
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a novel biosynthetic pathway in Escherichia coli for the production of caffeine
Mengmeng Li a,
Ying Suna,
Si-an Pana,
Wei-wei Denga,
Oliver Yub and
Zhengzhu Zhang*a
a,
Ying Suna,
Si-an Pana,
Wei-wei Denga,
Oliver Yub and
Zhengzhu Zhang*a
aState Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei 230036, Anhui, People's Republic of China. E-mail: 11720502@ahau.edu.cn; 1441665707@qq.com; 1908852722@qq.com; dengweiwei@ahau.edu.cn; zzz@ahau.edu.cn
bWuxi NewWay Biotechnology, 100 Konggang Road, Wuxi, Jiangsu 214145, China. E-mail: oliver.yu@conagen-inc.com
First published on 15th December 2017
Abstract
Caffeine (Cf, 1,3,7-trimethylxanthine), a major secondary metabolite of many higher plants, is widely used in popular non-alcoholic beverages, and in the pharmaceutical and health industries. Currently, this valuable chemical is mainly manufactured by chemical synthesis. In this study, we developed a novel approach for de novo caffeine production in metabolically engineered Escherichia coli. Xanthine-to-caffeine conversion was first achieved by the expression of a plant-derived gene encoding tea caffeine synthase (TCS1). Caffeine accumulation was then increased using two metabolic strategies: higher-level expression of the target enzymes, and enhancement of xanthine and S-adenosyl-L-methionine biosynthesis. The final strain (BL21/pRSF-eCS1-SAM2-vgb-eGUD1) produced up to 21.46 ± 1.03 mg L−1 caffeine from 20 g L−1 of glucose in shake flask culture, yielding caffeine up to 2.96 mg g−1 glucose, which represents the highest titer of caffeine produced by fermentation reported to date. This novel microbial conversion also represents an innovative approach to produce value-added methylxanthine chemicals from cheap carbon sources.
Introduction
As one of the most popular purine alkaloids, caffeine (1,3,7-trimethylxanthine) represents a characteristic secondary metabolite derived from purine nucleotides in many higher plants. Previous studies have shown that caffeine plays an important role in chemical defense and allelopathy during plant growth.1,2 In addition to these physiological functions, caffeine has many valuable pharmacological effects. Caffeine has a desirable stimulatory effect on the central nervous system. A moderate amount of caffeine intake is beneficial for human health, as strong evidence reveals that caffeine is associated with a reduced risk of many diseases such as Parkinson's disease, Alzheimer's disease, type 2 diabetes, depression, and cancer of the liver and endometrium.3–5Caffeine is currently the most routinely ingested bioactive compound, and exists in a variety of caffeine-containing beverages include tea, coffee, soft drinks, and energy drinks, as well as in caffeinated food products. However, despite the growing demand for healthy green caffeine production, this valuable chemical is mainly manufactured by chemical synthesis, which is associated with environmental pollution.6 Industrial and natural green production of caffeine is still dependent on whole plant extraction, which is limited by the limited low caffeine in plants and the long growing cycle.7,8 In recent years, with the rapid development of genetic engineering and metabolic engineering, microbial platforms have shown increasing potential to produce plant-derived metabolites on an industrial scale. Compared with the traditional process, the microbial production of natural chemicals has the potential for a shorter production cycle and higher productivity.9 Correspondingly, research has increasingly focused on a diverse range of chemicals with the potential to be produced by microbial fermentation. Many bio-based products have been successfully synthesized from renewable resources by heterologous microorganisms, including biodiesel, aromatic compounds, and amino acids.10–15 These examples demonstrated that the microbial platform is economical and environmentally friendly approach for the production of plant-derived compounds on an industrial scale. Consequently, synthesis of caffeine by metabolic engineering of microbes is of great scientific and commercial interest.
Currently, tea and coffee plants are the major materials used for studies on caffeine biosynthesis. A number of studies have demonstrated that the major biosynthetic route for caffeine is almost the same in tea and coffee plants.16 Tea plants have relatively high concentrations of caffeine, theobromine, and theophylline. Some xanthine-based alkaloids can be synthesized via the synthesis of caffeine. The major caffeine biosynthetic pathway in tea plants is essentially the same as that in other purine alkaloid-accumulating plants, which begins with xanthosine and proceeds via three successive N-methylations of xanthosine, 7-methylxanthine, and theobromine. A number of genes encoding tea caffeine synthase have been cloned. In addition, previous studies with radiolabelled precursors indicated the existence of several minor pathways in tea leaves that also play a role in caffeine metabolism. Among these pathways, the 7-methylxanthine → paraxanthine → caffeine pathway and the theophylline → 3-methylxanthine → theobromine → caffeine pathway represent salvage routes for tea caffeine biosynthesis in young leaves (Fig. 1). However, the genes encoding the N-methyltransferases involved in these minor pathways have not been identified.16,17
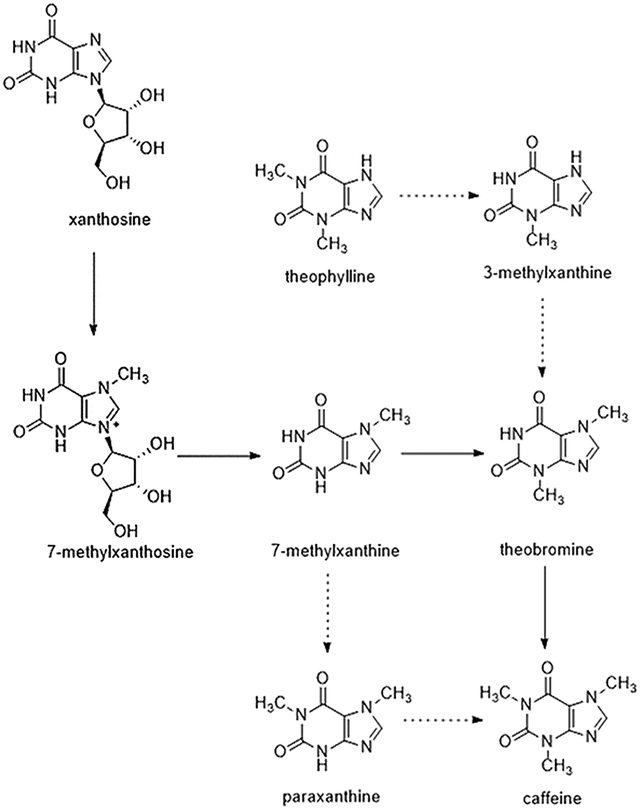 | ||
| Fig. 1 Caffeine biosynthetic pathway in tea plants (Camellia sinensis). Metabolic map of the caffeine biosynthetic pathway in tea (Camellia sinensis) plants. The solid line indicates the major N7 → N3 → N1 methylation order pathway and the dashed lines indicate the two salvaged routes for tea caffeine biosynthesis in young leaves. | ||
In a recent study, the biological production of caffeine from the fermentation of glucose was first demonstrated in metabolically engineered Saccharomyces cerevisiae.18 In that study, the major caffeine biosynthetic route via theobromine was heterologously constructed in eukaryotic cells, and a xanthine-to-xanthosine conversion pathway was completed via the overexpression of two target genes to secure a sufficient supply of the purine ring skeleton of xanthosine. These metabolic strategies resulted in the production of 270 μg L−1 of caffeine, which was accumulated in a 0.3 L bench-scale batch fermentation by the final engineered strain. Although the results are significant for methylxanthine production by fermentation, the low conversion rate of xanthine to xanthosine and the low production yield might limit its further industrial application.
In the present study, our objective was to metabolically engineer Escherichia coli to produce caffeine from a single glucose source. E. coli is a typical and well-studied model microorganism with several advantages for heterologous protein expression, such as a clear genetic background, short culture cycle, low production cost, robustness, and easy handling.19 In addition, our previous study20 successfully demonstrated the functional expression of plant-derived enzymes involved in caffeine synthesis in E. coli. Therefore, these factors render E. coli a suitable host for caffeine production. Finally, a novel methylation order of caffeine biosynthesis was achieved for the first time in E. coli. Additionally, a novel guanine-to-caffeine conversion was metabolically engineered to enable the production of caffeine directly from simple nutrient sources (Fig. 2), which represents an innovative approach to produce these methylxanthine chemicals from a single cheap carbon source in large quantities by fermentation.
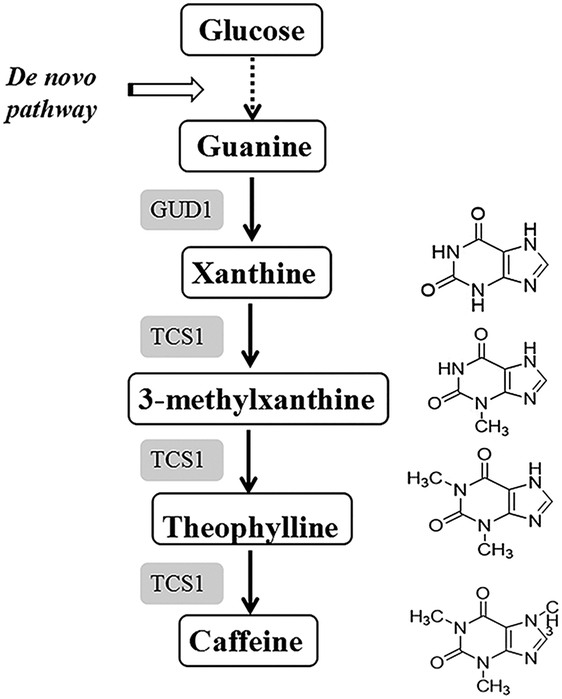 | ||
| Fig. 2 Engineered metabolic network for caffeine production from glucose in E. coli. Glucose is converted to guanine via the central purine metabolism of E. coli. Guanine is converted to xanthine by an endogenous E. coli guanine deaminase and the heterologous expression of S. cerevisiae guanine deaminase encoded by GUD1. Xanthine is converted to caffeine via 3-methylxanthine and to theophylline by the tea caffeine synthase encoded by TCS1. Dashed lines indicated multi-step reactions and solid lines indicated one-step reactions. Abbreviations: GUD1: guanine deaminase from S. cerevisiae, TCS1: tea caffeine synthase from C. sinensis. | ||
Experimental
Bacterial strains and plasmids
All bacterial strains and plasmids used in this study are listed in Table 1. For caffeine production in E. coli, E. coli trans T1 was used for DNA manipulation and E. coli BL21(DE3) was used as the wild-type production host. E. coli expression vectors pRSF-Duet-1, with a T7 promoter, and pMAL-c5X, with a Tac promoter, were used to introduce the caffeine-producing genes into E. coli.| Relevant characteristics | Sources or references | |
|---|---|---|
| Strains | ||
| E. coli trans-T1 | Trans-T1 phage-resistant chemically competent cell | Transgen Biotech |
| E. coli BL21(DE3) | Wild-type strain BL21(DE3), F- ompT hsdS(rB-mB-) gal dcm(DE3) | Transgen Biotech |
| BL21/pMAL-CaXMT | BL21(DE3) harboring plasmid pMAL-CaXMT | Previous work20 |
| BL21/pMAL-eXMT | BL21(DE3) harboring plasmid pMAL-eXMT | This work |
| BL21/pMAL-TCS1 | BL21(DE3) harboring plasmid pMAL-TCS1 | Previous work20 |
| BL21/pMAL-eCS1 | BL21(DE3) harboring plasmid pMAL-eCS1 | This study |
| BL21/pMAL-eXMT-eCS1 | BL21(DE3) harboring plasmid pMAL-eXMT-eCS1 | This study |
| BL21/pMAL-eXMT-eCS1 | BL21(DE3) harboring plasmid pMAL-CaXMT-TCS1 | This study |
| BL21/pRSF-TCS1 | BL21(DE3) harboring plasmid pRSF-TCS1 | This study |
| BL21/pRSF-eCS1 | BL21(DE3) harboring plasmid pRSF-eCS1 | This study |
| BL21/pRSF-eCS1-SAM2-vgb | BL21(DE3) harboring plasmid pRSF-eCS1-SAM2-vgb | This study |
| BL21/pRSF-eCS1-SAM2-vgb-GUD1 | BL21(DE3) harboring plasmid pRSF-eCS1-SAM2-vgb-GUD1 | This study |
| BL21/pRSF-eCS1-SAM2-vgb-eGUD1 | BL21(DE3) harboring plasmid pRSF-eCS1-SAM2-vgb-eGUD1 | This study |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Plasmids | ||
| pMAL-c5X | Ampr, E. coli expression vector, containing tac promoter, mcs, E. coli origin from pACYC177 | New England BioLabs® Inc. |
| pRSF-Duet-1 | Kanr, E. coli expression vector, containing T7 promoter-1 and T7 promoter-2, two multiple cloning sites, RSF origin and lacI gene | Novagen |
| pMAL-CaXMT | pMAL-c5X containing CaXMT | Previous work20 |
| pMAL-eXMT | pMAL-c5X containing eXMT | This study |
| pMAL-TCS1 | pMAL-c5X containing TCS1 | Previous work20 |
| pMAL-eCS1 | pMAL-c5X containing eCS1 | This study |
| pMAL-CaXMT-TCS1 | pMAL-c5X containing CaXMT and TCS1 | Previous work20 |
| pMAL-eXMT-eCS1 | pMAL-c5X containing eXMT and eCS1 | This study |
| pRSF-TCS1 | pRSF-Duet-1 containing TCS1 | This study |
| pRSF-eCS1 | pRSF-Duet-1 containing eCS1 | This study |
| pRSF-eCS1-SAM2-vgb | pRSF-Duet-1 containing eCS1, SAM2, and vgb | This study |
| pRSF-eCS1-SAM2-vgb-GUD1 | pRSF-Duet-1 containing eCS1, SAM2, vgb, and GUD1 | This study |
| pRSF-eCS1-SAM2-vgb-eGUD1 | pRSF-Duet-1 containing eCS1, SAM2, vgb and eGUD1 | This study |
Medium and culture conditions
Lysogeny broth (LB; Difco) was used for strain construction and fermentation cultures; this broth comprised polypeptone (10 g L−1), yeast extract (5 g L−1), and NaCl, (5 g L−1) in distilled water. To select recombinant plasmids and derived strains, either kanamycin (50 mg L−1) or ampicillin (100 mg L−1) was added as appropriate. Cell growth was monitored according to the OD600 value.For caffeine production in E. coli, a single colony of each E. coli-derived strain was pre-cultured in 3 mL of LB medium with shaking at 37 °C and 200 rpm overnight. A-liquots of seed cultures were cultivated in 40 mL of LB medium in 150 mL flasks with shaking at 37 °C and 200 rpm for 2–3 h until the OD600 value reached 0.6–0.8. For target gene expression, isopropyl β-D-1-thiogalactopyranoside (IPTG; 1 mM final concentration) was added, and the main cultures were cultivated at 16 °C and 110 rpm for 20 h.
DNA manipulation and construction of recombinant strains
Polymerase chain reaction (PCR) amplification, purification, digestion, ligation, transformation, and other routine molecular manipulations were performed according to established protocols. The tea caffeine synthase gene TCS1 from Camellie sinensis, the coffee xanthosine methyltransferase gene CaXMT from Coffea arabica, the methionine adenosyltransferase gene SAM2 from Saccharomyces cerevisiae, the Vitreoscilla hemoglobin gene vgb, and the guanine deaminase gene, GUD1, from Saccharomyces cerevisiae were codon optimized and then commercially synthesized (Genscript Bio Tech Co., Lid, NJ) to permit high-efficiency expression in E. coli.These target genes were amplified from their synthesized fragment using PrimeSTAR Max DNA Polymerase (Takara) with their corresponding primers. The expression vector was linearized by restriction enzyme digestion. The resulting gene fragment was then coloned into the linearized expression vector using the ClonExpress II One Step Cloning Kit (Vazyme Bio Tech Co., Lid), yielding the target recombinant plasmid. Specially, both CaXMT and eXMT (in all the gene names, ‘e’ represents the codon optimized version) were inserted into the expression plasmid pMAL-c5X using the introduced restriction site Not I, while TCS1 and eCS1 was constructed using BamH I, respectively. Meanwhile, TCS1 and eCS1 were inserted into expression plasmid pRSF-Duet-1 using the restriction site Nde I on multiple clone site 2 (MCS2), SAM2 and vgb was constructed using Not I in MCS1, GUD1 and eGUD1 were constructed with BamH I in MCS1. To construct E. coli recombinant strains, a purified recombinant plasmid was transferred into competent E. coli cells by chemical transformation, and the recombinant strains were screened and verified through colony PCR and DNA sequencing.
Fermentation
To determine the in vivo activities of the enyzmes involved in caffeine synthesis, xanthosine at more than 1 g L−1 was supplemented in the main culture. All strains were cultivated at 30 °C and 200 rpm for 96 h. Samples of the cultures were subjected to centrifugation at 12000 × g for 10 min at various times, and the culture supernatants were analyzed by high-performance liquid chromatography (HPLC).
For de novo caffeine production in batch cultures, the E. coli cells were cultivated in LB medium containing 2% glucose at 30 °C with shaking at 200 rpm for 96 h. Samples of the cultures were subjected to centrifugation at 12000 × g for 10 min at various times, and the culture supernatants were analyzed by HPLC.
Analytical methods
To determine the extracellular caffeine content, 100 μL of supernatant was diluted 10-fold with distilled water. After being mixed by vortexing, the diluted sample was filtered through a Millipore filter (0.22 μm) and stored at −20 °C. For the identification and quantification of the caffeine content, an HPLC system (Waters; America) equipped with a reverse-phase C18 column and ultraviolet detector at a flow rate of 1 mL min−1 was used. The mobile phase was as follows: 0–3 min, A: 95–95%; 3–8 min, A: 95–90%; 8–11 min; A: 90–85%; 11–13 min, A: 85–70%; 13–15 min, A: 70–60%; 15–20 min, A: 60–60%; 20–22 min, A: 60–95%; 22–28 min, A: 95–95%. The temperature was maintained at 30 °C and the injection volume was 5 μL. In addition, a triple-quadrupole tandem mass spectrometer (QQQ-MS/MS) system was used to further identify the intermediates and caffeine products.18Results and discussion
Engineering the xanthosine-to-caffeine conversion pathway from caffeine-accumulating plants in E. coli
To establish the de novo biosynthesis of caffeine in a non-native purine alkaloids producer, we first attempted to reconstruct the major caffeine biosynthetic pathway. Our previous study showed that beginning with xanthosine as the precursor, the expression of a heterologous four-step pathway comprising the coffee xanthosine methyltransferase (CaXMT, AB048793) from Coffea arabica21,22 and tea caffeine synthase (TCS1, AB031280) from Camellia sinensis23,24 enabled the conversion of xanthosine into caffeine via 7-methylxanthine and theobromine by fermentation in E. coli and S. cerevisiae.20 In this study, CaXMT and TCS1 were selected to complete the main route of caffeine biosynthesis in E. coli. The vector pMAl-c5X was selected as the E. coli expression vector. Additionally, for highly efficient and functional expression of the caffeine biosynthesis pathway in E. coli system, the target sequences of genes encoding for CaXMT and TCS1 were codon optimized with corresponding ribosome-binding site sequences, generating eXMT and eCS1.The content of endogenous xanthosine in non caffeine-producing hosts is the key factor for caffeine biosynthesis. In a previous study, caffeine biosynthesis was achieved by feeding xanthosine both in vivo and in vitro.20 Based on these findings, the contents of endogenous purine metabolites related to caffeine biosynthesis in the background E. coli strain BL21(DE3) with glucose as the only carbon source were examined. However, only xanthine (35.46 ± 3.73 mg L−1) was detected in E. coli by HPLC analysis and further confirmed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Fig. 3), suggesting that the wild-type E. coli is not naturally capable of synthesizing xanthosine using endogenous enzymes.
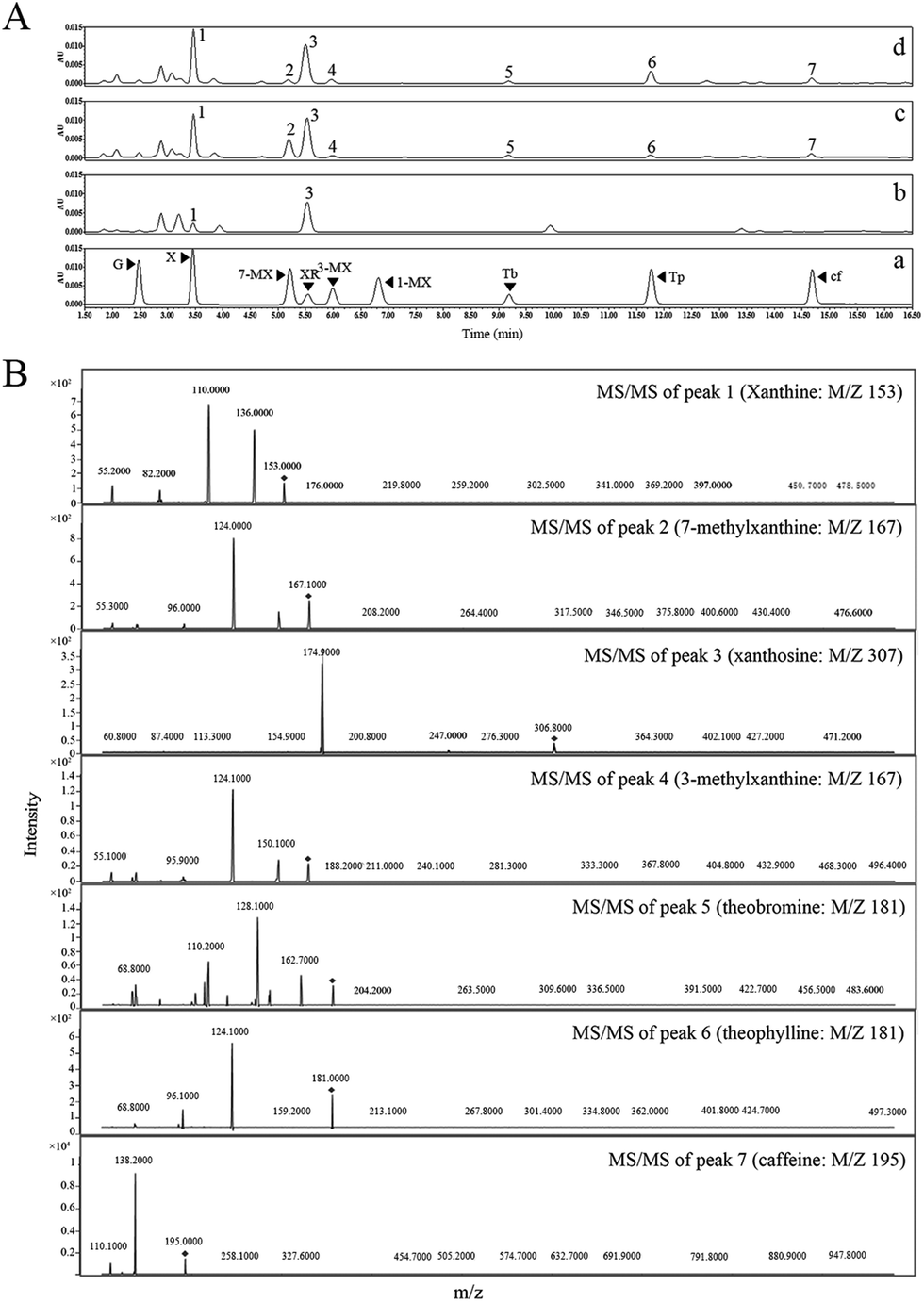 | ||
| Fig. 3 HPLC and LC-MS/MS analysis after expression of different types of CaXMT and TCS1 in E. coli with xanthosine supplementation. (A) HPLC analysis after expression of wild-type and codon-optimized CaXMT and TCS1. (a) A trace showing the authentic standards run in parallel. (b) A trace showing the HPLC analysis of the fermentation products in the derived strain, BL21/pMAL-CK. (c) A trace shows the HPLC analysis of the fermentation products in the derived strain, BL21/pMAL-CaXMT-TCS1. (d) A trace showing the HPLC analysis of the fermentation products in the derived strain, BL21/pMAL-eXMT-eCS1. (B). MS/MS spectra of peaks 1–7 formed in vivo and the authentic standards. Black arrowheads represent the reaction products. Abbreviations: Cf, caffeine; 1-MX, 1-methylxanthine; 3-MX, 3-methylxanthine; 7-MX, 7-methylxanthine; Tb, theobromine; Tp, theophylline; X, xanthine; XR, xanthosine; G, guanine. | ||
Consequently, three recombinant strains, BL21/pMAL-c5X, BL21/pMAL-eXMT-eCS1, and BL21/pMAL-CaXMT-TCS1 were cultivated in LB medium supplement with xanthosine at more than 1 g L−1 to verify the influence of different gene combinations on caffeine production. After HPLC analysis, no methylxanthine products could be detected in the control strain, BL21/pMAL-c5X, suggesting that the background strain BL21(DE3) did not have the capability to produce caffeine by fermentation. However, the recombinant strains, BL21/pMAL-CaXMT-TCS1 and BL21/pMAL-eXMT-eCS1, were able to produce measurable levels of caffeine after 96 h of cultivation (Fig. 3). Moreover, the titer of caffeine (7.81 ± 0.89 mg L−1) produced by the recombinant strain, BL21/pMAL-eXMT-eCS1, was greater than that produced by BL21/pMAL-CaXMT-TCS1 (4.63 ± 0.51 mg L−1) under the same fermentation conditions. The result showed that both genes were functionally expressed in the E. coli host and that the codon-optimized genes were expressed more strongly and efficiently than the wild-type genes. Therefore, eXMT and eCS1 were selected as the target enzymes for de novo biosynthesis of caffeine in subsequent engineered strains.
Identifying a novel xanthine-to-caffeine conversion pathway in E. coli
Meanwhile, measurable levels of 3-methylxanthine, and theophylline by-products were detected together with 7-methylxanthine and theobromine intermediates in the two engineered strains (BL21/pMAL-CaXMT-TCS1 and BL21/pMAL-eXMT-eCS1) by HPLC analysis. In addition, large quantities of xanthine were detected in these E. coli-derived strains (Fig. 3). The results suggested that endogenous xanthine was also converted to 3-methylxanthine and theophylline by tea caffeine synthase (TCS1 and eCS1).To further determine the metabolic fate of endogenous xanthine and the in vivo activities of CaXMT and TCS1, the engineered strains, BL21/pMAL-CaXMT, BL21/pMAL-eXMT, BL21/pMAL-TCS1, BL21/pMAL-eCS1, BL21/pMAL-CaXMT-TCS1, and BL21/pMAL-eXMT-eCS1 were cultivated in LB medium without any methylxanthine supplementation. After 96 h of cultivation, neither caffeine nor the other methylxanthines could be detected in BL21/pMAL-CaXMT and BL21/pMAL-eXMT, while measurable levels of 3-methylxanthine and theophylline were detected in BL21/pMAL-TCS1, BL21/pMAL-eCS1, BL21/pMAL-CaXMT-TCS1, and BL21/pMAL-eXMT-eCS1. Unexpectedly, a measurable level of caffeine was also produced in these strains. These experiments were repeated several times under the same fermentation conditions and using the same strains. All results were in good agreement, indicating that the results were valid and repeatable. Based on these results, it was hypothesized that the endogenous xanthine was converted to caffeine via 3-methylxanthine and theophylline by tea caffeine synthase in E. coli.
TCS1 has been widely identified and reported as an N-methyltransferases that catalyzes the N-3 and N-1 methylation of the xanthine skeleton to form caffeine. TCS1 is specific for several substrates, including xanthine, 3-methylxanthine, 7-methylxanthine, and theobromine. When xanthine was supplied as the only methyl acceptor, 3-methylxanthine was the major product.25–29 These activities have been verified at both the biochemical and molecular level. These investigations, combined with the findings reported in this study, demonstrated that both methylation orders of N3 → N1 → N7 and N7 → N3 → N1 exist in engineered E. coli strains harboring CaXMT and TCS1. Specifically, xanthine was sequential methylated at the N3 → N1 → N7 positions to produce 3-methylxanthine, theophylline, and caffeine by TCS1.
To test our hypothesis and further verify the novel in vivo activity of TCS1, the engineered strains BL21/pMAL-TCS1 and BL21/pMAL-eCS1 were cultivated in LB medium supplemented with a variety of methylxanthine precursors, including 3-methylxanthine (final concentration of 0.1 g L−1), 7-methylxanthine (final concentration of 0.1 g L−1), theobromine (final concentration of 0.1 g L−1) or theophylline (final concentration of 0.1 g L−1), respectively. Meanwhile, these strains were also cultivated in LB medium without any substrate supplementation to further determinate the fate of endogenous xanthine. After HPLC analysis, theobromine and caffeine were produced with feeding 7-methylxanthine; caffeine was produced after feeding with theobromine; theophylline and caffeine were produced with feeding on 3-methylxanthine; and 3-methylxanthine, theophylline, and caffeine were produced in the absence of substrate supplementation. Caffeine was also produced with feeding on theophylline. After further quantitative analysis, the results indicated that the conversion rates of xanthine, 3-methylxanthine, 7-methylxanthine, theobromine, and theophylline to caffeine by BL21/pMAL-eCS1 were 2.75, 2.02, 5.49, 3.38, and 12.54 times higher than that by BL21/pMAL-TCS1 (Fig. 4). All these results indicated that TCS1 played a key functional role in the novel xanthine-to-caffeine conversion. This study also demonstrated the conversion of theophylline to caffeine. Theophylline was methylated at the N7 position by TCS1 to produce caffeine in E. coli, and the N7 methylation was specific for theophylline. It also demonstrated that optimizing the codon sequence could improve the activity of target enzymes.
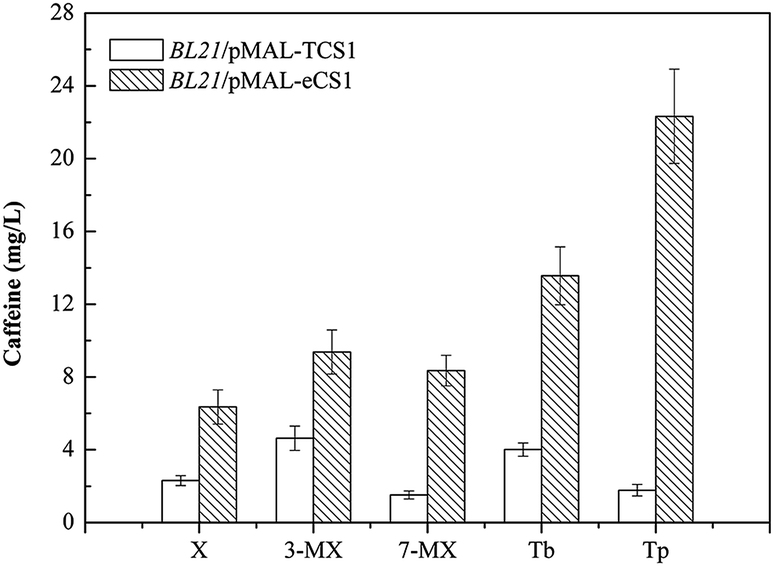 | ||
| Fig. 4 Caffeine production in different recombinant strains with different substrate supplementation. Two E. coli derived strains were cultivated with different methylxanthine substrates in 40 mL of LB medium in 150 mL baffled flasks at 30 °C and 200 rpm for 96 h. Their end products were detected and quantified by HPLC analysis. Abbreviations: 3-MX, 3-methylxanthine; 7-MX, 7-methylxanthine; Tb, theobromine; Tp, theophylline; X, xanthine. Data are expressed as means and standard deviations of three biological replicates. All results were consistent. | ||
Previously, Misako17 proposed that the alternative xanthine → 3-methylxanthine → theophylline → caffeine pathway was limited in caffeine synthesis in tea plants because of insufficient xanthine and theophylline because of purine and caffeine catabolism, further suggesting that sufficient xanthine was a determining factor for the alternative xanthine-to-caffeine conversion pathway in caffeine synthesis. Additionally, the first and highest reported de novo production of caffeine by fermentation was achieved in S. cerevisiae overexpressing several enzymes from coffee (Coffea arabica) plants. Meanwhile levels of 3-methylxanthine and theophylline byproducts were also detected during the fermentation. The observation implied that xanthine plays a crucial role in caffeine biosynthesis in the reconstructed heterologous pathway incorporating plant-derived caffeine synthase.18 We also verified the activity of CCS1 (encoding coffee caffeine synthase) from coffee plants in the E. coli system under the same culture conditions. The results showed that CCS1 could catalyze the conversion of xanthine to caffeine via 3-methylxanthine and theophylline. These findings further illustrated that the novel in vivo activity of caffeine synthase was activated by a sufficient supply of xanthine, 3-methylxanthine, or theophylline.
De novo biosynthesis of caffeine in engineered E. coli strains
In the current study, we found that E. coli can inherently produce xanthine under typical growth conditions. Based on the results presented above, we next established the de novo biosynthesis of caffeine through the novel xanthine-to-caffeine conversion in E. coli. The derived strain BL21/pMAL-eCS1 was cultivated in 40 mL LB medium without any substrate supplementation. The metabolites were then collected and analyzed at various times. Measurable levels of 3-methylxanthine, theophylline, and caffeine were detected by HPLC analysis. After further quantitative analysis, we determined that this strain produced 6.42 ± 0.24 mg L−1 caffeine (Fig. 5A). According to our review of the literature, this is the first report of de novo biosynthesis of caffeine via a novel xanthine-to-caffeine conversion in E. coli.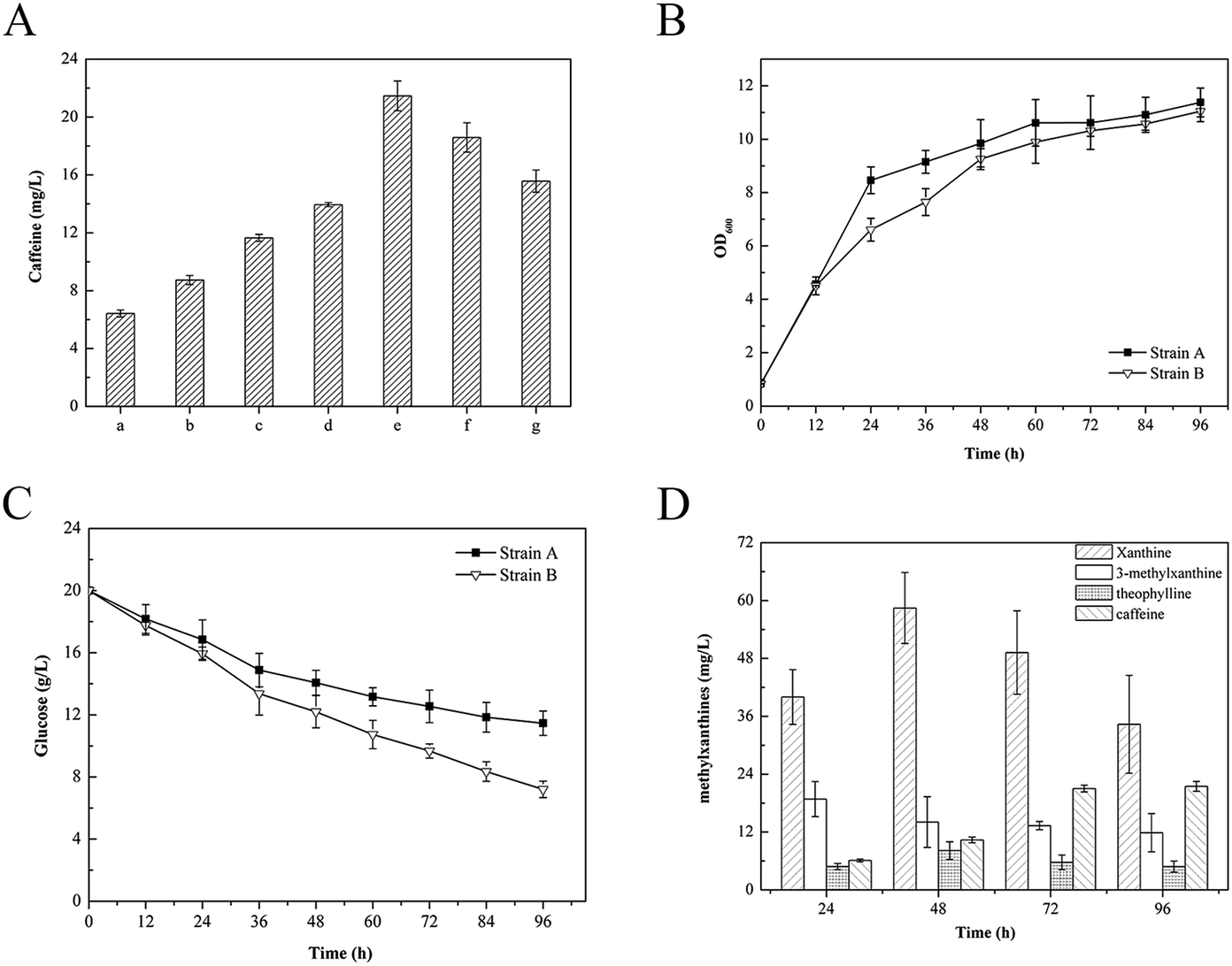 | ||
| Fig. 5 De novo caffeine production in different E. coli strains. (A) Production of caffeine in different E. coli strains. Five E. coli strains, (a) BL21/pMAL-eCS1, (b) BL21/pRSF-TCS1, (c) BL21/pRSF-eCS1, (d) BL21/pMAL-eCS1-SAM2-vgb, and (e) BL21/pRSF-eCS1-SAM2-vgb-eGUD1, were cultivated in 40 mL LB medium containing 20 g L−1 glucose in 150 mL baffled flasks at 30 °C and 200 rpm for 96 h. (f) Strain (e) cultivated with 10% glycerin. (g) Strain (e) cultivated with 20 g L−1 sucrose. (B) Time course of cell growth in the background strain and the final engineered strain (e) with 20 g L−1 glucose. Strain A: BL21(DE3), strain B: BL21/pRSF-eCS1-SAM2-vgb-eGUD1. (C) Time course of glucose consumption in the background strain and the final engineered strain with 20 g L−1 glucose. Strain A: BL21(DE3), strain B: BL21/pRSF-eCS1-SAM2-vgb-eGUD1. (D) Time course of intermediates production in BL21/pRSF-eCS1-SAM2-vgb-eGUD1 with 20 g L−1 glucose. Abbreviations: Cf, caffeine; 3-MX, 3-methylxanthine; 7-MX, 7-methylxanthine; Tb, theobromine; Tp, theophylline; X, xanthine. The data are expressed as means and standard deviations of three biological replicates. All results were consistent. | ||
Strategies for enhancing xanthine-to-caffeine conversion in E. coli
To improve caffeine biosynthesis, we further employed two metabolic engineering strategies to enhance xanthine-to-caffeine conversion in E. coli.First, for greater and more efficient TCS1 activity, the eCS1 gene was overexpressed in another E. coli expression vector, pRSF-Duet-1, with a stronger T7 promoter. TCS1 was also inserted into the vector pRSF-Duet-1 as a control. The constructed recombinant plasmids pRSF-TCS1 and pRSF-eCS1 were transformed into BL21(DE3), yielding two derived strains, BL21/pRSF-TCS1 and BL21/pRSF-eCS1. When the BL21/pRSF-TCS1 and BL21/pRSF-eCS1 strains were cultivated in LB medium, production of 8.73 ± 0.31 mg L−1 and 11.65 ± 0.24 mg L−1 caffeine was confirmed by HPLC analysis, respectively. These results demonstrated that the T7 promoter increased prokaryotic expression of the heterologous genes (Fig. 5A). Hence, in subsequent experiments, we used the expression vector pRSF-Duet-1.
Previous studies have shown that overexpression of SMA2 encoding S. cerevisiae methionine adenosyltransferase (MAT) and vgb encoding Vitreoscilla hemoglobin (VHb) in microbes is an efficient means of producing S-adenosyl-L-methionine (SAM), which is the only methyl donor involved in the biosynthesis of caffeine.30–32 Thus, the SMA2 and vgb genes were inserted into the vector pRSF-eCS1 to increase the SAM pool for caffeine biosynthesis. The reconstructed plasmid pRSF-eCS1-SMA2-vgb was then transformed into BL21(DE3) yielding the derived strain BL21/pRSF-eCS1-SAM2-vgb. This derived strain was also cultured in LB medium. After HPLC analysis, 13.95 ± 0.14 mg L−1 caffeine was produced, although no statistical difference in caffeine production yields was observed between the BL21/pRSF-eCS1 and BL21/pRSF-eCS1-SAM2-vgb strains (Fig. 5A), indicating that the SAM pool synthesized by the central metabolism in E. coli was sufficient to support the methyl demand and that it is not a limiting step for caffeine biosynthesis.
Increasing the xanthine pool by overexpression of xanthine-biosynthesis enzymes in E. coli
For a higher caffeine yield, increasing the xanthine pool is an important strategy. To generate the xanthine pool supply, adenine and guanine derivatives metabolism are essential activities in microorganisms. Furthermore, because the adenine derivatives can be converted into guanine nucleotides irreversibly, the guanylic nucleotide pool could be converted into xanthine by a series of guanine deaminases. Therefore, in an effort to increase caffeine production, we further expressed genes involved in xanthine synthesis in E. coli. In this study, S. cerevisiae was chosen as the donor of guanine deaminase because its functionality has been demonstrated.33 In particular, both of the wild-type GUD1 encoding S. cerevisiae guanine deaminase and a codon-optimized version of this enzyme (eGUD1) were constructed and expressed with eCS1, SAM2, and vgb, yielding two derived strains, BL21/pRSF-eCS1-SAM2-vgb-GUD1 and BL21/pRSF-eCS1-SAM2-vgb-eGUD1, respectively. After the cultivation of these derived strains in LB medium containing 20 g L−1 glucose, the strains produced distinct caffeine yields. The codon-optimized genes had a significant effect on the production of caffeine (Fig. 5A). The results also confirmed that the codon-optimized genes significant improved the in vivo activities of these enzymes. This result is consistent with the findings of previous studies.34,35Finally, the engineered E. coli strain harboring the best gene combination (BL21/pRSF-eCS1-SAM2-vgb-eGUD1) produced a relatively high yield of caffeine (21.46 ± 1.03 mg L−1) from 20 g L−1 glucose in flask batch culture, which was almost 100-fold higher than the de novo caffeine production reported in engineered S. cerevisiae (Fig. 5B). The results demonstrate that compared with the increase in the methyl donor pool, the enhanced xanthine skeleton content played a key role in improving caffeine biosynthesis in the metabolically engineered E. coli. Meanwhile, compared with the glucose consumption in the background strain BL21(DE3), the finally engineered strain (BL21/pRSF-eCS1-SAM2-vgb-eGUD1) achieved a yield of 2.96 mg-caffeine/g-glucose (Fig. 5C). In addition, during the fermentation, the formation of intermediated xanthine, 3-methylxanthine, and theophylline increased at the beginning, and then slowly declined, while the formation of caffeine continued increase at all times points (Fig. 5D). A yield of 21.46 ± 1.03 mg L−1 also represents the highest reported caffeine production in an engineered microorganism. Thus, our engineered strain may provide a simple and cost-effective approach for industrial-scale production of plant-derived caffeine and other methylxanthines from widely available carbon sources, including glucose and ethanol, by fermentation. To further improve the caffeine titer and reduce production cost, the engineered strain BL21/pRSF-eCS1-SAM2-vgb-eGUD1 was cultivated in 40 mL LB medium containing 10% glycerin or 20 g L−1 sucrose. After HPLC analysis, 18.59 ± 1.01 mg L−1 and 15.57 ± 0.77 mg L−1 caffeine were produced with either glycerin or sucrose supplementation, respectively (Fig. 5A). The results showed that glucose was the most appropriate carbon source in the engineered strain.
In this study, a novel caffeine biosynthetic pathway was demonstrate for the first time, and E. coli was engineered to be a caffeine producer by reconstruction of this novel biosynthetic pathway. Caffeine accumulation was improved by enhancing xanthine and SAM biosynthesis, which increased the yield of caffeine by 234% compared with strain BL21/pMAL-eCS1. This results showed that a sufficient supply of substrates is a key factor for caffeine production. Guanine is not only the initial substrate for caffeine production in our engineered strain, but also is one of the four main nucleobases found in the nucleic acids DNA and RNA. Guanine is involved in many cellular metabolisms and has several functions in cellular systems. Studies have shown that most of the guanine is converted to guanosine and then GMP in E. coli. Purine nucleotide metabolism is tightly regulated for the balance of metabolic networks;36,37 therefore, it may be difficult to prevent the metabolic flux of guanine shifting toward non-caffeine pathways. Therefore, to further meet the demand of industrial caffeine production by the engineered strain, an effective strategy would be to maximize carbon metabolic flow and rapidly guide it toward the target metabolites by identifying the essential nodes of central metabolism during fermentation and modifying the key factors for higher production. Currently, on the basis of the results of corresponding proof experiments and reports in literature,38,39 this might be achieved by enhancing the guanine pool from GMP and blocking the conversion of xanthine to uric acid; for example, by overexpressing a gene encoding a 5′-nucleosidase and deactivating xanthien oxidase in E. coli. In addition, the expression of the target genes was reliant on IPTG induction and antibiotic resistance markers, which were unavoidable for the selection and maintenance of the plasmids in cell culture; this may also impede further application of the engineered strain. The goal of future studies should be genome integration of this two-enzyme pathway to reduce the metabolic burden and further improve engineered E. coli caffeine production.
Conclusions
We demonstrated the novel in vivo activity of tea caffeine synthase in E. coli. A xanthine-to-caffeine conversion was achieved by expressing TCS1 in metabolically engineered E. coli BL21(DE3). We showed that the construction of the novel caffeine biosynthesis pathway is sufficient for E. coli to produce caffeine from glucose. In addition, higher-level expression of the target enzymes and enhanced xanthine and SAM biosynthesis had a strong and efficient effect on the accumulation of caffeine production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China project 31570692; the Changjiang Scholars and Innovative Research Team in University (IRT_15R01); the Chinese National 863 Project (Award 2013AA102801 to O. Y.); and the Jiangsu International Collaboration program (BZ2014014).Notes and references
- H. Ashihara, H. Sano and A. Crozier, Phytochemistry, 2008, 69, 841–856 CrossRef CAS PubMed.
- P. Mohanpuria, V. Kumar and S. K. Yadav, Food Sci. Biotechnol., 2010, 19, 275–287 CrossRef CAS.
- D. M. E. Gonzalez and M. V. Ramirez-Mares, Trends Endocrinol. Metab., 2014, 25, 489–492 CrossRef PubMed.
- B. J. Gurley, S. C. Steelman and S. L. Thomas, Clin. Ther., 2015, 37, 275–301 CrossRef CAS PubMed.
- J. A. Vernarelli, D. C. Mitchell, B. J. Rolls and T. J. Hartman, Eur. J. Nutr., 2015, 54, 59–65 CrossRef CAS PubMed.
- X. Wang, X. Wan, S. Hu and C. Pan, Food Chem., 2008, 107, 1086–1091 CrossRef CAS.
- X. X. Sun, X. L. Shen, R. Jain, Y. H. Lin, J. Wang, J. Sun, J. Wang, Y. J. Yan and Q. P. Yuan, Chem. Soc. Rev., 2015, 44, 3760–3785 RSC.
- E. Leonard, W. Runguphan, S. O'Connor and K. J. Prather, Nat. Chem. Biol., 2009, 5, 292–300 CrossRef CAS PubMed.
- Y. Li and B. A. Pfeifer, Curr. Opin. Plant Biol., 2014, 19, 8–13 CrossRef CAS PubMed.
- T. Hirasawa and H. Shimizu, Curr. Opin. Biotechnol., 2016, 42, 133–146 CrossRef CAS PubMed.
- T. Kubota, A. Watanabe, M. Suda, T. Kogure, K. Hiraga and M. Inui, Metab. Eng., 2016, 38, 322–330 CrossRef CAS PubMed.
- M. J. Lee, I. R. Brown, J. Rokas, F. Stefanie and M. J. Warren, Metab. Eng., 2016, 36, 48–56 CrossRef CAS PubMed.
- B. Pereira, H. Zhang, M. De Mey, C. G. Lim, Z. Li and G. Stephanopoulos, Biotechnol. Bioeng., 2016, 113, 376 CrossRef CAS PubMed.
- J. Plassmeier, Y. Li, C. Rueckert and A. J. Sinskey, Metab. Eng., 2016, 33, 86 CrossRef CAS PubMed.
- E. C. Tatsis and S. E. O'Connor, Curr. Opin. Biotechnol., 2016, 42, 126 CrossRef CAS PubMed.
- H. Ashihara, K. Mizuno, T. Yokota and A. Crozier, Progress in the chemistry of organic natural products, Springer International Publishing, vol. 105, 2017, pp. 1–88 Search PubMed.
- K. Misako and M. Kouichi, Front Biosci, 2004, 9, 1833 CrossRef.
- M. McKeague, Y. H. Wang, A. Cravens, M. W. Win and C. D. Molke, Metab. Eng., 2016, 38, 191–203 CrossRef CAS PubMed.
- T. Matsumoto, T. Tanaka and A. Kondo, Bioresour. Technol., 2017, 245, 1362–1368 CrossRef CAS PubMed.
- L. Jin, M. W. Bhuiya, M. M. Li, X. Q. Liu, J. Han, W. W. Deng, M. Wang, O. Yu and Z. Z. Zhang, PLoS One, 2014, 9, e105368 Search PubMed.
- K. Mizuno, A. Okuda, M. Kato, N. Yoneyama, H. Tanaka, H. Ashihara and T. Fujimura, FEBS Lett., 2003, 534, 75–81 CrossRef CAS PubMed.
- H. Uefuji, S. Ogita, Y. Yamaguchi, N. Koizumi and H. Sano, Plant Physiol., 2003, 132, 372–380 CrossRef CAS PubMed.
- M. Kato, K. Mizuno, T. Fujimura, M. Iwama, M. Irie, A. Crozier and H. Ashihara, Plant Physiol., 1999, 120, 579–586 CrossRef CAS PubMed.
- M. Kato, K. Mizuno, A. Crozier, T. Fujimura and H. Ashihara, Nature, 2000, 406, 956–957 CrossRef CAS PubMed.
- J. Q. Jin, M. Z. Yao, C. L. Ma, J. Q. Ma and L. Chen, Plant Physiol. Biochem., 2016, 100, 18–26 CrossRef CAS PubMed.
- Y. Kodama, T. Shinya and H. Sano, Biochimie, 2008, 90, 547–551 CrossRef CAS PubMed.
- M. Kato, N. Kitao, M. Ishida, H. Morimoto, F. Irino and K. Mizuno, Z. Naturforsch., C: J. Biosci., 2010, 65, 245–256 CAS.
- A. A. McCarthy and J. G. McCarthy, Plant Physiol., 2007, 144, 879–889 CrossRef CAS PubMed.
- N. Yoneyama, H. Morimoto, C. X. Ye, H. Ashihara, K. Mizuno and M. Kato, Mol. Genet. Genomics, 2006, 275, 125–135 CrossRef CAS PubMed.
- H. Chen, J. Chu, S. Zhang, Y. Zhuang, J. Qian, Y. Wang and X. Hu, Appl. Microbiol. Biotechnol., 2007, 74, 1205–1212 CrossRef CAS PubMed.
- Y. Gu, X. Wang, C. Yang, W. Geng, J. Feng, Y. Wang, S. Wang and C. Song, Appl. Biochem. Biotechnol., 2016, 178, 1–13 CrossRef PubMed.
- G. Han, X. Hu, T. Qin, L. Ye and X. Wang, Enzyme Microb. Technol., 2016, 83, 14–21 CrossRef CAS PubMed.
- C. Saint-Marc and B. Daignan-Fornier, Yeast, 2004, 21, 1359–1363 CrossRef CAS PubMed.
- M. Robinson, R. Lilley, S. Little, J. S. Emtage, G. Yarranton, P. Stephens, A. Millican, M. Eaton and G. Humphreys, Nucleic Acids Res., 1984, 12, 6663 CrossRef CAS PubMed.
- M. Gouy and C. Gautier, Nucleic Acids Res., 2011, 10, 7055 CrossRef.
- R. K. Mehra and W. T. Drabble, J. Gen. Microbiol., 1981, 123, 27–37 CAS.
- L. M. Meng and P. Nygaard, Mol. Microbiol., 1990, 4, 2187–2192 CrossRef CAS PubMed.
- E. Hamel and M. Cashel, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 3250–3254 CrossRef CAS.
- J. T. Maynes, R. G. Yuan and F. F. Snyder, J. Bacteriol., 2000, 182, 4658–4660 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |