DOI:
10.1039/C7RA10516A
(Paper)
RSC Adv., 2017,
7, 56537-56542
A postsynthetic ion exchange method for tunable doping of hydroxyapatite nanocrystals†
Received
22nd September 2017
, Accepted 8th December 2017
First published on 15th December 2017
Abstract
Hydroxyapatite (HAp) is the main inorganic component of human bones and teeth. The doping of HAp nanocrystals plays an important role in tissue engineering, drug delivery, biomarkers and artificial bones. In this article, a postsynthetic metal ion exchange method was developed for doping hydroxyapatite (HAp) nanocrystals in organic solutions. It can be mono- or multi-ion doped with Mg2+, Sr2+, Zn2+, Mn2+, Fe3+ or Cu2+ etc., despite the significant radius variation of Ca2+ and doping ions. The doping ratio can be tuned in a wide range, e.g. Fe3+ of 0–20%, which is much higher than the ion exchange performed in aqueous solution. The structure and morphology of the HAp nanocrystals were preserved after postsynthetic doping, suggesting potential biological applications.
1. Introduction
Hydroxylapatite, also called hydroxyapatite (HAp), is the main inorganic component of human bones and teeth. As a material, thanks to its good properties of biocompatibility, bioactivity and biodegradability, nanosized HAp plays an important role in tissue engineering, drug delivery, biomarkers and artificial bones.1–8 HAp is composed of Ca2+, OH− and PO43− with the chemical formula of Ca10(PO4)6(OH)2 (or written as Ca5(PO4)3(OH)). HAps in biology systems contain not only Ca2+, but also various of metal ions, e.g., Mg2+, Na+, K+, Sr2+, Zn2+, Fe3+/2+, Cu2+, Mn2+ etc. with positive biological effect. Fortunately, the ions of pure HAp can be artificially replaced by other cations or anions. For examples, Ca2+ can be replaced by metal ions of Zn2+, Sr2+, Mn2+, Fe3+, Tb3+ or Eu3+,9,10 OH− replaced by F−, Cl− or Br−,11,12 and PO43− replaced by SeO32− or CO32−, etc.13,14 The doping may improve the performance of HAp nanocrystals in acid resistance, optical, antibacterial and anticancer properties.9–18 Doped Cu2+ can offer antibacterial properties; Zn2+ is involved in the formation of various proteins and nucleic acids in human body; Sr2+ can promote the formation of new bones; Fe3+/2+ not only exists in the blood, but also in bones and enamel; rare earth ions can serve as fluorescent centers and so on. Therefore, understanding the doping mechanisms is of fundamental importance in developing nanomaterials with desired functionalities.
To date, various chemical doping strategies have been developed for nanocrystals,19,20 e.g. co-nucleation doping,21 growth doping,22 dopant-nucleation,23 diffusion doping,24 ion-exchange doping,25 etc. Among them, the most widely adopted method is the co-nucleation doping. It features in the dopant and host precursors being loaded into the synthesis system simultaneously at the beginning of the reaction, where the doping and nucleation steps could not be decoupled and the doping continues during the following growth stage. Although there has been successfully HAp doping examples via this method,9–18 there remains some critical challenges. First, the doping ions would affect the nucleation and growth kinetics or crystallization dynamics, leading to the change of the final morphology of the nanocrystals. Therefore, on one hand, the size and shape of the nanocrystals may be tuned by the concentration of the doping metal ions; on the other hand, it sets up an obstacle toward HAp nanocrystals of the same morphology and size with different doping ratios. Second, the doping ions may form impurity phases during the growth of HAp nanocrystals. Third, the ion exchange of HAp nanocrystals performed in aqueous containing solutions requires the PH value near neutral or alkaline, for the HAp may decompose in acidic solution.26–29 However, many salts in water exhibit acidity because of their hydrolysis, such as FeCl3 and CuCl2, etc. The last but not the least, only the ionic radius of the cation falls within 0.09–0.13 nm, approaching that of Ca2+ of 0.1 nm, the Ca2+ can be easily replaced to form heavy doped HAps.30 For examples, there are many reports on Sr-HAp with the doping ratio exceeding 50%, and even 100% Sr-HAp was reported, for the radius of Sr2+ is 0.12 nm;31 The radius of Mg2+ is 0.072 nm, 10% is believed to be the upper limit;32 the radius of Fe3+ is even smaller of 0.055 nm, thus rarely >5% Fe3+ doped HAp was reported.33 All these limitations will affect the structure stability, ion releasing and thus the bio-properties of the doped HAp nanocrystals in service.
To solve the above challenges, we developed a postsynthetic strategy to doping HAp nanocrystals in non-aqueous organic solution. It was demonstrated that with this route, the doping ratio can be feasibly tuned by the concentration, temperature and time. It can be mono- or multi-metal ion doping. The doped HAp nanocrystals was highly pure without byproduct impurities. The morphology and size of the doped HAp nanocrystals were almost the same as the undoped ones. It is a practical route toward heavy doping which can hardly be achieved in aqueous ion exchange because of the unwanted hydrolysis and acidity. The doping ratio of Fe3+ can easily boost up to >20%. Also, the as-described method can make metal ions doped into the HAp crystal more effectively without destroying the crystal structure.
2. Experimental section
2.1 Materials
Ca(NO3)2·4H2O (AR, >99%), Na3PO4·12H2O (AR, >98%), FeCl3·6H2O (AR, ≥99%), C18H33NaO2 (CP, >98%), CuCl2·3H2O (AR, ≥99%), Mn(NO3)2·4H2O (AR, ≥97.5%), Fe(NO3)3·9H2O (AR, ≥98.5%), Sr(NO3)2 (AR, ≥99.5%), Zn(NO3)2·6H2O (AR, ≥99%), Mg(NO3)2·6H2O (AR, ≥99%), NaIO4 (AR, ≥99.5%), Na2CO3 (AR, >99.8%), PEG20000 (AR, >90%), ethyl acetate (AR, ≥99.5%), acetonitrile (AR, ≥99.8%), hexane (AR, ≥97%), ethanol (AR, ≥99.7%) and cyclohexane (AR, ≥99.7%) were obtained from Sinopharm Chemical Reagent Co. The oleic acid (OA, 90%) was purchased from Alfa Aesar. 1-Octadecene (ODE, 90%) and oleylamine (OLA, 90%), octadecylamine (ODA, ≥97.0%) were purchased from Aldrich. All chemicals were used as received without further purification.
2.2 Synthesis of oleate complex
The metal–oleate complex was synthesis by reacting metal–salt and sodium oleate. In a typical synthesis of iron–oleate, 2.6 g ferric chloride was dissolved in 4 mL deionized water, 7.3 g sodium oleate was dissolved in the mixture of 12 mL deionized water and 16 mL ethanol, then they were mixed together in a flask and 32 mL hexane was put in at the same time, the resulting reddish brown mixture was heated to 65 °C for four hours, and cooled to room temperature, then the supernatant organic layer containing the iron–oleate complex was washed three times with 30 mL distilled water to remove the Na+ and Cl−. It was heated to 80 °C for eight hours in an oven to evaporate the hexane. Finally, the iron–oleate complex in a waxy solid form was obtained for further use. The other metal–oleate complexes were prepared in the same way.
2.3 Synthesis of hydrophobic HAp nanorods
HAp nanorods were prepared by a hydrothermal method. Firstly, in a 40 mL Teflon-lined autoclave, 4 mL oleic acid, 1 mL oleylamine and 16 mL ethanol was mixed together to form a homogeneous solution, and an aqueous solution of Ca(NO3)2·4H2O (0.25 M, 7 mL) was added to produce a calcium–oleate precursor, then the Na3PO4·12H2O (0.15 M, 7 mL) solutions were add in with continued agitation. The mixture was stirred for 5 min, sealed, and hydrothermally treated at 150 °C for 10 hours. The HAp product was collected by centrifugation and washed with cyclohexane and ethanol several times.
2.4 Synthesis of Ca–PO4–CO3 (CHAp) nanowires
In a 40 mL Teflon-lined autoclave, octadecylamine (0.5 g) was dissolved in 16 mL ethanol, then 4 mL oleic acid and aqueous Ca(NO3)2·4H2O (0.25 M, 7 mL) were added in the mixture. Then, an aqueous solutions of Na2CO3 (0.25 M, 7 mL) and Na3PO4·12H2O (0.15 M, 3.5 mL) were added in. The mixture was agitated for another 5 min, then sealed and hydrothermally treated at a temperature of 150 °C for 12 h. The obtained nanowires were collected by centrifugation, washed three times with cyclohexane and ethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) for further ion exchange.
:2) for further ion exchange.
2.5 Doping HAp or CHAp nanocrystals by postsynthetic ion exchange
In a flask 500 mg HAp nanocrystals (hydrophobic HAp nanorods; CHAp nanowires), 5 g oleate and 5 mL ODE were mixed. It was then treated at a controlled temperature of 100–300 °C for 1 hour under the protection of argon. After cooled to room temperature naturally, the product was washed and centrifuged with ethanol and cyclohexane for three times. The multi-ion exchange is carried out in the same way. For example, 500 mg hydrophobic HAp nanocrystals with 2.5 g copper oleate and 2.5 g zinc oleate were mixed with 5 mL ODE. It was then treated at a temperature of 200 °C for 1 hour under the protection of argon. After cooled to room temperature naturally, the product was washed and centrifuged with ethanol and cyclohexane for three times to yield HAp:Zn2+, Cu2+ nanorods.
2.6 Co-nucleation doping of Fe3+ with the HAp synthesis
The process is similar to synthesis of hydrophobic HAp nanorods, firstly, in a 40 mL Teflon-lined autoclave, 4 mL oleic acid, 1 mL oleylamine and 16 mL ethanol was mixed together to form a homogeneous solution, a certain mount Fe(NO3)3·9H2O (0.25 M, 1.6 mL) and Ca(NO3)2·4H2O (0.25 M, 5.6 mL) was added, then the Na3PO4·12H2O (0.15 M, 7 mL) solutions were add in with continued agitation. The mixture was stirred for 5 min, sealed, and hydrothermally treated at 150 °C for 10 hours. The HAp product was collected by centrifugation and washed with cyclohexane and ethanol for three times.
2.7 Synthesis of amphiphilic HAp nanocrystals and ion exchange in water
The method of synthesis amphiphilic HAp is similar to hydrophobic HAp with slight modification. Typically, 0.45 g PEG20000 was dissolved in an aqueous solution of Ca(NO3)2·4H2O (7 mL, 0.25 M), then 7.2 mL OA and 16 mL ethanol was added and mixed. Then Na3PO4·12H2O (7 mL, 0.15 M) was added and stirred for 5 min. Finally, the mixture was sealed and reacted at 110 °C for 10 h. The white precipitate was collected by centrifugation. After washed with cyclohexane and ethanol (v/v = 1:2) for twice, it was further washed with water twice for further ion exchange in water. 50 mg HAp and proper amount of Fe(NO3)3·9H2O was add in a 5 mL deionized water to form a 0.005, 0.01 or 0.1 M solution, and the mixture was ultrasonic for an hour, then aged for 24 hours. Finally, the HAp was washed with deionized water three times and freeze dried.
2.8 Hydrophilic treatment of HAp for cytotoxicity assay
To carry out the cytotoxicity assay experiment, the HAp nanorods were treated to be hydrophilic. Hydrophobic HAp nanorods (undoped HAp nanorods; HAp:Fe3+ nanorods postsynthetic ion exchanged at 150 °C with the doping ratio of 5.1%) of 200 mg were dispersed in 10 mL cyclohexane. Then 20 mL ethyl acetate and acetonitrile (v/v = 1:1) was added. A 15 mL water solution containing 0.8 g NaIO4 was added into the former mixture drop by drop while stirring for 2 h. The mixture is divided into two layers, the upper of cyclohexane is colorless and discarded, the bottom is centrifuged, washed with ethanol and water for three times, yielding the hydrophilic HAp that can be dispersed in water.
2.9 Cytotoxicity assay
The human cervical carcinoma (HeLa, KeyGEN BioTECH) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, KeyGEN BioTECH) with fetal bovine serum (10%, FBS, Gibco), penicillin (80 U mL−1), and streptomycin (0.08 mg mL−1) at 37 °C with 5% CO2. The cytotoxicity of HAp nanocrystals to HeLa cells was studied by using a LDH-Cytotoxicity Colorimetric Assay Kit (BioVision). HeLa cells were cultured in 96-well plates for 24 h, and the culture medium was discard. Afterwards, 200 μL of DMEM medium (without FBS) containing different concentrations (20, 40, 80, 160, and 320 μg mL−1) of HAp nanocrystals was added into corresponding wells, respectively. Cells cultured in DMEM medium (no FBS) were set as low control, and cells in DMEM medium (no FBS) containing 1% Triton X-100 served as high control. After 24 h, 100 μL of supernatant from each well was pipetted to another well and mixed with 100 μL of the reaction mixture (containing 2.2 μL catalyst solution and 97.8 μL dye solution, BioVision) was added into each well. After being incubated at room temperature for 20 min away from light, the samples were measured using a microtiter plate reader under the wavelength of 490 nm (PowerWave XS2, BioTek). The percentage of cell viability was calculated using the formula as follows: cell viability = 100% − (test sample − low control)/(high control − low control) × 100%.
2.10 Characterization
The power X-ray diffraction (XRD) patterns were recorded using a D/max 2500 VL/PC diffractometer equipped with graphite monochromatized Cu Kα radiation (λ = 0.15406 nm), the samples were ground into powder and measured in a voltage of 40 kV at a rate of 2° min−1 and angle range of 20–60°. The phase was identified by comparison to the standard HAp X-ray diffraction pattern. The morphologies and sizes of the obtained samples were observed with a JEM-200CX transmission electron microscope (TEM), with accelerating voltage of 80 kV. The functional groups of the samples were characterized using a Fourier-transform infrared spectrophotometer (Vector 22 spectrometer) with the KBr pellet technique, FTIR spectra were the range of 4000–400 cm−1. The elemental analysis of the samples was determined by energy dispersive spectrometer (EDS). The high-resolution transmission electron microscopy (HRTEM) images were taken on JEOL-2100F with accelerating voltage of 200 kV. Inductively coupled plasma (ICP) measurements were performed on an induction coupled plasma quantometer (Jarrell-Ash 1100 +2000).
3. Results and discussion
3.1 Postsynthetic doping mechanism
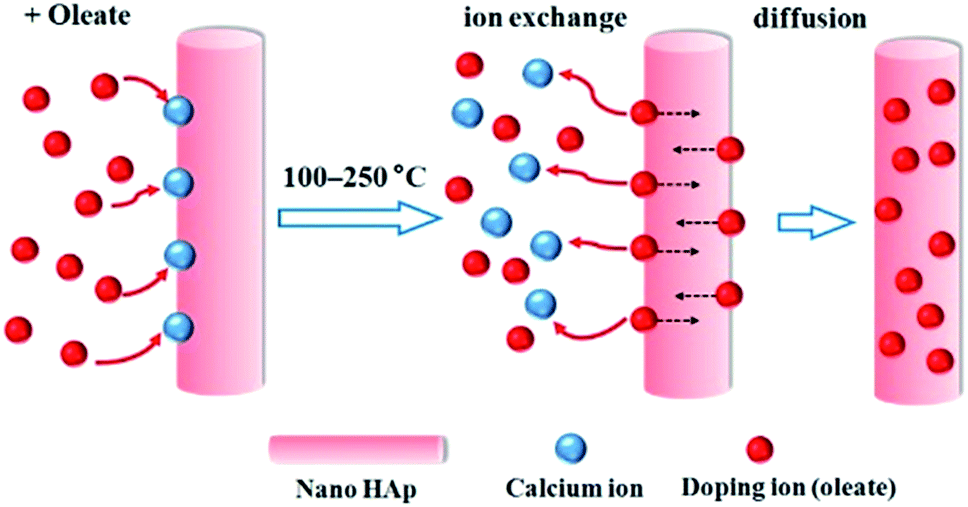
A schematic illustration of the doping mechanism is shown in Fig. 1. Briefly, the un-doped HAp nanocrystals were firstly synthesized, washed and purified. Then the doping processed is performed in the organic solvent with a high boiling point such as octadecene (ODE). The doping metal is introduced in the organic soluble form, e.g. metal oleate. No water is introduced or formed during the doping. Therefore the doping temperature can be freely tuned below the thermal decomposing of the metal oleate. The doping ions first exchange with the Ca2+ at the surface of HAp nanocrystals and then diffuse into the crystal lattice of HAp.
 |
| | Fig. 1 A schematic illustration of the metal ion doping mechanism for HAp nanocrystals. | |
Fig. 2a shows typical pure and doped HAp nanocrystals. They are well dispersed in cyclohexane, forming transparent solutions with various colors which are from the doped ions. It is worth noting that the color of the metal ions may also affected by the coordination field. For the Fe3+ doped HAp, the surface Fe3+ could be modified by oleic acid; while Fe3+ inside the HAp is thermodynamically preferred to occupy the CaI position than the CaII position.34 The color deepens with the doping ratio increases, e.g. Fe3+ doping in this report and Co2+ doped HAps.35 Transmission electron microscope (TEM) measurement shows that the nanorod morphology is preserved for all the HAp nanocrystals doped with metal ions (Fig. 2b–i). The EDS spectra indicate the HAp nanocrystals are successfully doped with metal ions (Fig. S1†).
 |
| | Fig. 2 (a) From left to right the undoped, Mg2+, Sr2+, Zn2+, Cu2+, Mn2+ and Fe3+ doped HAp nanocrystals, respectively. Their corresponding TEM images (b–h), and (i) a typical high-resolution transmission electron microscopy (HRTEM) image of the undoped HAp nanocrystals, showing the nanocrystals growth along the [002] orientation. ((b–h) Scale bar: 100 nm). | |
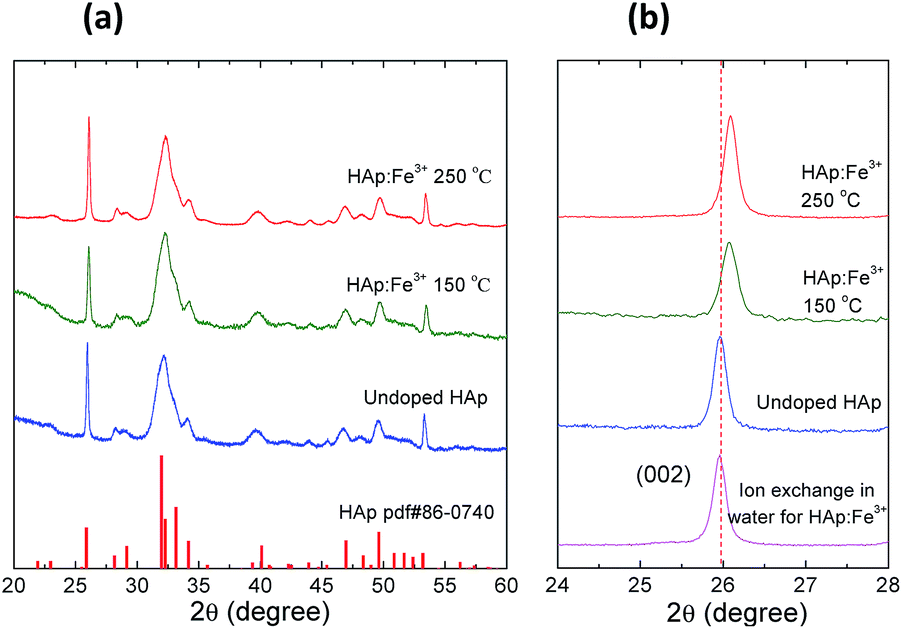
To investigate the doping mechanism, the Fe3+ doped HAp (abbreviated as HAp:Fe3+) nanocrystals was selected as a representative prototype. Fig. 3 shows the XRD patterns of the undoped and Fe3+ doped HAp nanocrystals at different temperatures. The XRD peaks of the undoped HAp nanocrystals match well with that of the standard HAp (PDF #86-0740), indicating the hexagonal crystal structure belonging P63/m space group. The crystal structure of the doped HAp remains the unchanged. But a shift to the higher angle side was observed for the HAp samples doping at elevated temperature of 150 and 250 °C (Fig. 3). It is consistent with the fact that the radius of the Fe3+ is significantly smaller than that of Ca2+. So the ion exchange of Ca2+ with Fe3+ leads to the shrinking of the HAp crystal lattice, which is proportional to the doping temperature. It should be mentioned that when the temperature further increased to 280–300 °C, the Fe–oleate will decompose to yield Fe3O4 nanocrystals, which is proved by TEM and XRD (Fig. S2†).
 |
| | Fig. 3 (a) The XRD patterns of the undoped and Fe3+ doped HAp nanocrystals. (b) The enlarged XRD patterns with the 2θ between 24° to 28° of the corresponding samples. | |
The FTIR spectra of the undoped and Fe3+ doped HAp nanocrystals were measured (Fig. S3†). The band at 3570 cm−1 is the vibration of OH group in HAp. The bands at 2922 and 2852 cm−1 are assigned to the CH3 and CH2 groups, indicating the surface modification of oleate ligands, which enables the dispersibility of the HAp nanocrystals in nonpolar solvent. The peaks at around 1554 and 1458 cm−1 are assigned to the COO− group of surface oleates.36 The intense bands at 1095 and 1027 cm−1 are attributed to the P–O asymmetric stretching vibration modes, and peaks at 603 and 562 cm−1 correspond to the O–P–O bending modes. The Fe3+ doped and undoped HAp nanocrystals have almost the same FTIR vibration modes within 4000–400 cm−1, indicating that the crystal structure remained unchanged and the CH3, CH2, COO–, P–O and O–P–O vibration modes can hardly affected by the doped Fe3+. This shielding effect was generally observed in metal doped HAps, including Fe3+ doped HAps.33
3.2 Postsynthetic doping in organic and aqueous solutions
To understand the difference of the postsynthetic ion exchange performed in organic solvent and in aqueous solution, the HAp nanocrystals doped with Fe3+ were studied. Fig. 4a–d show typical TEM images of undoped and Fe3+ doped HAp nanocrystals with an increasing doping ratio, which is defined by the mole ratio of Fe3+/(Fe3+ + Ca2+). The doping ratio increased monotonically with the doping temperature, e.g. 150 °C, 200 °C and 250 °C yielding 5.1%, 9.1% and 20.5%, respectively (Fig. 4b–d). For the sample doped at 150 °C, the same as HAp synthesis temperature, the morphology and size show almost no change (Fig. 4b). In the ODE solution, the Ca2+ on the HAp surface exchanged with Fe3+. Since the concentration of Fe3+ is relatively high in solution, the surface Ca2+ was effectively exchanged, and Ca2+ vacancies on the surface may also eliminated by Fe3+. To reduce the chemical potential gradient, the Fe3+ ions diffuse from the surface into the nanocrystal, i.e. exchanged with the Ca2+ ions inside the nanocrystal. The diffusion rate increases at elevated temperature. The mass transfer may also happen between HAp nanocrystals during the doping procedure, known as the Ostwald ripening effect.37 However, the Ostwald ripening effect has minor impact on the overall morphology mutation and the rod shape retained. At higher doping temperature up to 250 °C, it undergoes only ∼16% and ∼8% increase of the HAp average diameter and length, respectively (Fig. 4d).
 |
| | Fig. 4 TEM images of (a) the undoped HAp nanocrystals, and HAp:Fe3+ nanocrystals with the doping ratio of (b) 5.1%, (c) 9.1%, (d) 20.5%. (e) Amphiphilic HAp nanocrystals, and ion exchanged with Fe(NO3)3 aqueous solutions yielding the HAp with the doping ratio of (f) 2.3%, (g) 5.8% and (h) 11.7%. The insets are images of the sample dispersed in cyclohexane (a–d) and in water (e–h). ((a–h) Scale bar: 100 nm). | |
To illustrate the advantage of this doping strategy, the comparison is carried out with the Fe3+ ion exchange in water. The Fe(NO3)3 aqueous solutions with concentration values of 0, 0.005, 0.01 and 0.1 M were used, with according ion exchange results shown in Fig. 4e–h, respectively. It presents that the doping ratio increases monotonically with the Fe(NO3)3 concentration. But at the same time, the nanorod morphology becomes out of shape and aggregation takes place. At Fe(NO3)3 concentration of 0.1 M, the doping ratio amounts to 11.7%, while the HAp nanocrystals completely degrade to amorphous aggregates and the XRD revealed a total crystal decomposing (Fig. S4†). This is due to the fact that HAp is sensitive to the acidity of the aqueous solution induced by the hydrolysis of Fe3+. The HAp sample ion exchanged with Fe(NO3)3 aqueous solution of 0.01 M has a doping ratio of 5.8%, comparable to the ODE solution ion exchanged sample of the doping ratio of 5.1%. However, the XRD measurement indicates that the former has no detectable shift of the XRD peak of (002) at 25.97°, while the later shifts to 26.07° (Fig. 3b). This difference may suggest that for the organic solution doping method, the Fe3+ ions diffuse effectively into the HAp nanocrystals. It could be anticipated that the body doping may have a lower release ratio of the doped ions in biological serving conditions than those of the surface adsorbed ions. On one hand, for the biologically toxic ions, the body doping would reduce the toxicity. On the other hand, for the bio-active or life nutritious doped ions, a long term releasing could be expected.
3.3 Cytotoxicity assay
The cytotoxicity assay was performed to study the undoped and doped HAp nanocrystals by using the Lactate Dehydrogenase (LDH) cytotoxicity assay. The human cervical carcinoma (HeLa) cells were cultured for 24 h at 37 °C in cell media containing 0–320 μg mL−1 of HAp:Fe3+ nanocrystals with the doping ratio of 5.1% (postsynthetic ion exchanged at 150 °C), and the undoped HAp nanocrystals with the same size and morphology served as a control experiment (Fig. S5†). Both of the HeLa cells treated with undoped HAp and HAp:Fe3+ nanocrystals show nearly 100% viability throughout the 0–320 μg mL−1 range, indicating their excellent biocompatibility.
3.4 Co-nucleation doping and postsynthetic doping
In the investigation of co-nucleation doping, the Fe3+ ions were introduced into the synthesis of HAp nanocrystals at the beginning of the reaction. It turned out that the resulting product has diverse morphologies (Fig. S6†). The doping of Ca–PO4–CO3 (CHAp,14) solid-solution nanowires with Fe3+ by the postsynthetic ion exchange also indicates that the method is very effective (Fig. S7†). Furthermore, and bimetallic ions co-doping HAp:Zn2+,Cu2+ nanocrystals were also successfully achieved by this method (Fig. S8†). These results further demonstrate the superiority and generality of the postsynthetic organic solution doping strategy.
3.5 The maximum doping ratio
The maximum of the HAp doping ratio depends on several factors. First, the doping should not destroy or change the crystal structure. So the maximum doping ratio various for different doping metals. The limit is also related the HAp size, since the surface atom ratio and specific surface energy increase with the HAp size decreasing. Given a closed reaction system, the maximum doping ratio is achieved at the chemical equilibrium point, when the Gibbs free energy change is zero:and ΔG can be written as:| | |
ΔG = RTln(Q/Kθ)
| (2) |
where Q is reaction quotient; Kθ is the standard equilibrium constant; [CM] is the concentration of the doping ions in solution. From the equilibrium state, increasing the [CM] makes ΔG < 0. It will drive for further doping. If the starting [CM] is high enough, the doping level can keep rising until the HAp structure limit. However, the [CM] has a maximum value in the state of the pure precursor. Therefore, the doping maximum depends on the lower value from the doping precursor limit and the structure controlled limit. The chemical equilibrium constant of the doping reaction can be tuned by the temperature:| | |
ln(K1θ/K2θ) = −ΔHrθ(1/T1 − 1/T2)/R
| (4) |
where ΔHrθ is the standard enthalpy change of the doping; R is the gas constant. In case the doping reaction is endothermic:higher doping level can be achieved in higher temperature. This is experimentally supported in this work. At the same time, the doping procedure is faster at higher temperature and/or higher doping ion concentration in solution.
4. Conclusion
In conclusion, a general postsynthetic ion exchange method was developed for doping of various metal ions into the HAp nanocrystals in organic solvent. This method is featured in not only its tunable doping ratio (for Fe3+ it is 0–20%, higher than that ion exchanged in aqueous solution), but also the preserved nanocrystal morphology, size, crystal structure, pure products and effective body doping. The merits of this technique will contribute to the applications of the doped functional HAp nanocrystals in artificial bones, tissue engineering, drug delivery and biomarker. The only obstacle is that organic solvents and precursors are involved in this method. It may hinder direct biomedical use. The surface of HAps needs to be treated as hydrophilic and biocompatible, as we did for the cell experiment. To carry out the doping in biocompatible none aqueous solvent is on going. We expect that it may further be applied to HAp anion doping, such as F−, CO32− or SeO32− etc., and extend to the doping of other nanocrystals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 51572120, 21376190, 21676215, 21533012 and 51503101), China Postdoctoral Science Foundation (2013M542378, 2014T70933), Shaanxi Provincial Postdoctoral Science Special Foundation (No. [2014]907), Shaanxi Provincial Science Foundation (2014SZS07-Z02, 2017SF-201), Scientific Research Program of Shaanxi Provincial Department of Education (15JS104), Six Talent Peaks Project in Jiangsu Province No. 184080H102231, and Nanjing Normal University Young Leading Talent Fund No. 184080H20210.
References
- J. Hui and X. Wang, Inorg. Chem. Front., 2014, 1, 215–225 RSC.
- S. V. Dorozhkin, Int. J. Chem. Mater. Sci., 2013, 1, 105–174 Search PubMed.
- K. Lin, C. Wu and J. Chang, Acta Biomater., 2014, 10, 4071–4102 CrossRef CAS PubMed.
- M. Vallet-Regí and J. M. González-Calbet, Prog. Solid State Chem., 2004, 32, 1–31 CrossRef.
- C. Heng, X. Zheng, M. Liu, D. Xu, H. Huang, F. Deng, J. Hui, X. Zhang and Y. Wei, Appl. Surf. Sci., 2016, 386, 269–275 CrossRef CAS.
- Y. Huang, G. Zhou, L. Zheng, H. Liu, X. Niu and Y. Fan, Nanoscale, 2012, 4, 2484–2490 RSC.
- X. Zheng, J. Hui, H. Li, C. Zhu, X. Hua, H. Ma and D. Fan, Mater. Sci. Eng., C, 2017, 75, 699–705 CrossRef CAS PubMed.
- S. V. Dorozhkin, Prog. Biomater., 2016, 5, 9–70 CrossRef CAS PubMed.
- J. Hui, X. Zhang, Z. Zhang, S. Wang, L. Tao, Y. Wei and X. Wang, Nanoscale, 2012, 4, 6967–6970 RSC.
- X. Zheng, M. Liu, J. Hui, D. Fan, H. Ma, X. Zhang, Y. Wang and Y. Wei, Phys. Chem. Chem. Phys., 2015, 17, 20301–20307 RSC.
- X. Zhang, J. Hui, B. Yang, Y. Yang, D. Fan, M. Liu, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 4120–4125 RSC.
- J. Hui and X. Wang, Chem.–Eur. J., 2011, 17, 6926–6930 CrossRef CAS PubMed.
- J. Sun, X. Zheng, H. Li, D. Fan, Z. Song, H. Ma, H. Xiu and J. Hui, Mater. Sci. Eng., C, 2017, 73, 596–602 CrossRef CAS PubMed.
- J. Hui, Q. Yu, Y. Long, Z. Zhang, Y. Yang, P. Wang, B. Xu and X. Wang, Chem.–Eur. J., 2012, 18, 13702–13711 CrossRef CAS PubMed.
- V. Uskoković, M. A. Iyer and V. M. Wu, J. Mater. Chem. B, 2017, 5, 1430–1445 RSC.
- W. Luo, Y. Liu and X. Chen, Sci. China Mater., 2015, 58, 819–850 CrossRef CAS.
- Z. C. Xiong, Y. J. Zhu, F. F. Chen, T. W. Sun and Y. Q. Shen, Chem.–Eur. J., 2016, 22, 11224–11231 CrossRef CAS.
- J. Hui, G. Xiang, X. Xu, J. Zhuang and X. Wang, Inorg. Chem., 2009, 48, 5614–5616 CrossRef CAS PubMed.
- D. J. Norris, A. L. Efros and S. C. Erwin, Science, 2008, 319, 1776–1779 CrossRef CAS PubMed.
- N. Pradhan, S. D. Adhikari, A. Nag and D. D. Sarma, Angew. Chem., Int. Ed., 2017, 56, 7038–7054 CrossRef CAS PubMed.
- R. Zeng, R. Shen, Y. Zhao, X. Li, Z. Sun and Y. Shen, Nanotechnology, 2014, 25, 135602 CrossRef PubMed.
- G. Galli, Nature, 2005, 436, 32–33 CrossRef CAS PubMed.
- N. Pradhan, D. Goorskey, J. Thessing and X. Peng, J. Am. Chem. Soc., 2005, 127, 17586–17587 CrossRef CAS PubMed.
- V. A. Vlaskin, C. J. Barrows, C. S. Erickson and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 14380–14389 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- M. Miyake, K. Watanabe, Y. Nagayama, H. Nagasawa and T. Suzuki, J. Chem. Soc., Faraday Trans., 1990, 86, 2303–2306 RSC.
- J. A. Gómez del Rio, P. J. Morando and D. S. Cicerone, J. Environ. Manage., 2004, 71, 169–177 CrossRef PubMed.
- S. Saxena and S. F. D'Souza, Environ. Int., 2006, 321, 199–202 CrossRef PubMed.
- A. Yasukawa, T. Yokoyama, K. Kandori and T. Ishikawa, Colloids Surf., A, 2007, 299, 203–208 CrossRef CAS.
- T. Suzuki, T. Hatsushika and Y. Hayakawa, J. Chem. Soc., Faraday Trans., 1981, 77, 1059–1062 RSC.
- A. Bigi, S. Panzavolta and N. Roveri, Biomaterials, 1998, 19, 739–744 CrossRef CAS PubMed.
- D. Laurencin, N. Almora-Barrios, N. H. de Leeuw, C. Gervais, C. Bonhomme, F. Mauri, W. Chrzanowski, J. C. Knowles, R. J. Newport, A. Wong, Z. H. Gan and M. E. Smith, Biomaterials, 2011, 32, 1826–1837 CrossRef CAS PubMed.
- H. R. Low, N. Phonthammachai, A. Maignan, G. A. Stewart, T. J. Basto, L. L. Ma and T. J. White, Inorg. Chem., 2008, 47, 11774–11782 CrossRef CAS PubMed.
- M. Jiang, J. Terra, A. M. Rossi, M. A. Morales, E. B. Saitovitch and D. E. Ellis, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 224107 CrossRef.
- K. P. Tank, K. S. Chudasama, V. S. Thaker and M. J. Joshi, J. Nanopart. Res., 2013, 15, 1644 CrossRef.
- B. Q. Lu, Y. J. Zhu and F. Chen, Chem.–Eur. J., 2014, 20, 1242–1246 CrossRef CAS PubMed.
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: TEM images, XRD patterns, EDS spectra, cytotoxicity assay. See DOI: 10.1039/c7ra10516a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Jianchun Bao
*b,
Jianchun Bao