
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of poly(n-hexyl methacrylate)-b-poly(methyl methacrylate) via anionic polymerization with t-BuOK as the initiator at ambient temperature
Anna Zheng,
Ling Su ,
Zheng Li,
Guijin Zou,
Xiang Xu* and
Yong Guan*
,
Zheng Li,
Guijin Zou,
Xiang Xu* and
Yong Guan*
Key Laboratory of Special Functional Polymeric Materials and Related Technology of the Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yguan@ecust.edu.cn; xiangxu@ecust.edu.cn
First published on 23rd November 2017
Abstract
To break through the industrial development bottlenecks of anionic polymerization in the field of polar monomers, poly n-hexyl methacrylate (PHMA) was prepared at 0 °C, 30 °C, and 60 °C with potassium tert-butoxide (t-BuOK) as the initiator in tetrahydrofuran (THF) and the conversions were almost complete. Gel permeation chromatography (GPC) analysis of poly n-hexyl methacrylate indicated that t-BuOK existed in numerous states when dissolved in THF, including different types of association and ion-pairs. A series of block copolymers of n-hexyl methacrylate (n-HMA) and methyl methacrylate (MMA), PMMA-b-PHMA (Mn 8200–16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200) and PHMA-b-PMMA (Mn 19100–9500) were synthesized at 0 °C, and conversions were close to 100%. The results of dynamic mechanical analysis (DMA) indicated that there were two glass transition temperatures, −10 to 0 °C for the PHMA block and 100 to 110 °C for the PMMA block. Furthermore, the competing behaviors of n-HMA and MMA in copolymerization were investigated by one-shot feeding of two types of monomers at 0 °C, 30 °C, and 60 °C. The kinetics curves and 13C-NMR analysis of the copolymers indicated that the polymerization of n-HMA in the copolymerization was significantly dominant over that of MMA at set temperatures. The results in this study provided the possibility to realize anionic polymerization of methacrylate esters on a commercial scale.
200) and PHMA-b-PMMA (Mn 19100–9500) were synthesized at 0 °C, and conversions were close to 100%. The results of dynamic mechanical analysis (DMA) indicated that there were two glass transition temperatures, −10 to 0 °C for the PHMA block and 100 to 110 °C for the PMMA block. Furthermore, the competing behaviors of n-HMA and MMA in copolymerization were investigated by one-shot feeding of two types of monomers at 0 °C, 30 °C, and 60 °C. The kinetics curves and 13C-NMR analysis of the copolymers indicated that the polymerization of n-HMA in the copolymerization was significantly dominant over that of MMA at set temperatures. The results in this study provided the possibility to realize anionic polymerization of methacrylate esters on a commercial scale.
Introduction
Anionic polymerization has unique advantages in synthesis of block copolymer for fast initiation and propagation, almost no chain transition and termination.1,2 Based on this, a series of block copolymers have been synthesized in industrial scale like SBS, SIS and (SB)4R.3–7 However, a bottleneck was encountered in synthesis of block copolymers containing methacrylate involving severe side reactions.8–11To solve the problem, extreme low temperature and complex inhibiting ligands were used in recent studies. Varshney et al.12 prepared poly(methyl methacrylate)-b-poly(tert-butyl acrylate) (PMMA-b-PtBA) di-block copolymer at −78 °C in THF. Kim et al.13 synthesized polystyrene-b-poly(methyl methacrylate) di-block copolymer (PS-b-PMMA) with high molecular weight in flow tube reactor using THF as the solvent at −78 °C. Ren et al.14 prepared poly(1,3-pentadiene-b-poly(methyl methacrylate)) di-block copolymer (PEP-b-PMMA) at −78 °C. Lee et al.15 obtained the polystyrene-b-polyisoprene-b-poly(methyl methacrylate) tri-block copolymer (PS-PI-PMMA) by anionic polymerization, in which the PS-b-PI di-block was prepared in toluene at room temperature at first and then the reactor was cooled down to −78 °C for PMMA block. Baskaran et al.16 researched the polymerization behaviors of MMA at −40 °C in THF and −78 °C in toluene/THF (9/1 v/v) using 1,1-diphenylhexyl lithium (DPHLi) as the initiator and lithium perchlorate (LiClO4) as the additive. Wang et al.17 elevated the temperature to 0 °C for polymerization of MMA with DPHLi as the initiator and LiCl as the additive in pyridine. Kitayama et al.18 successfully prepared PMMA-b-polybutylacrylate (PMMA-b-PBA) copolymer using tert-butyllithium/bis(2,6-di-tert-butylphenoxy)ethylaluminum (t-BuLi/EtAl(ODBP)2) as the composite initiators. Vinogradova et al.19 adopted t-BuOK as the initiator and directly synthetized PMMA in ethylene oxide at 0 °C without any ligands. However, the initiation efficiency was relatively low.
In order to further understand the initiation and side reaction mechanisms of anionic polymerization and broaden the types of monomers, researchers in our laboratory performed a lot of studies and made some progress. For example, Guan et al.20–22 researched the initiation mechanism of n-butyllithium (n-BuLi) during the association and dissociation processes. Zheng et al.23,24 synthesized a compound called “P-Complex”, which can be dissolved in polar solvent and control side reactions in anionic polymerization of MMA and its copolymers. Shu et al.25 successfully prepared PMMA-b-polybutylmethacrylate di-block copolymer (PMMA-b-PBMA) at 40 °C using DPHLi as the initiator and t-BuOK as the additive through counter ion exchange from Li+ to K+. Li et al.26 used cyclohexane as the solvent to prepare PS-PI-PMMA tri-block copolymer by two step-polymerization. PS-b-PI di-block copolymer was polymerized at 50 °C at first by sequential feeding of monomers and then the system was cooled down to 0 °C for the polymerization of MMA.
Based on these findings, we used t-BuOK as the initiator and THF as the solvent. PHMA and PMMA-b-PHMA/PHMA-b-PMMA di-block copolymers were prepared at ambient temperature with nearly complete conversion. Furthermore, the initiation mechanism of t-BuOK was investigated. The results in this study contribute to further understanding of initiation mechanism of anionic polymerization and provide the possibility to realize anionic polymerization of methacrylate esters on a commercial scale.
Experimental
Materials
Argon (Ar, 99.99999%) was purified by flowing through two connected cylinders filled with 4 Å molecular sieves. Tetrahydrofuran (THF, AR, Sinopharm Chemical Reagent Co., Ltd., China) was refluxed with sodium at 66 °C for 24 h, distilled and soaked with sodium. N-Hexyl methacrylate (n-HMA, AR, Shanghai Aladdin Bio-Chem Technology Co., Ltd., China) and methyl methacrylate (MMA, AR, Lingfeng Chemical Reagent Co., Ltd., China) were purified by distillation in a vacuum system at 80 °C and 30 °C, respectively, after stirring with calcium hydride for 48 h and soaked with 4 Å molecular sieves for more than 24 h before use. N-Butyl lithium (n-BuLi, 2.5 mol l−1 in n-hexane, J&K Chemical Co., Ltd., Shanghai, China) was used as received. 1,1-Diphenylethylene (DPE, 98%, TCI, Japan) was used as received. Potassium tert-butoxide (t-BuOK, 99%, Sinopharm Chemical Reagent Co., Ltd., China) was used as received. The moisture of all the reagents through refining was less than 10 ppm.Characterization methods
The moisture of the reagents was determined by moisture meter (KF831, Switzerland). The determination range is from 10−5 g to ∼10−3 g and the measurement accuracy is ±3 × 10−6 g.The molecular weight and its distribution (MWD) were determined by multi-detector gel permeation chromatography (Water 1515 system, Waters corporation, USA), equipped with an 18 angles laser scattering detector (LS signal) and a refractive detector (RI signal) using THF as the eluent at a flow rate of 1.0 ml min−1 at 25 °C.
1H-NMR and 13C-NMR spectra were measured by a BRUKER AV400 spectrometer with deuterated chloroform (CDCl3) as solvent and tetramethylsilane (TMS) as the internal reference.
Dynamic mechanical spectrometer (Q800, TA Co., USA) was used to study the dynamic mechanical properties of the copolymer. The samples were made into rectangular films with size of 20 × 10 × 2 mm, tested with single cantilever bending mode and heated from −100 °C to 150 °C, at heating rate of 5 °C min−1 and frequency of 1 Hz in a nitrogen atmosphere.
Synthesis of PHMA
All the polymerizations were carried out in glass reactors which were flamed under vacuum, then purged with Ar and passed through 4 Å molecular sieves. THF, DPE and n-BuLi were transferred into the glass reactor using rubber septa and stainless glass syringes. A known amount of t-BuOK was added into the glass reactor and dissolved completely through stirring. The solution was then cooled or heated to the set temperature and kept for 15 min, followed by addition of monomers demanded. The product was quenched by degassed methanol and the final solution was concentrated before being precipitated into an excess of 90/10 (v/v) methanol/water mixture under stirring. The crude product was vacuum dried at 80–100 °C for 12 h.Synthesis of copolymers of MMA and n-HMA
The glass reactors were treated in the same way as that of homopolymerization. THF was transferred into the glass reactor by using rubbery septa and stainless glass syringes. A known amount of t-BuOK was added into the glass reactor and dissolved fully through stirring. The solution was then cooled or heated to the set temperature and kept for 15 min, followed by addition of the first monomers. After a certain amount of time, a small amount of solution was taken out for homopolymer analysis and then the second set of monomers were added. Post-treatment of the copolymer obtained was the same as above.Competing behaviors of MMA and n-HMA
The glass reactors were treated in the same way as that of homopolymerization. THF was transferred into the glass reactor using rubbery septa and stainless glass syringes. A known amount of t-BuOK was added into the glass reactor and dissolved fully through stirring. The solution was then cooled or heated to the set temperature and kept for 15 min, followed by addition of the two types of monomers at the same time. A certain amount of reaction solution was taken out at set intervals and the conversion of the each monomer calculated through 1H-NMR.Results and discussion
The initiation mechanism of t-BuOK
The results of homopolymerization are summarized in Tables 1 and 2. The conversions of samples 8 and 9 were less than 10%, because severe side reactions between the negative carbonyl and the counter ion Li+, which causes inactivation of the active centre. In sharp contrast, all the polymerizations initiated by t-BuOK proceeded with conversion more than 90% and were almost complete for samples 1–6, indicating the reaction centres were active during polymerization. It should also be noted that all the polymerizations showed a broad MWD, which narrowed with the rise in temperature. Moreover, the actual molecular weight was much higher than the calculated value based on the mole ratio, which differs from the classical theory of anionic polymerization.| Samplea | Temp (°C) | t-BuOK (mmol) | nHMA (mmol) | Mn × 10−3 | Mn × 10−4 | MWDc | Conversion (%) |
|---|---|---|---|---|---|---|---|
| Calb | Obsc | ||||||
| a 45 ml of THF, reaction time 30 min.b Mn was calculated by mole ratio of n-HMA to t-BuOK.c Mn and MWD were determined by GPC. | |||||||
| 1 | 0 | 5.2 | 26 | 0.90 | 1.26 | 1.88 | >99.9 |
| 2 | 0 | 5.2 | 52 | 1.80 | 2.37 | 1.95 | >99.9 |
| 3 | 0 | 5.2 | 78 | 2.70 | 3.31 | 1.94 | >99.9 |
| 4 | 0 | 2.6 | 26 | 1.80 | 1.49 | 1.83 | >99.9 |
| 5 | 0 | 10.4 | 26 | 0.45 | 0.97 | 1.82 | >99.9 |
| 6 | 30 | 5.2 | 26 | 0.90 | 1.04 | 1.70 | >99.9 |
| 7 | 60 | 5.2 | 26 | 0.90 | 0.56 | 1.54 | 96.0 |
GPC curves of samples 1–3 are shown in Fig. 1. The molecular weight of the polymer in GPC main peak increased almost proportionally to the amount of monomers. There also existed two small peaks, whose molecular weights were in the range of 1000–2000 and could be removed through 2–3 purification cycles (we dissolved the vacuum dried polymer with suitable amount of THF. Cyclohexane was added drop wise to the solution with stirring until turbidity. Then, we continued to add a small amount of cyclohexane to precipitate the product with relatively bigger molecular weight.27). The new GPC curves are shown in Fig. 2. As apparently observed, the two small peaks disappeared and the MWDs became narrower.
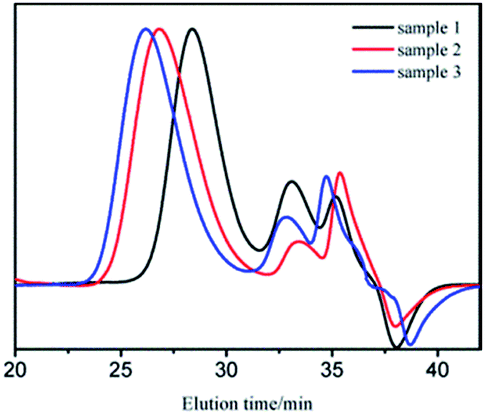 | ||
| Fig. 1 GPC curves of sample 1, 2, and 3. | ||
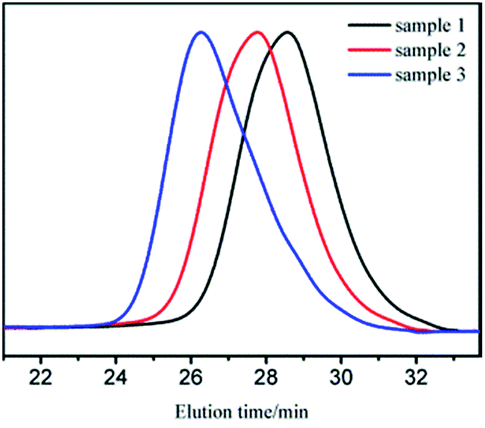 | ||
| Fig. 2 GPC curves of sample 1, 2, 3 after purification (MWDs were 1.40, 1.43, and 1.52 for sample 1, 2, and 3, respectively). | ||
It was already revealed that t-BuOK possessed several states in THF containing association state.19 Actually, it was found in previous studies that associated initiator could also initiate polymerization.20–22,28,29 Combined with the results above, we infer that t-BuOK exists in different types of associations and ion-pairs when dissolved in THF. All states of t-BuOK can initiate polymerization, but the reactivity for small association state and ion-pairs is much faster than that of big association state because of steric effect (Fig. 3). This explains why the actual molecular weight is much higher than the theoretical value as well as the broad MWDs. To verify the speculation, reactions were conducted at different temperatures and the results are illustrated in Fig. 4. With the increase of temperature, the main peak shifted to the lower molecular weight region and the two small peaks weakened gradually. This is attributed to the increase in the number of small association states or ion-pairs derived from dissociation of aggregated t-BuOK.
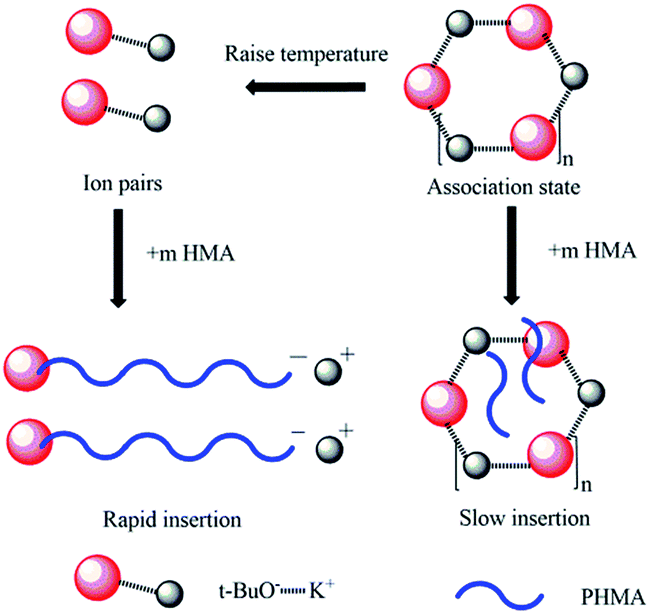 | ||
| Fig. 3 Initiation mechanism of t-BuOK in polymerization of n-HMA. | ||
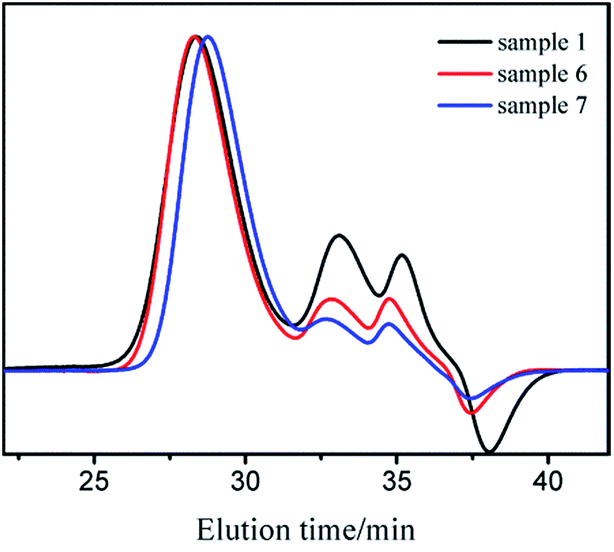 | ||
| Fig. 4 GPC curves of sample 1, 6, and 7. | ||
For sample 1, 4, and 5 (Table 1), the decrease of molecular weight is not proportional to the increasing amount of the initiator (Fig. 5). Moreover, the two small peaks declined clearly with increase in concentration of the initiator. In order to research the phenomenon further, the relationships between efficiency of initiator and mass concentration of sample 1–5 are arranged in Table 3. Apparently, the efficiency of initiator reduced with increase in concentration probably because t-BuOK is more likely to associate at higher concentrations. As mentioned above, the initiator in ion-pairs or small association states dominates the initiation process; therefore, while their proportion decreases, the initiation efficiency will decline.
 | ||
| Fig. 5 GPC curves of sample 1, 4, and 5. | ||
It was commonly perceived that cyclic ketone was formed during the polymerization of methacrylate by the bite back reaction, which would terminate the propagation of the polymer chain. However, conversions shown in Table 1 indicated that there are few side reactions in polymerization initiated by t-BuOK. It could be considered that the radius of the counter ion K+ is much larger than that of Li+. Therefore, the charge density of K+ is significantly lower than that of Li+, which accounts for relatively lower reactivity and less side reactions. This explains the high conversion of the polymerizations.
Sequential block copolymerization of n-HMA and MMA
Block copolymers of n-HMA and MMA were prepared by sequential feeding of monomers (Scheme 1). The results are concluded in Table 4. The conversions of the two synthetic routes were almost complete. GPC curves of sample 10–13 are shown in Fig. 6 (the part in product with low molecular weight was removed through several refining processes). For both of the synthetic routes, the homopolymer of the first monomer presented a unimodal peak, while the final block copolymer showed an extra small shoulder peak located nearly at the same position as the original homopolymer. This is because a small amount of active centres diminished due to the impurities in the following addition and sampling processes. As a result, a small part of homopolymer remained in the system. This also explains why the MWD of the copolymer is slightly broader than that of homopolymer. The results of DMA analysis are shown in Fig. 7. Two glass transition temperatures are observed, −10 to 0 °C for the PHMA block and 100 to 110 °C for the PMMA block. This demonstrated the existence of micro-phase separation of the block copolymers.30,31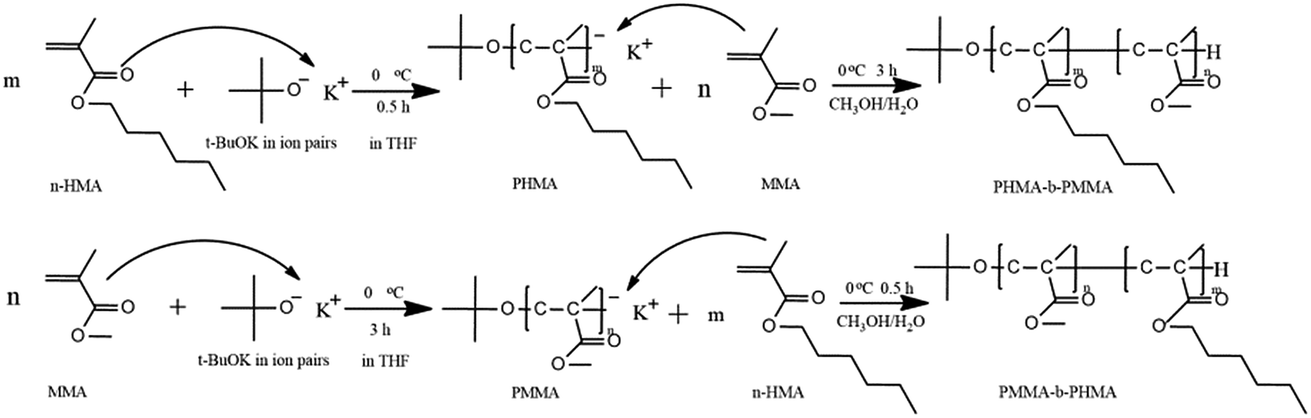 | ||
| Scheme 1 Reaction pathways in synthesis of block copolymers. | ||
| Samplea | Polymer | Time (h) | Conversion (%) | Mnb | MWDb | |
|---|---|---|---|---|---|---|
| MMA | n-HMA | |||||
| a MMA 28 mmol, HMA 42 mmol, t-BuOK 5.2 mmol, 50 ml of THF.b Mn and MWD were determined by GPC. | ||||||
| 10 | PMMA | 3 | 99.0 | — | 8200 | 1.38 |
| 11 | PMMA-block-PHMA | 3 + 0.5 | — | >99.9 | 24400 |
1.41 |
| 12 | PHMA | 0.5 | >99.9 | — | 19100 |
1.44 |
| 13 | PHMA-block-PMMA | 0.5 + 3 | — | >99.9 | 28600 |
1.45 |
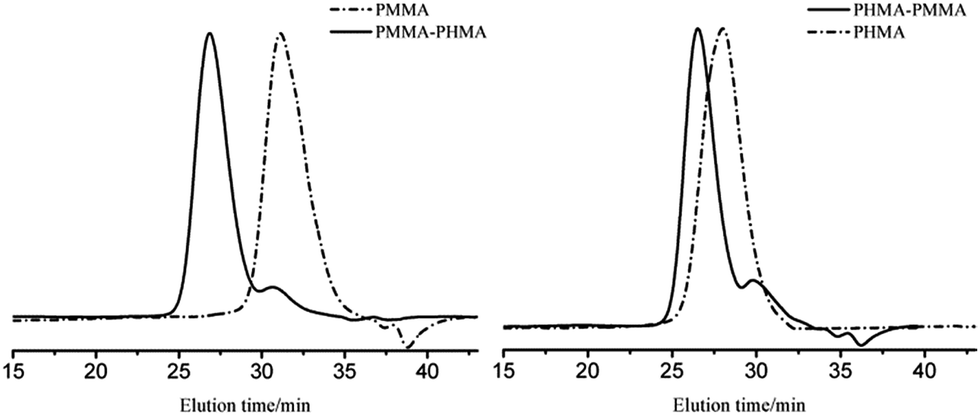 | ||
| Fig. 6 GPC curves of block copolymers of MMA and n-HMA. | ||
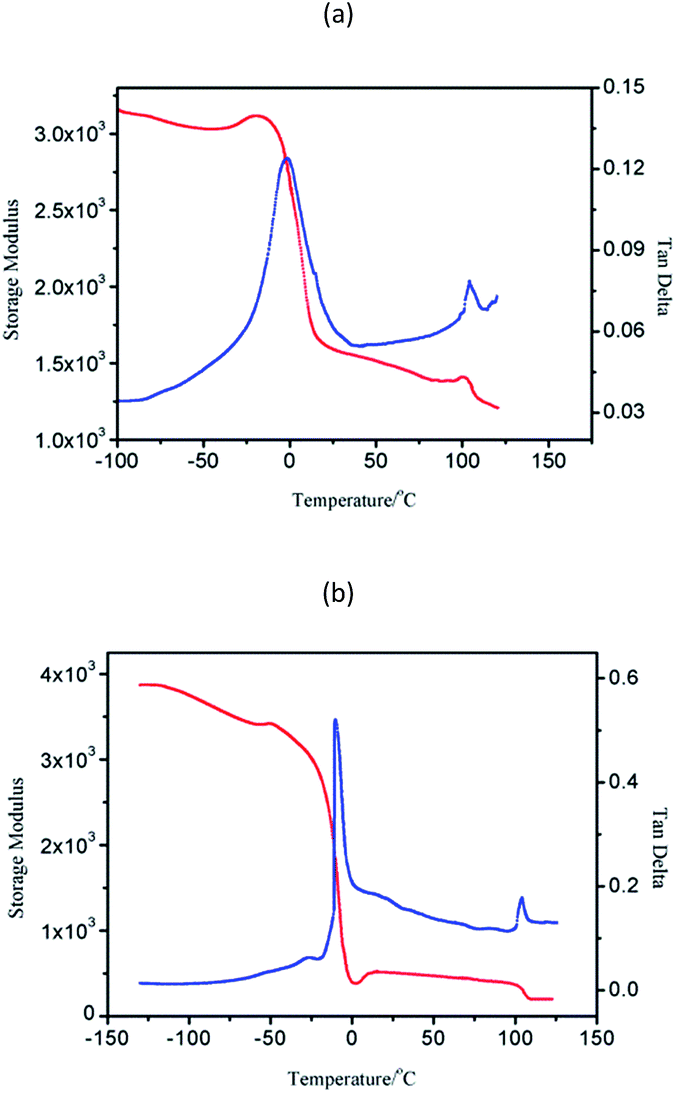 | ||
| Fig. 7 Dynamic mechanical curves of PMMA-PHMA (a) and PHMA-PMMA (b). | ||
Competing behaviors of n-HMA and MMA
The results of polymerization with one-shot feeding of the two types of monomers are summarized in Table 5; the conversions of the three samples were all nearly complete. The polymerization kinetics curves determined by 1H-NMR (Fig. 8) suggested that n-HMA tended to complete polymerization preferentially. At 0 °C, the conversion of n-HMA was nearly 100% when the reaction was carried out for 15 min while that of MMA was only about 15%. The polymerization rate increased clearly with increase in temperature. The results of 13C-NMR analysis of sample 14–16 are shown in Fig. 9. As shown in the figure, peak splitting and displacement clearly did not exist. The spectra of the copolymers were very simple and almost identical to those of mixtures of the corresponding homopolymers, which indicated that the copolymer consisted of long blocks of each monomer and no evident alternate polymerization. This result is consistent with the kinetic curves and it provides a new method to synthesize some block copolymers. | ||
| Fig. 8 Time courses of sample 14 (a), sample 15 (b), sample 16 (c). | ||
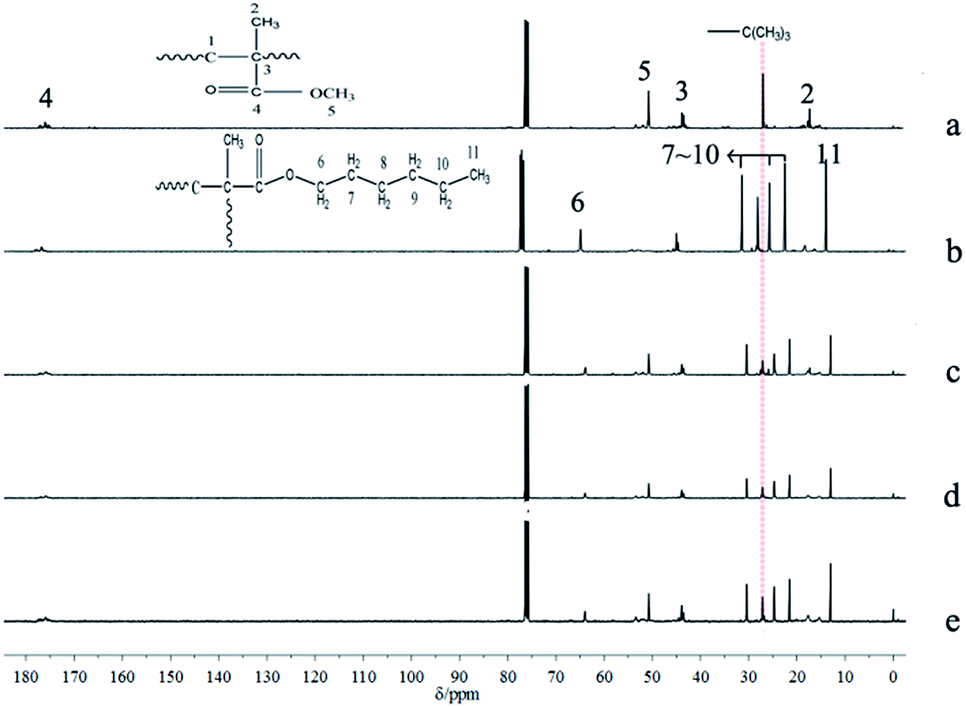 | ||
| Fig. 9 13C NMR spectra of homopolymer of n-HMA and MMA and their copolymers. PMMA initiated by t-BuOK (a), PHMA initiated by t-BuOK (b), sample 14 (c), sample 15 (d), sample 16 (e). | ||
Conclusion
A series of PHMA were prepared through anionic polymerization using t-BuOK as the initiator and THF as the solvent. The conversion was nearly up to 100% even at 60 °C. GPC analysis of PHMA indicated that t-BuOK exits in numerous states when dissolved in THF, including different types of association and ion-pairs. All the states can initiate polymerization with different reactivity because of the steric effect. Based on this, the sequential block copolymers were synthesized with almost complete conversion. Furthermore, the kinetics curves and 13C-NMR of the copolymer prepared by one-shot feeding of two types of monomers indicated that the polymerization of n-HMA in the copolymerization was significantly dominant over that of MMA at set temperatures, although the total conversion could almost reach 100%. The results in this study not only indicated the initiation mechanism of t-BuOK but also provided the possibility to realize anionic polymerization of methacrylate esters at a commercial scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (50933002, 51373052, 51573043).References
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 192–228 Search PubMed.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- S. Dong, Y. Long, J. Wang and A. Zheng, Chem. Res. Chin. Univ., 2014, 35, 2698–2705 Search PubMed.
- J.-m. Wang, B. Chen, S.-j. Tang, D. Shan and A.-n. Zheng, Chin. J. Polym. Sci., 2015, 33, 1096–1103 CrossRef CAS.
- K. Knoll and N. Nießner, Macromol. Symp., 2015, 132, 231–243 CrossRef.
- J. Wang, D. Shan, L. Yang, W. Liu, B. Chen and A. Zheng, Adv. Polym. Technol., 2016, 35, 1163–1169 Search PubMed.
- X. Yuan, J. Wang, D. Shan and A. Zheng, Polym. Mater.: Sci. Eng., 2014, 30, 1–6 CAS.
- D. Baskaran, Prog. Polym. Sci., 2003, 28, 521–581 CrossRef CAS.
- D. Baskaran and A. H. E. Müller, Prog. Polym. Sci., 2007, 32, 173–219 CrossRef CAS.
- W. E. Goode, F. H. Owens and W. L. Myers, J. Polym. Sci., Part A: Polym. Chem., 2010, 47, 75–89 Search PubMed.
- D. M. Wiles and S. Bywater, J. Phys. Chem., 1964, 68, 1983–1987 CrossRef CAS.
- S. K. Varshney, C. Jacobs, J. P. Hautekeer, P. Bayard, R. Jerome, R. Fayt and P. Teyssie, Macromolecules, 2002, 23, 51–56 Search PubMed.
- J. S. Kim, J. O. Kweon, J. H. Lee and S. T. Noh, Macromol. Res., 2015, 23, 100–110 CrossRef CAS.
- L. Ren, K. Liu, Q. He, E. Ou, Y. Lu and W. Xu, RSC Adv., 2016, 6, 51533–51543 RSC.
- S. Lee and T. Chang, Eur. Polym. J., 2011, 47, 800–804 CrossRef CAS.
- D. Baskaran and S. Sivaram, Macromolecules, 1997, 30, 1550–1555 CrossRef CAS.
- J. S. Wang, R. Jerome and P. Teyssie, Macromolecules, 1995, 28, 2990–2992 CrossRef CAS.
- T. Kitayama, M. Tabuchi, T. Kawauchi and K. Hatada, Polym. J., 2002, 34, 370–375 CrossRef CAS.
- L. V. Vinogradova, V. N. Zgonnik, A. A. Il'Ina, D. Docheva and C. Tsvetanov, Macromolecules, 1992, 25, 6733–6738 CrossRef CAS.
- Y. Guan, N. Wang, D. Hu, J. Zhang, H. Cao and A. Zheng, Pastoral Care in Education An International Journal of Personal Social & Emotional Development, 2012, vol. 42, p. 732 Search PubMed.
- D. H. Hu, S. H. Wu, T. Y. Li, A. N. Zheng and Y. Guan, Acta Polym. Sin., 2013, 013, 563–569 CrossRef.
- A. Zheng, B. Chen, D. Hu, Y. Guan, D. Wei, S. Li and D. Ji, Chin. J. Chem., 2013, 31, 393–400 CrossRef CAS.
- B. Chen, J. Wang, M. Shu, B. Zou, Y. Guan and A. Zheng, Chin. J. Chem., 2014, 32, 1128–1134 CrossRef CAS.
- A. Zheng, J. Zhang, Y. Guan, F. Hu, D. Wei and S. Wang, US Pat., 8546503 B2, USA, 2013.
- M. Z. Shu, B. Chen, B. Zou, F. Y. Chen, A. N. Zheng and Y. Guan, J. Assoc. Am. Med. Coll., 2015, 71, 835–844 Search PubMed.
- Z. Li, J. Chen, L. Su, B. Zou, P. Zhan, Y. Guan and A. Zheng, RSC Adv., 2017, 7, 9933–9940 RSC.
- T. Öztürk and E. Meyvacı, J. Macromol. Sci., Part A: Pure Appl.Chem., 2017, 54, 1–7 CrossRef.
- A. Z. Niu, J. Stellbrink, J. Allgaier, L. Willner, A. Radulescu, D. Richter, B. W. Koenig, R. P. May and L. J. Fetters, J. Chem. Phys., 2005, 122, 6878 Search PubMed.
- A. Z. Niu, J. Stellbrink, J. Allgaier, L. Willner, D. Richter, B. W. Koenig, M. Gondorf, S. Willbold, L. J. Fetters and R. P. May, Macromol. Symp., 2004, 215, 1–15 CrossRef CAS.
- T. Öztürk, M. Göktaş and B. Hazer, J. Appl. Polym. Sci., 2010, 117, 1638–1645 Search PubMed.
- T. Öztürk, M. Yavuz, M. Göktaş and B. Hazer, Polym. Bull., 2016, 73, 1497–1513 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |