. C. Ford, M. Overduin, I. D. Kerr, T. R. Dafforn and A. J. Rothnie, Biochem. J., 2014, 461, 269–278 CrossRef CAS PubMed.
Footnote |
| † N. V. and W. M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |