
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic poly(ether ether ketone)s capable of crosslinking via UV irradiation to improve gas separation performance
María Puertas-Bartoloméa,
Michelle E. Doseb,
Paula Boscha,
Benny D. Freeman b,
James E. McGrathc,
Judy S. Rifflec,
Angel E. Lozanoade,
José G. de la Campaa and
Cristina Álvarez*a
b,
James E. McGrathc,
Judy S. Rifflec,
Angel E. Lozanoade,
José G. de la Campaa and
Cristina Álvarez*a
aInstitute of Polymer Science and Technology, Consejo Superior de Investigaciones Científicas, ICTP-CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: cristina.alvarez@ictp.csic.es
bThe University of Texas at Austin, Department of Chemical Engineering, Center for Energy and Environmental Resources, 10100 Burnet Road, Building 133, Austin, TX 78758, USA
cMacromolecules and Interfaces Institute and Department of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
dSMAP, UA UVa-CSIC, University of Valladolid, Paseo Belen 7, 47011 Valladolid, Spain
eIU CINQUIMA, University of Valladolid, Paseo Belen 7, 47011 Valladolid, Spain
First published on 5th December 2017
Abstract
The effect of UV-crosslinking on the gas transport properties of two poly(ether ether ketone)s derived from difluorobenzophenone and two bisphenol derivatives, with four (TMBP-DFB) or six (HMBP-DFB) methyl groups, has been studied. The crosslinking reaction was conducted on dense membranes, using polychromatic light, with wavelengths higher than 350 nm, at room temperature and in presence of air. Both polymers were able to produce crosslinked membranes, with gel fractions close to 75%, but a shorter irradiation time was required for HMBP-DFB. A DFT quantum mechanical study has stated that HMBP-DFB radical formation is much easier than for TMBP-DFB, which would support the fastest kinetics of the crosslinking process for HMBP-DFB. The crosslinked membranes have shown greatly improved gas transport properties, especially for the O2/N2 gas pair, where the Robeson upper bound line of 1991 was clearly surpassed. The improvement in selectivity has been ascribed to the better molecular-sieving characteristics of crosslinked membranes.
Introduction
Permeability and selectivity are the two important parameters that determine the characteristics of a gas separation membrane.1 Polymer membranes must have good separation capacity for a particular gas in a mixture and high permeability, as well as good mechanical properties and high thermal stability.2,3 However, conventional polymer membranes suffer from a well-known trade-off between these two parameters, because as the permeability of polymers increases, their ability to differentiate between gases decreases. This behaviour was first evidenced by Robeson in 1991 and then updated in 2008, showing the progress achieved in the gas separation field.4–7Glassy aromatic polymers, such as polyimides and polysulfones, are very commonly employed for gas separation membranes because they offer high selectivity and good mechanical properties. Based on the solution-diffusion mechanism, the more permeable gases are those with small molecular diameter and selectivity is mainly controlled by size based diffusion rate differences. Furthermore, it has been assumed that local to short scale cooperative segmental motions, which take place in glassy state, are responsible for the opening of the channels that connect free volume elements and then allow the passage of the gas molecules through the membrane.8 Therefore, glassy polymers have limited size-selectivity for practical applications because there is a wide size distribution of free volume elements (holes) and, moreover, the holes fluctuate in size due to the thermal motions of the polymer chains. In this context, the most effective way of improving permeability/selectivity properties is to create polymer structures with very high stiffness, while simultaneously frustrating interchain packing in order to improve permeability.9
Crosslinking induced by ultraviolet (UV) irradiation is a simple route to modify the structure of dense membranes by the formation of a network, which increases the rigidity. One of the first indications that UV crosslinking could improve the environmental stability and the performance of gas separation membranes of polyimides was reported in the patent literature.10 Since then, other studies have shown the effect of UV irradiation on gas transport properties of benzophenone containing polyimides,11 poliarylates,12 polyarylene ethers,13 poly(imide-siloxane) segmented copolymers,14 ketone containing poly(2,6-dimethylphenylene oxide) copolymers15 and poly(2,6-dimethylphenylene oxide) and poly(arylene ether ketone) blends.16 In general, the most notable improvements in membrane performance have been achieved in H2/CH4, O2/N2, or CO2/CH4 separations due to an enhancement in the size-sieving capability of the membranes. Besides, the method is useful to improve the membrane stability and to reduce undesirable phenomena, such as physical aging17 and plasticization18, which have a crucial impact on membrane durability.11,19,20
This work examines the effect of UV irradiation on the gas transport properties of two glassy amorphous poly(ether ether ketone)s (PEEKs) films based on difluorobenzophenone (DFB) and 3,3′,5,5′-tetramethyl-4,4′-dihydroxybiphenyl (TMBP) or 2,2′,3,3′,5,5′-hexamethyl-4,4′-dihydroxybiphenyl (HMBP). The monomers derived from bisphenol were chosen because the methyl groups reduce the efficiency of polymer chain packing, thereby increasing gas permeability and, moreover, they provide an abundant source of benzylic methyl groups for crosslinking reactions. UV light was used to excite the benzophenone moiety that subsequently abstracts a hydrogen radical from a benzylic methyl group. The two radicals created are combined to form a covalent bond, thus causing crosslinking. A DFT quantum mechanical study has been performed in order to estimate the ability of formation of radicals from methylene groups. The improvement in the size-sieving capability and gas selectivity of the dense membranes, as a function of the irradiation time, has been also examined.
Experimental
Materials
3,3′,5,5′-tetramethyl-4,4′-dihydroxybiphenyl (TMBP) and 2,2′,3,3′,5,5′-hexamethyl-4,4′-dihydroxybiphenyl (HMBP) were purchased from Akron Polymer Systems and purified by sublimation at 200 °C before use. 4,4′-Difluorobenzophenone (DFB) was purchased from Sigma-Aldrich, recrystallized from heptane and sublimated at 100 °C prior to use. Anhydrous (99.5%) N-methyl-2-pyrrolidinone (NMP) was purchased from Sigma-Aldrich.Polymers' synthesis
Polymers were synthesized using a nucleophilic aromatic substitution procedure previously reported.21 Typically, 5.0 mmol of TMBP or HMBP, 5.0 mmol of DFB and 10 mL of NMP were put into a 50 mL three necked flask, equipped with a mechanical stirrer and a Dean–Stark trap filled with toluene, and blanketed by a flux of dry nitrogen. The flask was immersed into a regulated oil bath, which was heated to 155 °C. After a homogeneous solution was obtained, 7.5 mmol of K2CO3 and 5 mL of toluene were added into the flask. The reaction was stirred at 155 °C for 3 h to azeotropically remove water and toluene, and then the bath was heated to 175 °C. Toluene and water were drained from the Dean–Stark trap, and the reaction was maintained for 24 h. The obtained viscous solution was diluted with additional NMP (10 mL) and the polymer was then precipitated onto rapidly stirred distilled water to produce a fibrous solid, which was filtered off and boiled several times in water to remove any residual salt and other by-products. Next, the polymer was washed with a mixture of distilled water and methanol (1/1), methanol and dried in a vacuum oven at 200 °C overnight. For both polymers quantitative yields were obtained.Film casting
Dense homogeneous membranes were prepared using a solution casting method. 0.500 g of polymer was dissolved in 10 mL of chloroform, and the solution was filtered through a 3.1 μm fiberglass Symta® syringe filter, poured onto a glass ring placed on a leveled glass plate, and maintained at room temperature overnight to remove most of the solvent. The membranes were peeled off of the glass and placed into a vacuum oven at 100 °C for 24 h, 120 °C for 2 h, 150 °C for 2 h, 180 °C for 1 h and 200 °C for 15 min. Finally, they were slowly cooled in the oven. The absence of residual solvent was confirmed by thermogravimetric analysis. The thickness of the circular membranes (area 70.8 cm2) ranged from 45 to 55 μm. The thickness percent uncertainty of a membrane was between 2 and 3%.UV irradiation
Circular membranes of 2.5 cm diameter were irradiated with polychromatic light in air, for different amounts of time on each side, using a 100 W high pressure Hg–Xe arc lamp (Hammatsu LC8 Lightningcure™) equipped with a 350 nm glass cut-off, working at 50% of nominal power. Samples were placed at 15 cm from the bulb of the UV lamp and irradiated during times between 30 and 60 min (per side). In this way, the gradient of crosslinking due to the thickness of the membranes was reduced.The degree of crosslinking was estimated from the gel fraction by eqn (1).
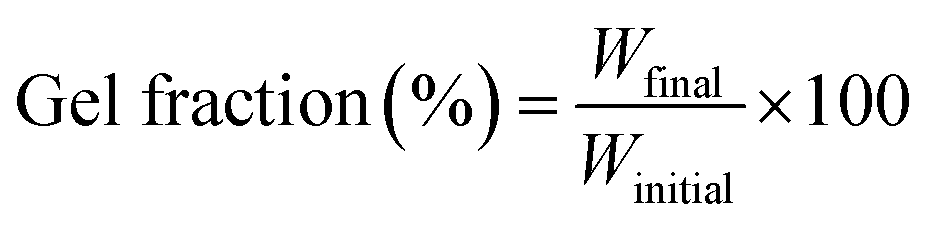 | (1) |
Techniques
1H and 13C NMR spectra were recorded on a Bruker Avance-300 spectrometer operating at 300 and 75 MHz in CDCl3. Fourier Transform Infrared (FTIR) spectra were registered on a Perkin Elmer Spectrum RX I FTIR spectrometer equipped with an ATR Accessory. Ultraviolet-visible spectra were acquired using a Perkin Elmer Lambda 35 UV/vis spectrometer. Inherent viscosities were measured at 25 °C with an Ubbelohde viscometer using NMP as solvent at 0.5 g dL−1 concentration. The polymers' solubility was determined by mixing 10 mg of polymer with 1 mL of solvent, followed by stirring for 12 h at room temperature. The samples that were not soluble were stirred for 5 additional hours at 60 °C. Thermogravimetric analysis (TGA) were performed on a TA-Q500 analyzer under nitrogen flux (60 mL min−1) at 10 °C min−1. Differential scanning calorimetry (DSC) was recorded on a TA Q-2000 instrument in nitrogen atmosphere (50 mL min−1) at 20 °C min−1. Wide-angle-X-ray scattering (WAXS) patterns were recorded in the reflection mode at room temperature, using a Bruker D8 Advance diffractometer provided with a Goebel Mirror and a PSD Vantec detector. CuKα (wavelength λ = 1.54 Å) radiation was used. A step-scanning mode was employed for the detector, with a 2θ step of 0.024° and 0.5 s per step. Density of membranes (ρ) was determined from Archimedes' principle using a top-loading electronic XS105 Dual range Mettler Toledo balance equipped with a density measurement kit. Six density measurements were made for each sample where the samples were sequentially weighed in air and into high purity isooctane at 25 °C. The density was calculated from eqn (2):
 | (2) |
Gas permeability measurements
The pure gas permeation properties for He, O2, N2 and CH4 were measured at 30 °C and an upstream pressure of 3 bar using a lab-made constant volume/variable pressure apparatus. The permeation of CO2 was measured in the upstream pressure range from 1 to 25 bar. The membranes were previously measured before being irradiated with UV light. After the membrane was mounted inside the permeation cell, both upstream and downstream chambers were exposed to high vacuum overnight to degas the membrane. The upstream pressure was adjusted to the desired value before starting the permeation experiments (t = 0 s), and then the increase of the permeate pressure was recorded as a function of time. All gases were allowed to permeate until the steady-state (ss) conditions were attained. The permeability coefficient (P) was calculated from eqn (3).
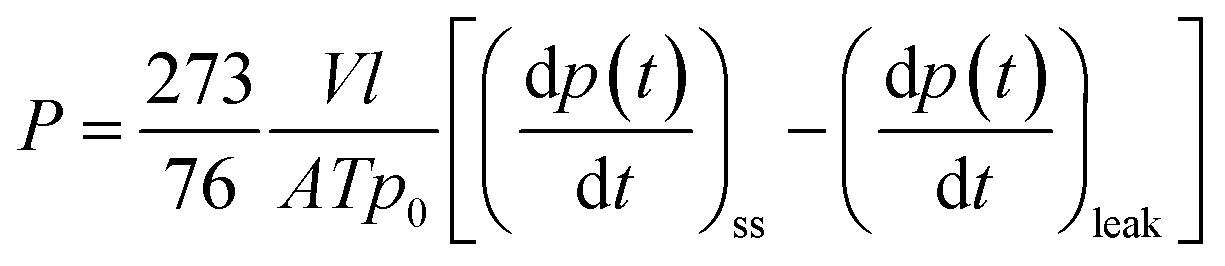 | (3) |
The diffusion coefficient (D) was obtained from the relation given in eqn (4).
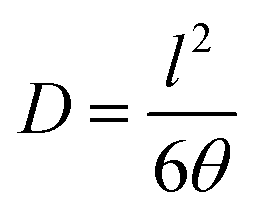 | (4) |
The solubility coefficient (S) was indirectly evaluated from S = P/D assuming the validity of the solution-diffusion permeation model. The ideal selectivity for a pair of gases A and B, which was calculated as the ratio for individual single gas permeability, can be decoupled into diffusivity selectivity and solubility selectivity (eqn (5)).
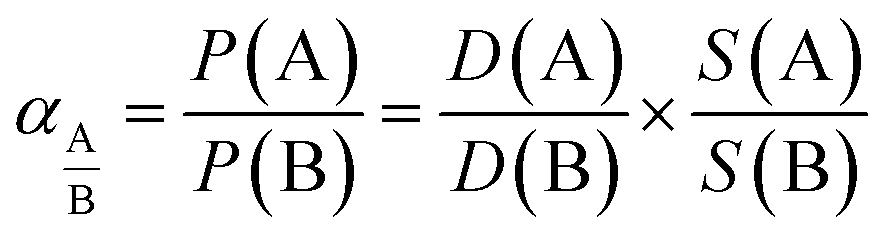 | (5) |
Computational methods
Computational chemistry was carried out by initially optimizing the structures at the AM1 level of theory.22 Subsequently, electronic energies and structures were calculated by full optimization, without any geometrical constraint, by using the Becke's three-parameter hybrid functional23 and the Lee et al. correlation functional with the 6-31G(d,p) basis set.24 The Restricted Hartree–Fock method, where the molecule is a closed shell system with all orbitals doubly occupied, was used for ground state molecules (RHF/B3LYP/6-31G(d,p)), while the unrestricted Hartree–Fock method, where the molecule is an open shell system with some of the electrons not paired, was used for radical species (UHF/B3LYP/6-31G(d,p)).25 Frequency calculations were used for all minimized structures to ensure that satisfactory minima were obtained. HOMO energies, zero-point electronic energies were determined by doing a single-point calculation with the hybrid B3LYP/6-31G+(d,p).26 The Gaussian 09 v.A1 and Gaussview v.5.08 program packages were used throughout this work.27,28 Molecular graphs and pictures were achieved with the GaussView and Arguslab programs.28,29Results and discussion
Synthesis and characterization of polymers
Two aromatic PEEKs were synthesized via nucleophilic aromatic substitution.30 The reaction proceeded by formation of bisphenolates, and subsequent ipso ether addition on the fluorine-substituted carbon atoms of the DFB, yielding a Meisenheimer complex, and posterior elimination of fluorine ions to give aromatic ether linkages. The synthetic scheme and the chemical structure of both polymers are shown in Fig. 1.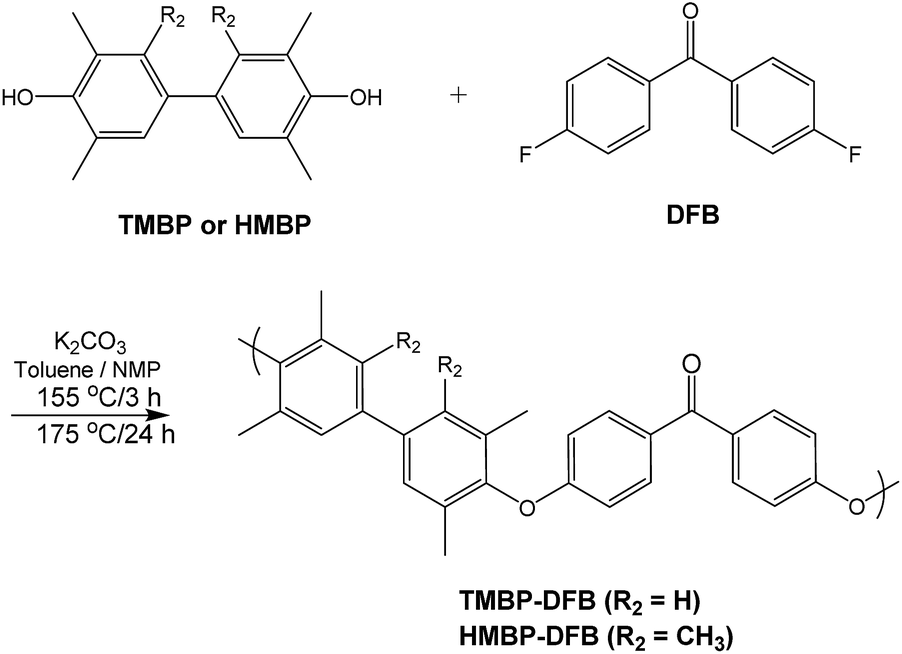 | ||
| Fig. 1 Synthesis of the poly(ether ether ketone)s. | ||
Chemical structures of polymers were confirmed by 1H and 13C NMR. As an example, Fig. 2 and 3 show the 1H NMR spectra of both polymers with the assignment of the signals. The integrations of the signals correspond well to the total number of protons.
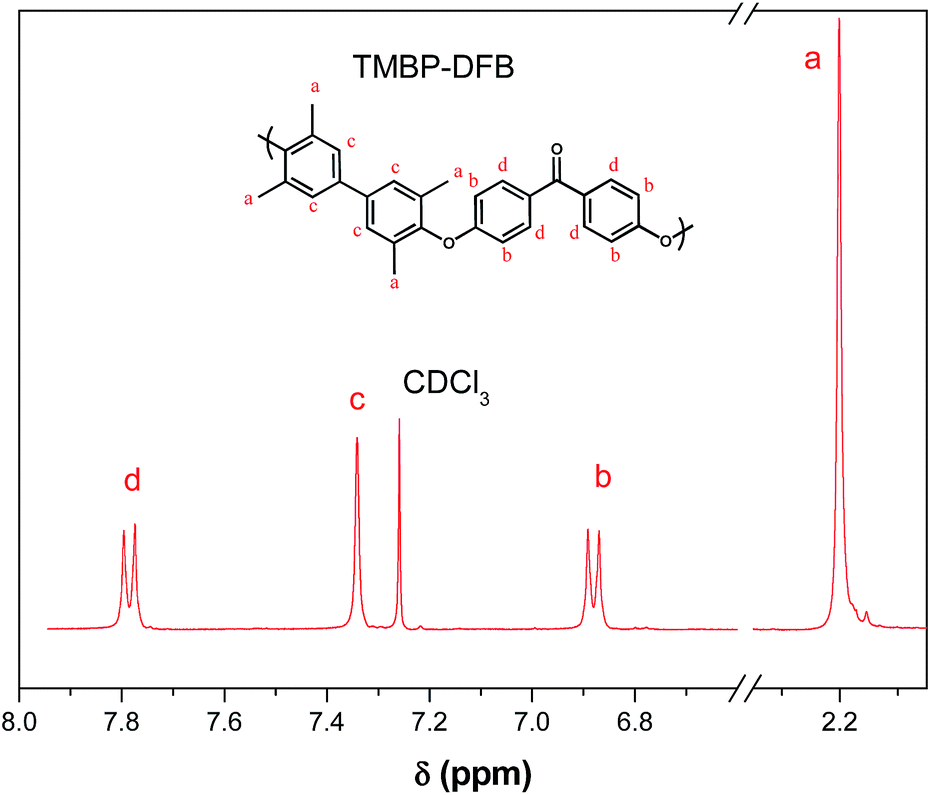 | ||
| Fig. 2 1H-NMR spectrum of TMBP-DFB in CDCl3. | ||
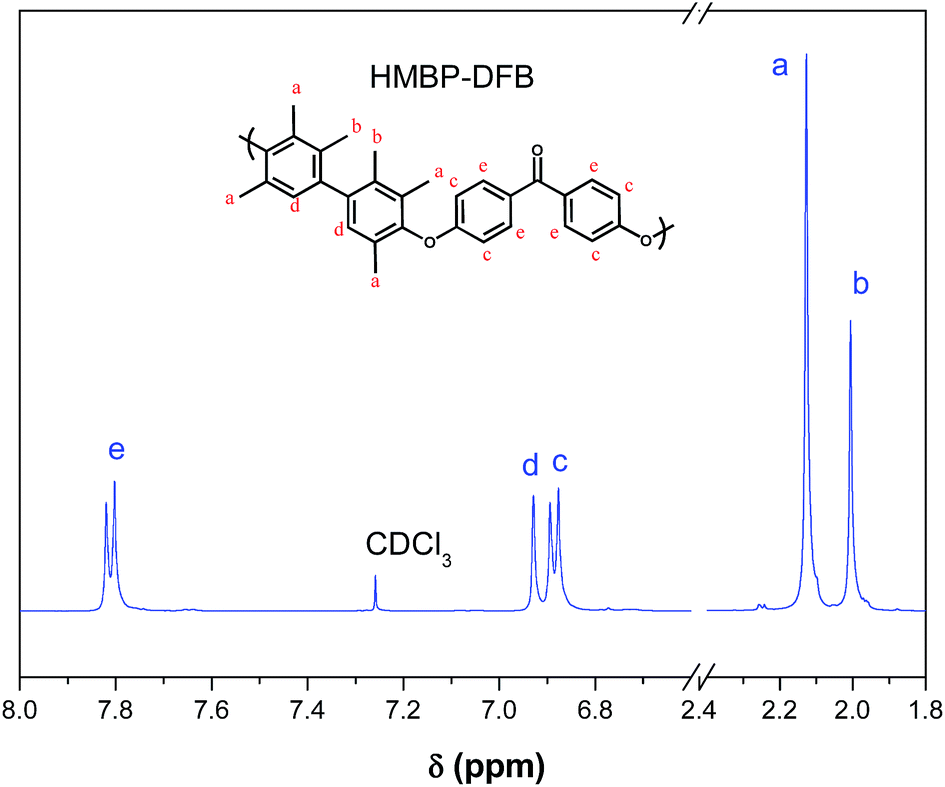 | ||
| Fig. 3 1H-NMR spectrum of HMBP-DFB in CDCl3. | ||
Despite the bulkiness introduced by the methyl groups on the diamines moieties, the solubility of polymers was poor in most common organic solvents, as seen in Table 1. None were soluble in typical GPC solvents, such as DMF or THF, and molecular weights of polymers were then estimated by measuring the inherent viscosities, whose values are also listed in Table 1. The values for both polymers were high enough, in particular for TMBP-DFB, indicating that high molecular weight polymers have been attained. The two polymers were soluble in CHCl3 and NMP, which allowed for the preparation of films by casting. The mechanical properties of the films were good, with elastic moduli near to 1.5 GPa and tensile strength values in the range of 90–110 MPa, to be used as gas separation membranes.
Additional thermal and physical properties of PEEKs are given in Table 2. Both polymers had thermal stabilities above 450 °C in nitrogen atmosphere, with high char yields, about 50%, as corresponds to highly aromatic structures. As expected, the glass transition temperatures, Tgs, were also high. No significant effect of the diamine structure on Tg was apparent, probably due to the conformational flexibility provided by the ether groups.
| Membranes (irradiation time) | Tda (°C) | R800b (%) | Tgc (°C) | ρ (g cm−3) | FFV | Gel fraction (%) |
|---|---|---|---|---|---|---|
| a Onset temperature at which decomposition begins.b Char yield at 800C.c Tg value was taken at the half-height of the heat capacity step from the second heating scan.d Estimated by considering that the repeat unit structure and molecular weight remain practically unchanged.e Density and gel fraction could not be measured because the membrane lost its integrity during immersion. | ||||||
| TMBP-DFB (0 min) | 460 | 46 | 263 | 1.118 | 0.165 | 0 |
| TMBP-DFB (30 min) | 450 | 45 | 267 | 1.135 | 0.152d | 20 |
| TMBP-DFB (60 min) | 455 | 35 | 273 | 1.228 | 0.083d | 75 |
| HMBP-DFB (0 min) | 455 | 58 | 267 | 1.101 | 0.168 | 0 |
| HMBP-DFB (30 min) | 450 | 37 | 278 | 1.211 | 0.084d | 75 |
| HMBP-DFB (45 min) | 455 | 42 | 285 | nme | — | nme |
Fractional free volume, FFV, was calculated from the experimental density of the membrane (ρ) using the relation (eqn (6)):31
| FFV = (V − 1.3Vw)/V | (6) |
The X-ray diffraction patterns revealed the amorphous nature of PEEKs, as seen in Fig. 4. The most probable intersegmental distance between the chains (d) was determined from the amorphous halo maximum using the Bragg equation (eqn (7)):
λ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinΘ sinΘ
| (7) |
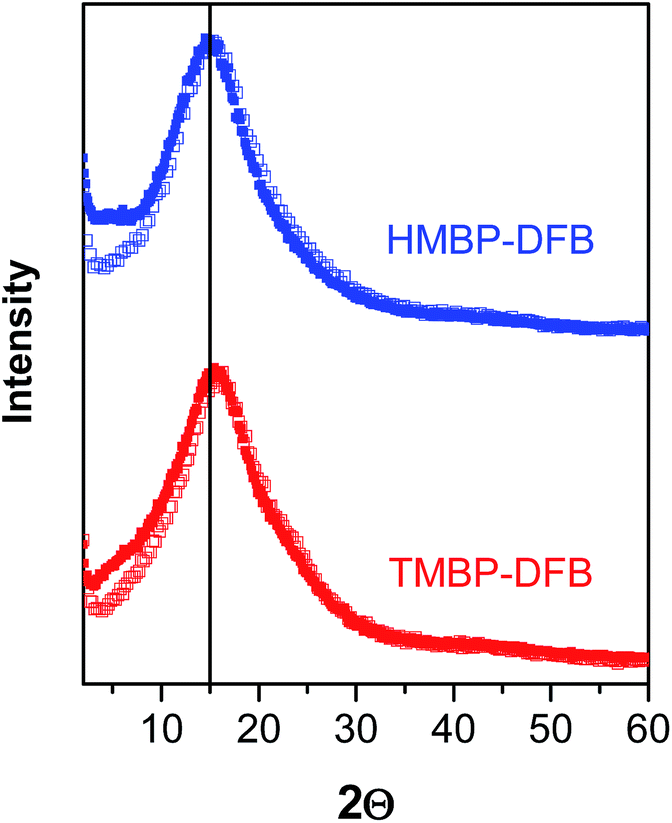 | ||
| Fig. 4 X-ray diffraction patterns of non-irradiated membranes (solid symbols) and irradiated membranes (open symbols) of PEEKs with gel fraction of 75%. | ||
UV crosslinking of the membranes
Membranes were irradiated in air, with ultraviolet light of wavelengths higher than 350 nm to selectively excite the n–π* absorption band of the benzophenone chromophore and then promote its photoreduction through abstraction of a vicinal H atom. Under these conditions, the undesirable secondary reactions, such as chain scission and/or photooxidation, should be minimized, even during long irradiation times, due to the lower energy of the light above 350 nm.34 The UV-vis spectra of both polymers showed an intense n–π* absorption band with no maxima at wavelengths between 350–500 nm, as seen in Fig. 5. The high absorbance values are due to the high concentration of benzophenone units in the membranes. According to the Beer–Lambert law, the fraction of incident light at a particular wavelength diminishes with depth in the film due to absorption. Thus, thick films are expected to have a crosslinking gradient. However, given the extremely high rate constant of benzophenone photoreduction (106–109 L mol−1 s−1) and the high photoreduction quantum yields (0.45–1) in good hydrogen donating media,35 the chromophore should rapidly react as irradiation proceeds, even for low values of incident light in the bottom of the film. In addition, membranes were irradiated in both sides to improve the homogeneity of the crosslinking reaction across the film. Exposure times were set long enough to assure maximum reduction of the benzophenone and minimal damage of mechanical properties of the membranes (irradiation times varying between 30–60 min).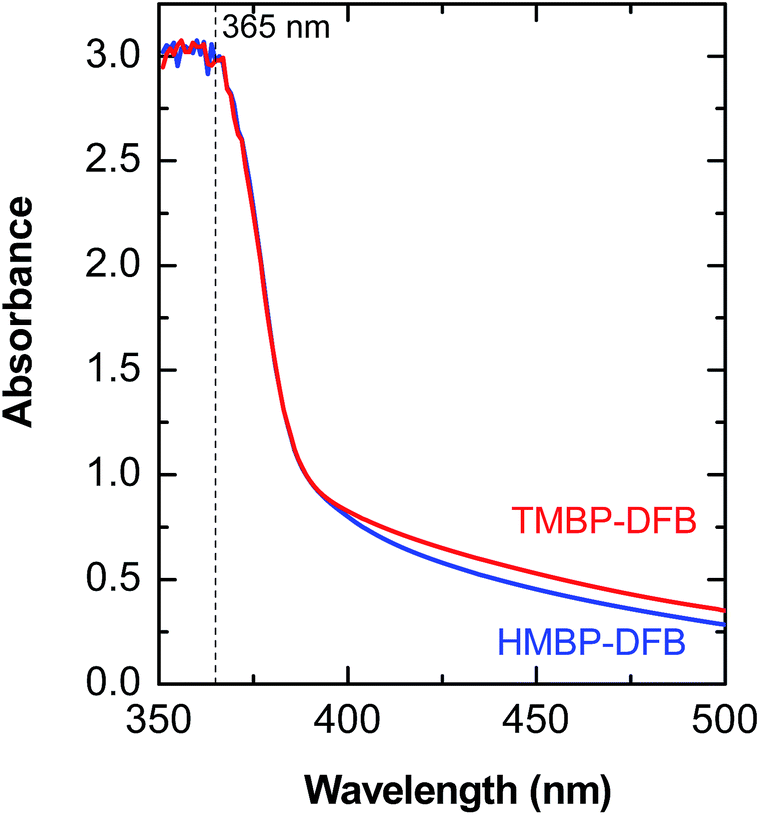 | ||
| Fig. 5 UV-vis spectra of the ketone containing films. The dashed line corresponds to the emission line for mercury (Hg) at 365 nm. | ||
Fig. 6 shows the most likely photoreduction mechanism and radical coupling reactions for the crosslinking of polymer chains: (A) the coupling between two benzylic radicals and (B) the coupling of benzylic radical with the ketyl carbon-centered radical. At the same time, benzylic radicals can also react with the oxygen in their surroundings to form peroxides, which tend rapidly to dissociate and yield carboxylic acids among other photooxidation products.36
 | ||
| Fig. 6 Expected mechanism of UV-induced crosslinking. | ||
The progress of crosslinking can be monitored by ATR-FTIR spectroscopy, by observing the decrease in the benzophenone carbonyl absorption band at 1652 cm−1 with the UV exposure time.20 Fig. 7 displays normalized ATR-FTIR spectra of TMBP-DFB and HMBP-DFB films as a function of irradiation time. After baseline correction, the spectra were normalized with respect to the band at 1592 cm−1, attributed to the stretching of substituted benzene moieties, which should remain unaffected by crosslinking. As shown in Fig. 7, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band intensity of benzophenone moiety, at 1655 cm−1, decreases with increasing UV irradiation time. Concomitantly, a broad band at around 1730 cm−1, which is attributed to carbonyl stretching from aldehydes and carboxylic acids generated from photooxidation, emerges and increases progressively in intensity with increasing UV irradiation time.37 These changes are also accompanied by the appearance of a new band at around 3300 cm−1 related to the stretching of hydroxyl groups generated via crosslinking and/or photooxidation. It should be noted that the benzophenone band of both membranes do not completely disappear even after 60 min irradiation, indicating that its photoreduction is not complete. Unfortunately, the quantitative analysis of the area under the peak at 1655 cm−1 could not be performed due to its overlapping with the broad peak at 1730 cm−1, which increases in intensity as the irradiation time increases. In addition, the peak at 1730 cm−1 was stronger for HMBP-DFB than for TMBP-DFB after 45 min of irradiation. This fact suggested a more rapid formation of photooxidation products in the case of HMBP-DFB, which was directly related to the loss of mechanical properties of the membranes upon longer irradiation time. Therefore, 60 min was set as the maximum irradiation time for TMBP-DFB and a maximum of 30 min of irradiation was used for HMBP-DFB.
O stretching band intensity of benzophenone moiety, at 1655 cm−1, decreases with increasing UV irradiation time. Concomitantly, a broad band at around 1730 cm−1, which is attributed to carbonyl stretching from aldehydes and carboxylic acids generated from photooxidation, emerges and increases progressively in intensity with increasing UV irradiation time.37 These changes are also accompanied by the appearance of a new band at around 3300 cm−1 related to the stretching of hydroxyl groups generated via crosslinking and/or photooxidation. It should be noted that the benzophenone band of both membranes do not completely disappear even after 60 min irradiation, indicating that its photoreduction is not complete. Unfortunately, the quantitative analysis of the area under the peak at 1655 cm−1 could not be performed due to its overlapping with the broad peak at 1730 cm−1, which increases in intensity as the irradiation time increases. In addition, the peak at 1730 cm−1 was stronger for HMBP-DFB than for TMBP-DFB after 45 min of irradiation. This fact suggested a more rapid formation of photooxidation products in the case of HMBP-DFB, which was directly related to the loss of mechanical properties of the membranes upon longer irradiation time. Therefore, 60 min was set as the maximum irradiation time for TMBP-DFB and a maximum of 30 min of irradiation was used for HMBP-DFB.
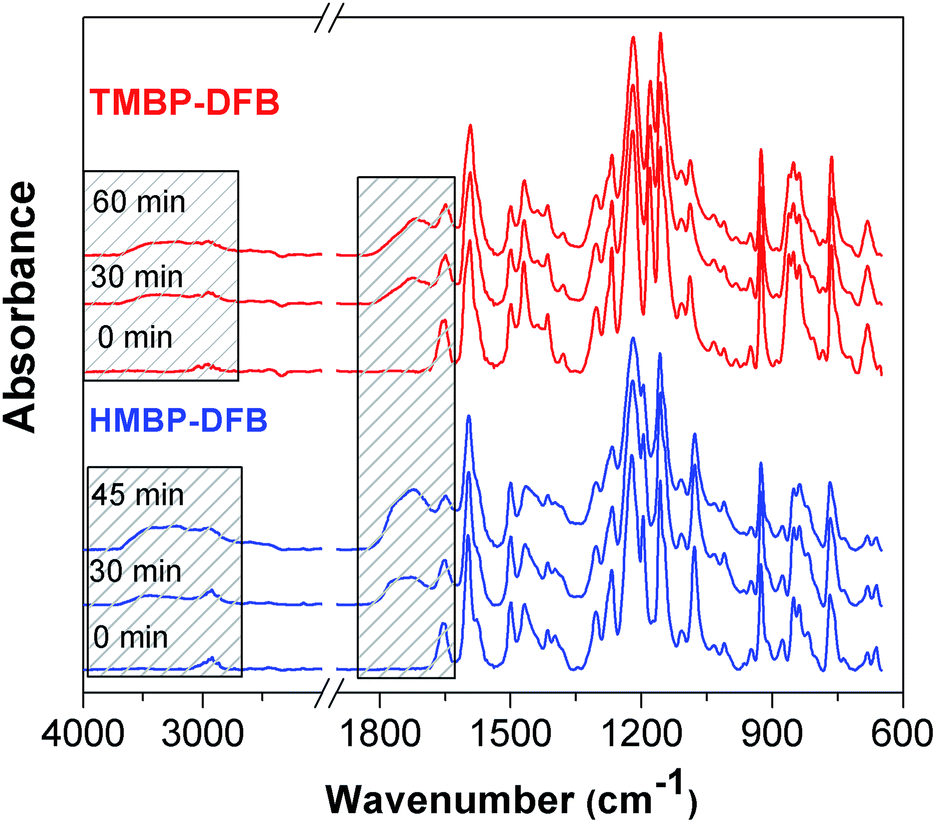 | ||
| Fig. 7 FTIR spectra of PEEKs films as a function of UV-irradiation time. The bands affected by the irradiation are shaded. | ||
The gel fraction of the irradiated membranes can be considered as a measure for assessing the extent of crosslinking. As expected, the gel fractions increased with increasing irradiation time (see Table 2). Thus, the gel fractions for TMBP-DFB samples went from 20% at tirr = 30 min to 75% at tirr = 60 min, confirming that the crosslinked thickness progressed with irradiation time. For HMBP-DFB membranes the exposure time required to achieve 75% gel fraction was considerably shorter, only 30 min. When HMBP-DFB membrane was irradiated for 45 min it became brittle, and the gel fraction could not be measured because it lost its integrity during immersion. Thereby, it was not tested as a gas separation membrane. This behavior could be consistent with the higher proportion of benzylic methyl groups in HMBP units, but could be also indicative of a higher reactivity of these groups to give radicals in comparison to those of TMBP-DFB. Therefore, we have performed quantum mechanical calculations to determine the ability of the different methyl groups to give –CH2˙ radicals. These calculations will be commented on below.
The density of both irradiated membranes with 75% gel fraction increased by 10% compared to the value of non-irradiated ones, as seen in Table 2. This increase indicates a densification of the polymer membranes upon photocrosslinking, which could be associated with a decrease in FFV. In fact, considering that the chemical structure of polymers hardly changes during crosslinking, the FFV of crosslinked films significantly decreased with the irradiation time (Table 2). The FFV decreased to half the original value for the indicated 10% increase in density. Moreover, the FFV was the same for the crosslinked membranes of both PEEKs with a 75% gel fraction. This densification of the membrane would be expected to be reflected in a decrease of the more probably intersegmental distances obtained by X-ray diffraction. However, no significant change in the position of the amorphous halo was visible, as seen in Fig. 4, where the patterns of non-irradiated membranes are compared to those of crosslinked ones with a 75% gel fraction. Only, a narrowing on the lower angle side of the amorphous halo of the crosslinked membrane patterns could be observed.
As it can be also seen in Table 2, the thermal stability of the membranes was not affected by crosslinking. In contrast, the glass transition temperatures of the irradiated membranes increased as the extent of crosslinking increased. The Tgs of crosslinked membranes with a 75% of gel fraction were about 10 °C higher than the values of non-crosslinked ones.
Quantum mechanical calculations on radical formation ability
As commented on above, the crosslinking of the membranes takes place through the reaction between radicals previously formed by UV irradiation. As the combination of two radicals occurs without a significant energy barrier, the process that controls the feasibility of crosslinking is the formation of the radicals. To assess the ability of the different groups to yield radicals, and therefore, to determine the influence of the number of methyls on the crosslinking process, a quantum mechanical study of the heat of formation of several models that represent the structure of the polymers and of their corresponding radicals, obtained by abstraction of one hydrogen radical, was performed. As the aromatic ketones are common in all structures, the models represent the moiety that is different in every case. With comparative purposes, the radical derived from tetramethyl bisphenol A (M-TMBPA), and two possible radicals formed from tetramethyl bisphenol F (M-TMBPF) were also studied, because these monomers have been previously employed in the same type of reaction.20,21 All the models are shown in Fig. 8. In the case of M-TMBP, only one monoradical is possible, corresponding to its formation on methyl groups placed in 3-position. However, in M-HMPB, three different radicals are possible, depending on the methyl groups which suffer the reaction.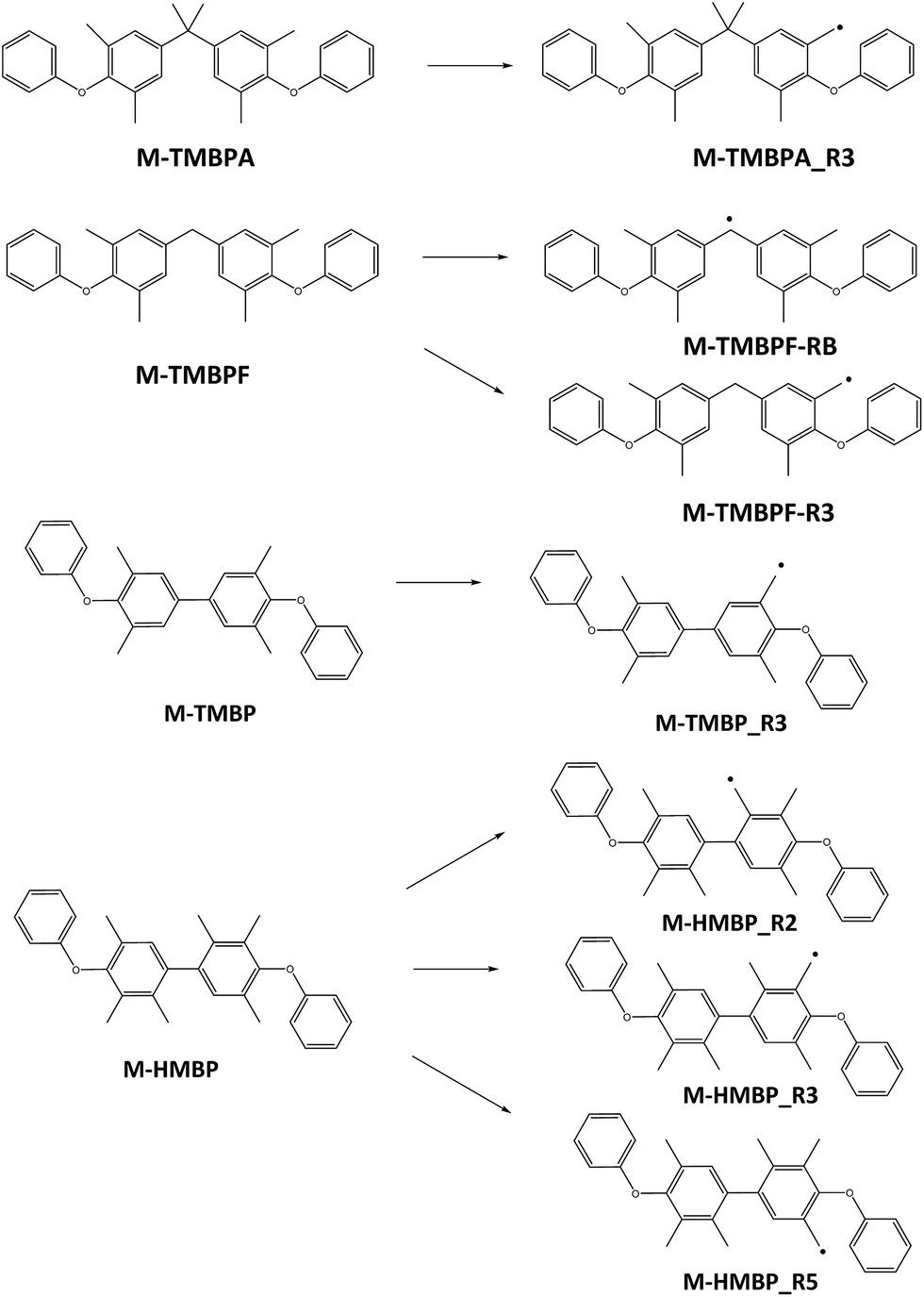 | ||
| Fig. 8 Structure of the possible radicals that can be obtained from the model structures. | ||
The second column of Table 3 shows the heat of formation, in eV, of the model compounds and their corresponding radicals. Column 3 shows the energy differences, in eV and in kcal mol−1, between the radicals and the model compounds (it should be taken into account that the loss of a hydrogen radical has also to be considered; energy of hydrogen radical = −13613 eV). From these data, it can be seen that the maximum energy difference corresponds to M-TMBP-R3 that, consequently, is more difficult to form during irradiation, but the energy is very similar to that of M-TMBPA-R3 and M-TMBPF-R3. This indicates that, for tetramethyl-substituted bisphenols, the reactivity is similar in all cases, irrespective of the bridge between the rings. Moreover, in the case of M-TMBPF, the energy difference is lower for the bridge radical, which consequently is more probable than any of the methylene radicals.
| Molecule | Energy (eV) | Energy difference (EM_R + EH) − EM | |
|---|---|---|---|
| (eV) | (kcal mol−1) | ||
| M-TMBPA | −36765.95 | — | — |
| M-TMBPA-R3 | −36748.22 | 4.117 | 94.939 |
| M-TMBPF | −34626.41 | — | — |
| M-TMBPF-RB | −34609.06 | 3.737 | 86.176 |
| M-TMBPF-R3 | −34608.67 | 4.127 | 95.169 |
| M-TMBP | −33556.63 | — | — |
| M-TMBP-R3 | −33538.88 | 4.137 | 95.400 |
| M-HMBP | −35695.25 | — | — |
| M-HMBP-R2 | −35678.57 | 3.067 | 70.725 |
| M-HMBP-R3 | −35678.54 | 3.097 | 71.417 |
| M-HMBP-R5 | −35678.53 | 3.107 | 71.648 |
As to the hexamethyl-substituted model (M-HMBP), the presence of additional methyl groups seems to favor the formation of radicals, irrespective of the methyl group affected. In fact, the energy of the three possible radicals is approximately 24 kcal mol−1 lower than the energy of the methylene radicals formed in the other models, and about 15 kcal mol−1 lower than for the bridge radical in the case of M-TMBPF.
Consequently, the theoretical results support the experimental ones, commented on above, where the polymer HMBP-DFB reached the same gel fraction as TMBP-DFB did in a shorter irradiation time, and the higher reactivity of the polymer cannot be solely attributed to the higher amount of methyl groups, but to the higher feasibility of formation of these radicals. Moreover, these results indicate that the HMBP-DBF should be the most reactive among any of the polymers considered here.
Gas transport properties
The single gas permeabilities of He, O2, N2, CH4 and CO2 for both linear PEEKs and UV-crosslinked analogs were determined using a constant volume-variable pressure permeator, and the results at a feed pressure of 3 bar and 30 °C are shown in Table 4. Gas permeabilities of linear PEEK membranes were lower for HMBP-DFB, except for He. This decrease in permeability was also accompanied by an improvement in selectivity. Taking into account that FFV is slightly higher in the latter polymer and, moreover, He is the gas molecule with smaller size, the lower permeabilities would be consistent with a distribution of FFV containing a higher number of smaller elements, which would be not accessible to the rest of gases with larger sizes. In contrast, P(CO2) is close to P(He) and P(CH4) is somewhat higher than P(N2) in the case of TMBP-DFB membrane, which is a typical behavior of polymers having high FFV.38–40 These results suggest that the methyl groups could be partially occupying the void space (internal free volume), which would cause a decrease in both the overall FFV and the size of accessible holes.41| Sample (time of irradiation) | Permeabilitya (barrer) | Ideal selectivity | |||||
|---|---|---|---|---|---|---|---|
| P(He) | P(O2) | P(N2) | P(CO2) | P(CH4) | P(O2)/P(N2) | P(CO2)/P(CH4) | |
| a 1 barrer = 10−10 cm3 (STP) cm per cm2 per s per cm Hg. | |||||||
| TMBP-DFB | 45 ± 1 | 8.3 ± 0.1 | 1.74 ± 0.04 | 41 ± 1 | 2.11 ± 0.06 | 4.8 ± 0.1 | 19.3 ± 0.7 |
| TMBP-DFB (30 min) | 38 ± 1 | 4.7 ± 0.1 | 0.72 ± 0.02 | 18.8 ± 0.6 | 0.57 ± 0.02 | 6.5 ± 0.2 | 33 ± 2 |
| TMBP-DFB (60 min) | 37 ± 1 | 2.73 ± 0.06 | 0.30 ± 0.01 | 8.3 ± 0.3 | 0.155 ± 0.006 | 9.1 ± 0.4 | 53 ± 3 |
| HMBP-DFB | 51 ± 1 | 6.38 ± 0.06 | 1.15 ± 0.02 | 28.4 ± 0.4 | 1.06 ± 0.02 | 5.5 ± 0.1 | 26.8 ± 0.6 |
| HMBP-DFB (30 min) | 41 ± 1 | 2.34 ± 0.03 | 0.274 ± 0.004 | 8.4 ± 0.2 | 0.125 ± 0.003 | 8.5 ± 0.2 | 67 ± 3 |
The UV-crosslinking process led to a drop in gas permeabilities in both polymers. The decrease was higher for gases with large kinetic diameter, and was much higher for CH4 gas. This is the typical behavior of glassy polymers with relatively low FFV. For the membranes with a 75% gel fraction, P(CO2) was almost five times lower than P(He) and P(CH4) was two times lower than P(N2). This decrease in permeability coefficients brought about a significant improvement of the O2/N2 and CO2/CH4 selectivities. These facts can be related to the above commented on decrease in FFV with crosslinking.
Since permeability is the product of diffusion, D, and solubility, S, coefficients, the selectivity can be separated in diffusivity selectivity and solubility selectivity and the values for linear and crosslinked membranes are listed in Tables 5 and 6. As it happens with the permeabilities, the diffusivities for all gases are considerably reduced with increasing irradiation times, and almost the same values are obtained for TMBP-DFB (60 min) and for HMBP-DFB (30 min), which have the same value of FFV. However, no effect of crosslinking was observed in solubility and solubility selectivity.
| Sample (time of irradiation) | Diffusivity, (D × 108 cm2 s−1) | Diffusivity selectivity | ||||
|---|---|---|---|---|---|---|
| D(O2) | D(N2) | D(CO2) | D(CH4) | D(O2)/D(N2) | D(CO2)/D(CH4) | |
| TMBP-DFB | 6.7 ± 0.2 | 1.85 ± 0.04 | 3.17 ± 0.07 | 0.52 ± 0.01 | 3.6 ± 0.1 | 6.1 ± 0.2 |
| TMBP-DFB (30 min) | 4.0 ± 0.2 | 0.70 ± 0.02 | 1.35 ± 0.04 | 0.139 ± 0.007 | 5.7 ± 0.3 | 9.7 ± 0.3 |
| TMBP-DFB (60 min) | 2.31 ± 0.05 | 0.30 ± 0.04 | 0.628 ± 0.006 | 0.0372 ± 0.0005 | 7.7 ± 0.2 | 16.9 ± 0.2 |
| HMBP-DFB | 5.2 ± 0.2 | 1.35 ± 0.06 | 2.50 ± 0.07 | 0.274 ± 0.008 | 3.8 ± 0.1 | 9.1 ± 0.4 |
| HMBP-DFB (30 min) | 1.92 ± 0.04 | 0.32 ± 0.07 | 0.72 ± 0.01 | 0.0318 ± 0.0009 | 5.1 ± 0.2 | 16.1 ± 0.6 |
| Sample (time of irradiation) | Solubility, (S × 103 cm3 (STP)cm−3 cmHg−1) | Solubility selectivity | ||||
|---|---|---|---|---|---|---|
| S(O2) | S(N2) | S(CO2) | S(CH4) | S(O2)/S(N2) | S(CO2)/S(CH4) | |
| TMBP-DFB | 12.2 ± 0.4 | 9.4 ± 0.3 | 129 ± 4 | 40 ± 2 | 1.30 ± 0.06 | 3.2 ± 0.2 |
| TMBP-DFB (30 min) | 11.8 ± 0.6 | 10.3 ± 0.4 | 139 ± 7 | 41 ± 1 | 1.14 ± 0.08 | 3.4 ± 0.4 |
| TMBP-DFB (60 min) | 11.8 ± 0.4 | 10 ± 2 | 132 ± 7 | 42 ± 2 | 1.2 ± 0.2 | 3.1 ± 0.2 |
| HMBP-DBF | 12.3 ± 0.3 | 8.5 ± 0.4 | 114 ± 3 | 39 ± 1 | 1.45 ± 0.07 | 2.9 ± 0.1 |
| HMBP-DBF (30 min) | 12.2 ± 0.6 | 8.6 ± 0.4 | 117 ± 4 | 38 ± 1 | 1.4 ± 0.4 | 3.1 ± 0.3 |
Gas diffusion coefficients for non-crosslinked and crosslinked membranes of TMBP-DFB and HMBP-DFB were compared by correlating data sets using eqn (8).42
|
lnD = α + βdg2
| (8) |
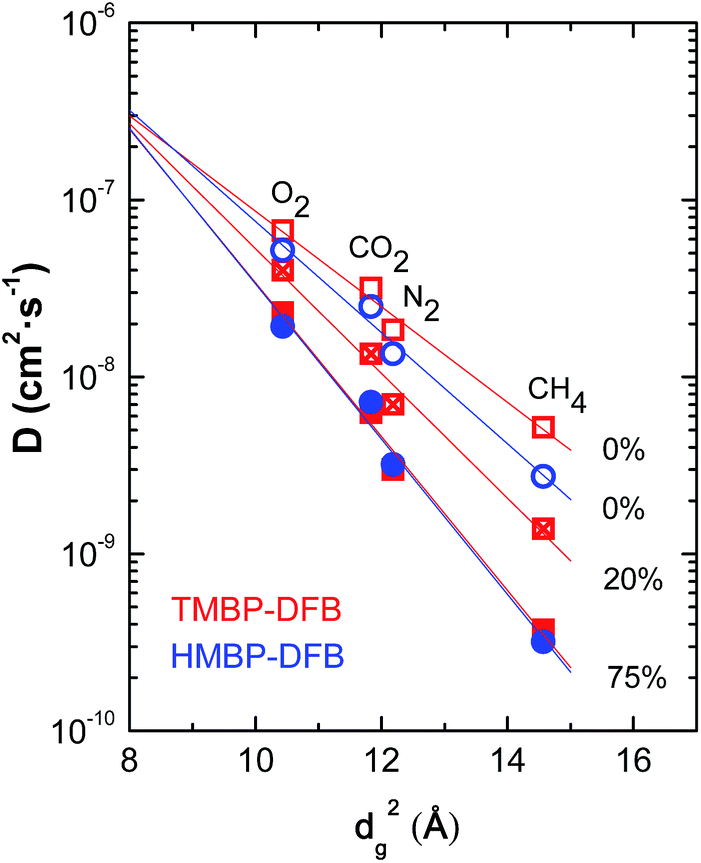 | ||
| Fig. 9 Dependence of diffusion coefficient on gas diameter squared for non-irradiated (open symbols) and irradiated (solid symbols) membranes. The numbers indicate the gel fraction of the membranes. The lines correspond to the linear fits to eqn (8). | ||
Fig. 10 presents the permeability to CO2 as a function of feed pressure for non-crosslinked membranes and crosslinked ones with a 75% gel fraction. The permeability of both linear polymers decreases with increasing pressure, mainly at low feed pressures, in a lower extension for HMBP-DFB, consistent with dual-sorption model.18 No plasticization behavior was observed in the measured pressure range. Both crosslinked membranes showed the same behavior and, moreover, the effect of the feed pressure on P(CO2) was significantly less pronounced.
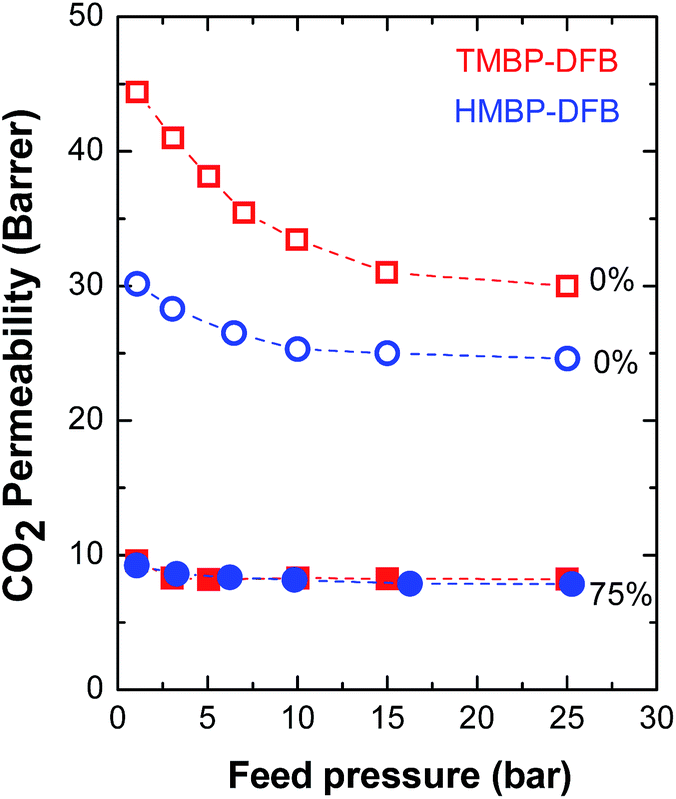 | ||
| Fig. 10 CO2 permeability as a function of feed pressure for non-irradiated (open symbols) and irradiated (solid symbols) membranes. The numbers indicate the gel fraction of the membranes. | ||
To evaluate the effect of crosslinking on the productivity of these membranes, Robeson upper bound plots for the O2/N2 and CO2/CH4 gas pairs are shown in Fig. 11.4,5 The bibliographic data of two different benzophenone-containing PEEKs, one derived from 4,4-methylene bis-(2,6-xylenol) (tetramethyl bisphenol-F or TMBPF)21 and other from 2,2-bis(3,5-dimethyl-4-hydroxyphenyl)propane (tetramethyl bisphenol-A or TMBPA),15 and of Matrimid have been also reported for comparison. Both PEEKs have a similar structure to these described in this paper, but bear a flexibilizing moiety in the bisphenol unit. Crosslinking of the films clearly moves the balance of selectivity versus permeability closer to the Robeson upper bound for both O2/N2 and CO2/CH4 gas pairs. The greatest improvement occurs for O2/N2, where a selectivity increase is observed in every case, much higher than for the reference PEEKs, even if the gel fraction was lower. This increase of gas productivity was so high that the UV-irradiated membranes surpassed the 1991 Robeson limit, with TMBP-DFB (75) nearly reaching the O2/N2 2008 upper bound. This result is outstanding for common glassy polymers.
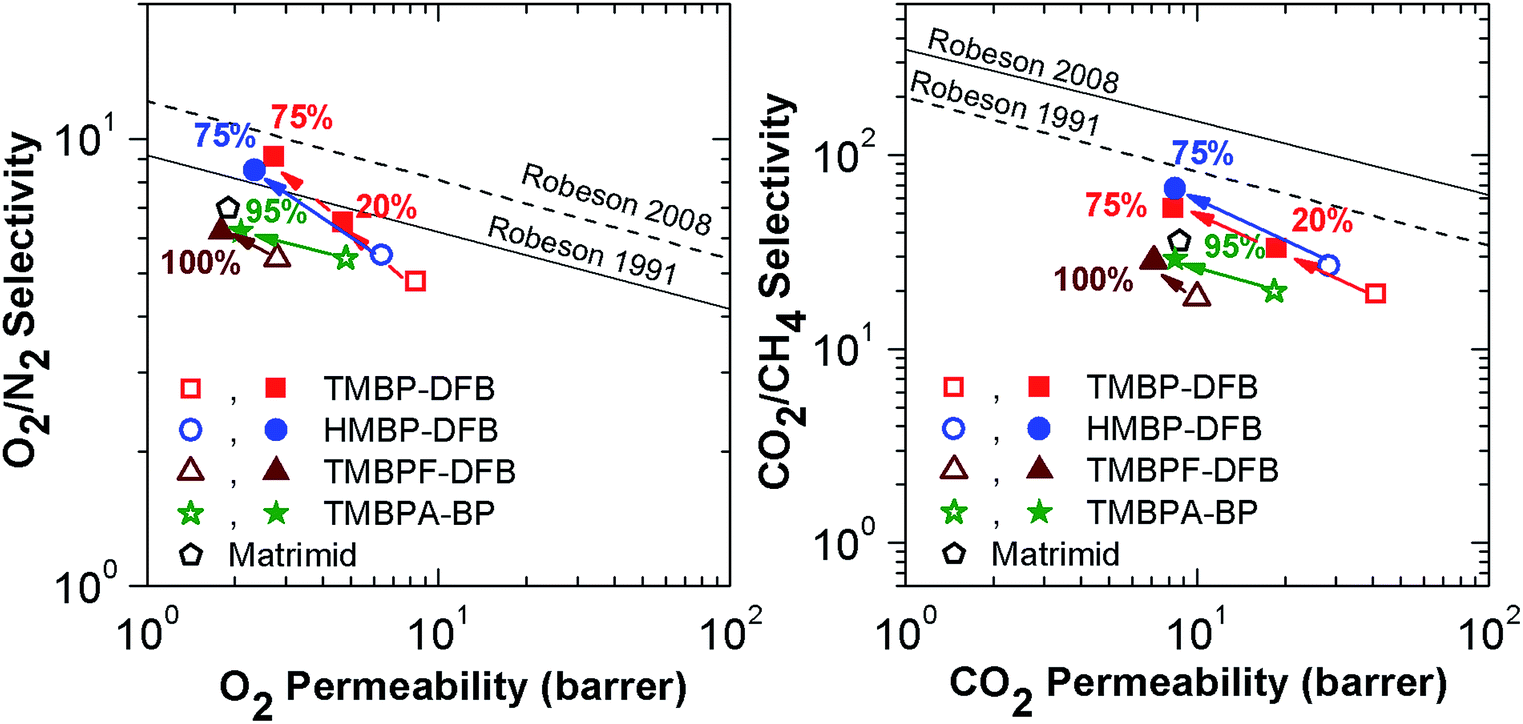 | ||
| Fig. 11 Upper bound plots for O2/N2 and CO2/CH4 gas pairs of TMBP-DFB and HMBP-DFB comparison of linear and crosslinked TMBP-DFB and HMBP-DFB, TMBPF-DFB21 (permeability at 10 bar and 35 °C) and TMBPA-BP15 (permeability at 10 bar and 35 °C) and Matrimid33 (permeability at 1 bar and 35 °C). Open symbols are used for non-irradiated membranes, while solid symbols represent the irradiated ones. The numbers indicate the gel fraction of the membranes. | ||
Conclusions
The UV-crosslinking of dense membranes of two poly(ether ether ketone)s derived from difluorobenzophenone and bisphenol derivatives, with four or six methyl groups, led to an significant improvement in the gas transport properties. Crosslinking and photooxidation reactions took place during the irradiation process using polychromatic light, with wavelengths above 350 nm, at room temperature and in presence of air. The two polymers were able to produce highly crosslinked membranes, with gel fractions close to 75% and similar FFV. However, the polymer with higher number of methyl groups on the biphenyl moiety (HMBP-DFB) required half the time to achieve the same gel fraction than the polymer with four methyl groups (TMBP-DFB).A DFT quantum mechanical study has stated that the higher crosslinking reactivity of HMBP-DFB is not only due to the higher amount of methyl groups, but also to the higher feasibility of formation of methylene radicals in this case.
The gas permeability in both membranes strongly decreased with the irradiation time. In particular the decrease in permeability was much higher for gases with larger size, because the crosslinking enhanced the size-sieving capability. This behavior was reflected in a considerable increase for O2/N2 and CO2/CH4 selectivities. In that way, UV irradiation moved the gas transport properties towards Robeson upper bound of 1991, even surpassing this limit for O2/N2. Moreover, crosslinking is expected to provide a good mechanical integrity to the membrane and is able to counteract the effect of degradation if the irradiation conditions are well controlled.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support provided by MINECO (MAT2013-45071-R, CTQ2012-31076 and MAT2016-76413-C2-R2) and by the National Science Foundation under grant number IIP-1237857. Authors wish to thank Judit González for helping with the characterization of polymers.References
- Y. Yampolskii, Macromolecules, 2012, 45, 3298–3311 CrossRef CAS.
- R. W. Baker and K. Lokhandwala, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- P. Bernardo and G. Clarizia, Chem. Eng. Trans., 2013, 32, 1999–2004 Search PubMed.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M Robeson, J. E McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- M. Calle, C. García, A. E. Lozano, J. G. de la Campa, J. de Abajo and C. Álvarez, J. Membr. Sci., 2013, 434, 121–129 CrossRef CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- R. A. Hayes, Polyimide gas separation membranes, US Pat., 4 717 393, 1988.
- K. Vanherck, G. Koeckelberghs and I. F. J. Vankelecom, Prog. Polym. Sci., 2013, 38, 874–896 CrossRef CAS.
- C. T. Wright and R. D. Paul, J. Membr. Sci., 1997, 124, 161–174 CrossRef CAS.
- J. McGrath, Y. Chen, R. Guo and B. Freeman, Crosslinked polymer compositions, gas separation membranes of such crosslinked polymer compositions, methods of making such membranes, and methods of separating gases using such membranes, US Pat., 9 533 254 B2, 2017.
- J. S. Park, K. L. Gleason, K. E. Gaines, S. J. Mecham, J. E. McGrath and B. D. Freeman, Energy Procedia, 2014, 63, 210–216 CrossRef CAS.
- J. R. Rowlett, Q. Liu, W. Zhang, J. D Moon, M. E. Dose, J. S. Riffle, B. D. Freeman and J. E. McGrath, J. Mater. Chem. A, 2016, 4, 16047–16051 CAS.
- A. Shaver, J. D. Moon, D. Savacool, W. Zhang, G. Narang, G. Miller, B. Vondrasek, J. J. Lesko, B. D. Freeman, J. S. Riffle and J. E. McGrath, Polymer, 2017, 114, 135–143 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and R. D. Paul, Polymer, 2009, 50, 5565–5575 CrossRef CAS.
- A. Bos, I. G. M. Pünt, M. Wessling and H. Strathmann, J. Membr. Sci., 1999, 155, 67–78 CrossRef CAS.
- M. S. McCaig and D. R. Paul, Polymer, 1999, 40, 7209–7225 CrossRef CAS.
- Q. Liu, Q. A. T. Shaver, Y. Chen, G. Miller, D. R. Paul, J. S. Riffle, J. E. McGrath and B. D. Freeman, Polymer, 2016, 87, 202–214 CrossRef CAS.
- B. J. Sundell, A. T. Shaver, Q. Liu, A. Nebipasagil, P. Pisipati, S. J. Mecham, B. D. Freeman and J. E. McGrath, Polymer, 2014, 55, 5623–5634 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5654 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- J. B. Foresman and Æ. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian Inc., Pittsburgh, 1996 Search PubMed.
- H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09 (Revision A.1), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView (Version 5), Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- M. A. Thompson, ArgusLab 4.0.1, Planaria Software LLC, Seattle, WA, 1997-2004 Search PubMed.
- A. E. Lozano, M. L. Jimeno, J. de Abajo and J. G de la Campa, Macromolecules, 1994, 27, 7164–7170 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- HyperChem(TM) Professional (Version 8.0.3), Hypercube, Inc., Florida, USA Search PubMed.
- M. D. Guiver, G. P. Robertson, Y. Dai, F. Bilodeau, Y. S. Kang, K. J. Lee, J. Y. Jho and J. Won, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4193–4204 CrossRef CAS.
- A.-K. Schuler, O. Prucker and J. Rühe, Macromol. Chem. Phys., 2016, 217, 1457–1466 CrossRef CAS.
- J. P. Wagner, R. J. Truman, A. E. Puchalski and R. Wake, J. Am. Chem. Soc., 1986, 108, 1127–1138 Search PubMed.
- S. Giancaterina, A. Rossi, A. Rivaton and J. L. Gardette, Polym. Degrad. Stab., 2000, 68, 133–144 CrossRef CAS.
- I. K. Meier and M. Langsam, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 83–89 CrossRef CAS.
- M. Calle, A. E. Lozano, J. de Abajo, J. G. de la Campa and C. Álvarez, J. Membr. Sci., 2010, 365, 145–153 CrossRef CAS.
- J. L. Santiago-García, C. Álvarez, F. Sánchez and J. G. de la Campa, J. Membr. Sci., 2015, 476, 442–448 CrossRef.
- C. Álvarez, A. E. Lozano and J. G. de la Campa, J. Membr. Sci., 2016, 501, 191–198 CrossRef.
- J. R. Wiegand, Z. P. Smith, Q. Liu, C. T. Patterson, B. D. Freeman and R. Guo, J. Mater. Chem. A, 2014, 2, 13309–13320 CAS.
- L. M. Robeson, Q. Liu, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2015, 476, 421–431 CrossRef CAS.
- L. M. Robeson, A. P. Smith, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2014, 453, 71–83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |