
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted rapid synthesis of graphene-analogue hexagonal boron nitride (h-BN) nanosheets and their application for the ultrafast and selective adsorption of cationic dyes from aqueous solutions
Aliyar Mahdizadeh,
Saeed Farhadi * and
Abedin Zabardasti
* and
Abedin Zabardasti
Department of Chemistry, Lorestan University, Khoramabad 68151-44316, Iran. E-mail: sfarhadi1348@yahoo.com; Fax: +98 06633120618; Tel: +98 06633120611
First published on 23rd November 2017
Abstract
Herein, graphene-analogue nanoscale hexagonal boron nitride (h-BN) with a sheet-like morphology is successfully synthesized by reacting borax (Na2B4O7·10H2O) and NH4Cl powder as inexpensive starting materials in air via a facile microwave-assisted method with a short reaction time (10 min). The as-prepared product is structurally characterized via X-ray powder diffraction (XRD), Fourier transformation infrared spectroscopy (FTIR), Raman spectroscopy, energy dispersive spectroscopy (EDS), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and BET surface area measurements. X-ray diffraction indicates the formation of hexagonal boron nitride (h-BN) with lattice constants of a = 2.50 and c = 6.67 Å. The purity of the BN nanosheets is confirmed via X-ray energy dispersive spectroscopy, X-ray photoelectron spectroscopy, and Raman spectroscopy. TEM and FE-SEM indicate that the as-synthesized product is composed of fine loosely aggregated plate-like particles with lengths in the range of 0.5 to 3.5 μm and thicknesses of 20–30 nm. EDX gives an average B/N atomic ratio of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. FT-IR displays strong B–N absorptions at 1396 and 805 cm−1. In addition, ultraviolet-visible (UV-vis) spectroscopy reveals that the as-prepared h-BN has an obvious band gap (5.92 eV). The as-prepared BN nanosheets also have a relatively large specific surface area of 177.07 m2 g−1. Due to these characteristics, the as-prepared BN nanosheets exhibit a very quick adsorption rate for cationic methylene blue (MB) and Rhodamine B (RhB) dye and their adsorption capacity is as high as 472.4 mg g−1. Furthermore, these BN nanosheets can also selectively adsorb MB from binary and ternary mixed dye solutions. Therefore, these BN nanosheets have wide promising applications in wastewater treatment and the recovery of valuable organic compounds from wastewater.
:1. FT-IR displays strong B–N absorptions at 1396 and 805 cm−1. In addition, ultraviolet-visible (UV-vis) spectroscopy reveals that the as-prepared h-BN has an obvious band gap (5.92 eV). The as-prepared BN nanosheets also have a relatively large specific surface area of 177.07 m2 g−1. Due to these characteristics, the as-prepared BN nanosheets exhibit a very quick adsorption rate for cationic methylene blue (MB) and Rhodamine B (RhB) dye and their adsorption capacity is as high as 472.4 mg g−1. Furthermore, these BN nanosheets can also selectively adsorb MB from binary and ternary mixed dye solutions. Therefore, these BN nanosheets have wide promising applications in wastewater treatment and the recovery of valuable organic compounds from wastewater.
1. Introduction
During the past few decades, a large amount of wastewater containing organic dyes and pigments, toxic metal ions, and cleaning agents has been discharged into the aquatic environment with the rapid development of modern industry, which will lead to devastating consequences to all living organisms.1,2 Especially, some colored organic dyes discharged from the plating, textile, and printing industries are resistant to biological degradation and generally stable to light, heat and oxidizing agents due to their complex aromatic structures.3–5 Additionally, synthetic organic dyes are not only highly visible and, even in small quantities, able to decrease gas solubility in water, but also toxic, carcinogenic and mutagenic for human beings.6–10 Thus, the removal of dye pollutants from effluent is quite desirous for water safety and human health protection. Currently, various physicochemical and biological treatment technologies such as coagulation, precipitation, filtration, oxidation, activated sludge and adsorption processes have been employed to remove dyes from aqueous solutions.11–15 Among them, adsorption is considered a promising method for wastewater purification due to its high efficiency, simple operation and low energy consumption. To date, many polymeric and inorganic adsorbents have been developed, such as activated carbon, carbon nanotubes, rice husk, metal oxides, clays, zeolites, chitosan, and metal–organic frameworks (MOFs).16–25 However, they frequently suffer from the disadvantages of limited adsorption capacity and slow adsorption rate, and generally limited by less adsorption sites, slow kinetics and nonequilibrium of adsorption. Therefore, it is necessary to develop new adsorbents with high adsorption capacity, high selectivity and ultrafast adsorption characteristic towards specific dyes.In recent years, two-dimensional (2D) graphene-like hexagonal boron nitride (h-BN) has attracted considerable attention due to its many unique physical and chemical properties, such as low density, high temperature stability, high thermal conductivity, low dielectric constant, excellent chemical inertness, and high resistance to thermal shock and corrosion.26 These properties make h-BN an interesting material for many applications such as lubricants, protective and optical coatings, and advanced ceramic composites.27–29 To date, many methods have been developed for the synthesis of hexagonal BN including classical high-temperature routes (e.g., reaction of boron oxide and ammonia, direct nitridation of boron with N2 at 1400–1900 °C and carbothermal reduction and nitridation of boron oxide at 1500 °C), vapor deposition methods (e.g., reaction of boron trichloride and ammonia), condensed-phase pyrolysis techniques (e.g., thermal decomposition of borazene or bisborazinylamine), solid-state metathesis, and pyrolysis and combustion synthesis.30–45 Most of these methods have to be carried out under high temperature or high N2 pressure and require long processing times. Furthermore, many of them also involve the use of complicated expensive instruments and some of them suffer from low production yields (less than 30%). These problems either pose difficulty in mass production or result in high production costs and thus hinder their practical application.
Over the past few decades, the microwave irradiation (MWI) technique has been developed for the fast synthesis of functional inorganic nanomaterials.46 This method has unique effects and significant merits such as easy workup, rapid volumetric heating, high reaction rate, short reaction time, energy saving and production of inorganic nanoparticles with narrow size distribution compared with other methods. Various inorganic nanomaterials have been synthesized for different applications using the MWI technique.47,48
Based on the aforementioned considerations, in this work, we develop a facile and rapid strategy for the preparation of graphene-like hexagonal boron nitride (h-BN) nanosheets by reacting borax (Na2B4O7·10H2O) with ammonium chloride (NH4Cl) as inexpensive B and N element precursors in a very short time (10 min) in air atmosphere. To the best of our knowledge, the microwave-assisted approach for the synthesis of BN nanostructures has not yet been reported. The morphology, structure and properties of the as-prepared h-BN nanosheets are fully characterized via various physical and spectroscopic methods and their adsorption performance evaluated towards some organic pollutants in aqueous solutions. This nanomaterial exhibits superior adsorption rate and selective adsorption ability for cationic dyes. Remarkably, it exhibits a large-scale adsorption capacity of 472.4 mg g−1 for MB. Hence, it is a promising and environmentally friendly adsorbent for the removal and separation of organic pollutants in dye-wastewater.
2. Experimental
2.1. Materials
Borax (Na2B4O7·10H2O, 98%), ammonium chloride (NH4Cl, 99%) and other chemicals, such as copper(II) oxide, hydrochloric acid and sodium hydroxide were purchased from Merck Chemical Corporation. Methylene blue (MB, C16H18ClN3S, 98%), Rhodamine B (RhB, C28H31ClN2O3, 98%) and methyl orange (MO, C14H14N3NaO3S, 98%) were purchased from Merck. All other chemicals were of analytical grade and were used as received.2.2. Preparation of BN nanosheets
To prepare BN nanosheets, a mixture of 10 g borax (Na2B4O7·10H2O) and 10 g ammonium chloride (NH4Cl) was ground to give an intimate mixture. The mixture was taken in a porcelain crucible which was placed in the middle of another larger porcelain crucible filled with CuO powder as a secondary microwave absorber. The reaction mixture was irradiated in a microwave oven (MW frequency: 2.45 GHz) at 900 W in an air atmosphere. The reaction was completed after an irradiation time of 10 min. The product was cooled to room temperature and washed three times with hydrochloric acid (HCl, 0.1 M). The final product was collected, washed with 100 mL of deionized hot water and then finally dried at 100 °C for characterization and used as an adsorbent.2.3. Characterization
X-ray diffraction (XRD) patterns were recorded with a X'Pert Pro diffractometer (PANalytical, Netherlands) with Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 40 mV between 10° and 80° (2θ) with a step size of 0.03°. Fourier transform infrared (FT-IR) spectra were recorded on a Shimadzu system FT-IR 8400 spectrometer using KBr pellets over the range of 4000–400 cm−1. Raman spectra were obtained on a Spex 1403 Raman spectrometer. Optical adsorption spectra were obtained on a Cary 100 Varian UV-vis spectrophotometer in the wavelength range of 200–800 nm. The morphology and composition of the product were determined on a MIRA3 TESCAN scanning electron microscope (SEM) equipped with an energy dispersive X-ray analyzer (EDX) for elemental analysis. The TEM image of the product was obtained at the accelerating voltage of 200 kV. The sample for TEM studies was well dispersed in ethanol to form a homogeneous suspension by sonication for 30 min. Then the ethanol dispersion was dropped on a carbon-coated copper grid. Moreover, to obtain the specific surface area and pore size distribution of the BN product, nitrogen physisorption isotherms were obtained on a Nova 2000 surface area and pore analyzer instrument at 77 K. The specific surface area was calculated using the BET (Brunauer–Emmett–Teller) model and the Barrett–Joyner–Halenda (BJH) method was used to calculate the pore size distribution from the adsorption branch. Magnetic measurements were carried out at room temperature using a vibrating sample magnetometer (VSM, Magnetic Daneshpajoh Kashan Co., Iran) with a maximum magnetic field of 10 kOe.2.4. Adsorption tests
To explore the adsorbent properties of the as-prepared BN nanomaterials, adsorption studies were carried out for the removal of MB, RhB and MO dyes from the aqueous solutions. The chemical structure and basic properties of these dyes are given in Fig. 1 and Table 1. Stock solutions of the dyes were prepared by dissolving accurately weighted quantities of the solid dyes in deionized water. In the adsorption experiment, 50 mL of dye solution with a known concentration (e.g. 25 mg L−1) was added to a 100 mL glass beaker and a definite amount of BN adsorbent (e.g. 60 mg) was dispersed in it via sonication. The dispersed solution was continuously stirred on magnetic stirrer at a constant speed of 500 rpm. After predetermined time intervals, sample solutions were withdrawn and centrifuged for 10 min to remove the suspended adsorbent particles, and filtered to analyze the residual concentrations of dye. The initial and final concentrations in the aqueous solutions were measured using a UV-vis spectrophotometer at 664, 554 and 463 nm for MB, RhB and MO dye, respectively. The effect of experimental parameters such as adsorbent dosage and time on the adsorption is discussed in detail. Upon analyzing the UV-vis data, the removal efficiency (%) and the adsorbed amount (qt, mg g−1) of the dye compounds were calculated using the following equation:| Removal efficiency (%) = [(C0 − Ct)/C0)] × 100 |
| qt = (C0 − Ct)V/m |
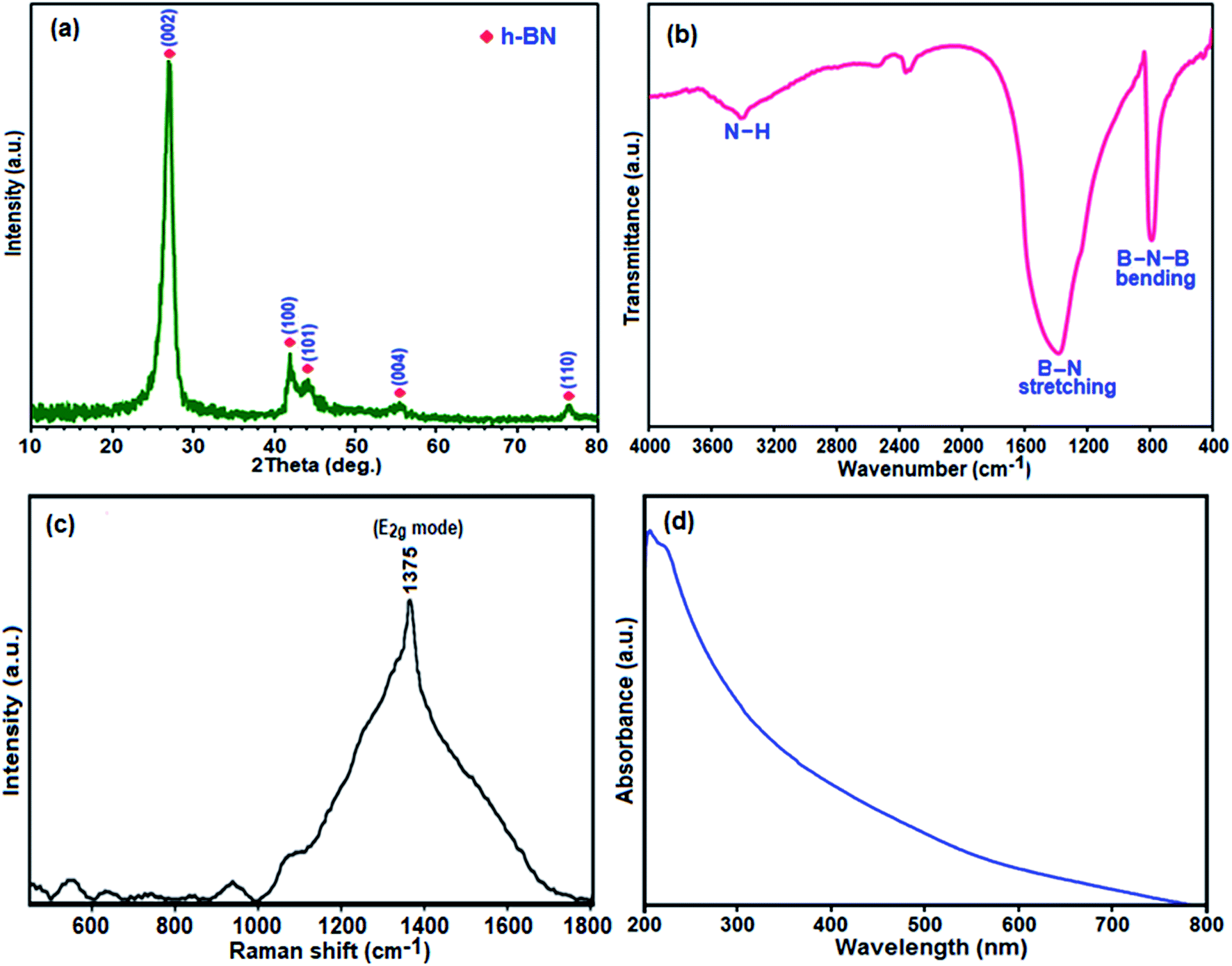 | ||
| Fig. 1 (a) XRD pattern, (b) FT-IR spectrum, (c) Raman spectrum, and (d) UV-vis absorption spectrum of the h-BN nanosheets. | ||
| Entry | Starting materials | Reaction conditions | Time (h) | Ref. |
|---|---|---|---|---|
| 1 | Boric acid (H3BO3) + PVA (1:4) + NH3(aq) |
Heated at 1200 °C in an N2 gas flow | 10 | 62 |
| 2 | NH4BF4 + NaN3 | Solvothermal in benzene and CS2, 260 °C, 250 MPa in N2 atmosphere | 24 | 63 |
| 3 | NaBH4 + FeCl3 + NaN3 | In stainless steel autoclave, heated at 750 °C | 12 | 64 |
| 4 | H3BO3 + NH4Cl | 950 °C under N2 in the presence of CuO + KCl | 4 | 65 |
| 5 | B2O3 + urea + Cu(NO3)2 | Heated at 1250 °C, under NH3 gas atmosphere | 5 | 66 |
| 6 | H3BO3 + urea | Heated at 900 °C in N2 atmosphere | 5 | 67 |
| 7 | B2O3 + guanidine hydrochloride | Heated at 1100 °C, under H2/N2 gas | 2 | 68 |
| 8 | B2O3 + urea (1:10) |
Heated at 1000 °C, under nitrogen/hydrogen (5%) gas flow | 3 | 69 |
| 9 | B2O3 + urea | Heated at 550 °C + 1100 °C in N2 atmosphere | 4 | 70 |
| 10 | Calcium hexaboride (CaB6) + NH4Cl | In stainless steel autoclave, heated at 600 °C under argon atmosphere | 48 | 71 |
| 11 | NaBH4 + NaN3 | In stainless steel autoclave, heated at 600 °C under argon atmosphere | 8 | 72 |
| 12 | H3BO3 + NH4Cl + Mg | In stainless steel autoclave, heated at 600 °C | 10 | 73 |
| 13 | Li2B4O7 + NaN3 + Mg | In stainless steel autoclave, heated at 500 °C | 12 | 74 |
| 14 | NaBH4 + CS(NH2)2 | In stainless steel autoclave, heated at 550 °C | 10 | 75 |
| 15 | Na2B4O7·10H2O + NH4Cl | Under MWI, at room temperature under atmospheric pressure and in air | 10 min | This work |
3. Results and discussion
3.1. Characterization of the BN nanosheets
In the present study, we developed a synthetic process to obtain porous BN nanosheets which relies on a fast microwave-assisted approach. Briefly, a homogeneous mixture of borax (Na2B4O7·10H2O) with NH4Cl as very cheap boron and nitrogen sources in the presence of CuO as a strong MWI absorber material was irradiated in an MW oven for 10 min under air atmosphere to yield a white product. For comparison, an Na2B4O7·10H2O and NH4Cl mixture was irradiated in the MW oven in the absence of CuO, and no BN was obtained even after 30 min. This control test confirms that the reaction mixture does not absorb microwaves directly and needs a secondary absorber (e.g. CuO). In addition, when the MW oven was opened after the reaction, some gases were released. It is possible that during the MW heating process, some NH4Cl decomposed, releasing a number of gaseous species including HCl and NH3. The escape of gases (gas bubbles) acts as fugitive templates for the final porous structure. The reaction between Na2B4O7·10H2O and NH4Cl takes place using the hot heating medium of CuO and the h-BN product was obtained within several minutes. The possible reaction equation for the synthesis of BN from Na2B4O7·10H2O and NH4Cl under MWI can be expressed as follows:| Na2B4O7·10H2O(s) + 4NH4Cl(s) → 4h-BN(s) nanosheets + 4HCl(g) + 17H2O(g) |
The phase purity and crystal structure of the as-prepared BN product were investigated via powder X-ray diffraction XRD studies. Fig. 1(a) shows the typical XRD pattern of the product. All the characteristic peaks at 2θ = 27.18°, 41.91°, 43.00°, 55.00°, and 77.00° can be indexed as the hexagonal phase of BN crystal with lattice constants of a = 2.50 Å and c = 6.67 Å which are very close to the reported value of h-BN (a = 2.51 Å and c = 6.69 Å; JPCDS, card no. 34-0421).49 Also, no impurity peaks were detected. The sharpness of the peaks confirms the formation of a highly crystalline single-phase BN nanomaterial with the P63/mmc space group. The interlayer spacing (d002) estimated according to Bragg's law is 0.335 nm, which is very close to that of bulk h-BN of ∼0.333 nm.50 The broadened peaks in the XRD pattern indicate the nanometer scale size of the as-prepared crystallites. The crystallite size of this BN nanomaterial was found to be about 20 nm by evaluating the line broadening of the most intense (002) peak using the classical Scherrer equation.51
To obtain further information on the types of chemical bonds in the obtained BN, the FTIR spectrum of the as-prepared product is presented in Fig. 1(b). In the FTIR spectrum, two strong characteristic bands were observed at around 1396 and 805 cm−1, which are similar to the reported result of h-BN.52 The former band results from the in-plane ring B–N stretching vibration (Eu mode) of the sp2-bonded BN, whereas the latter band is attributed to the out of plane B–N–B bending vibrations (A2u mode).53 Besides, the band centered at 3420 cm−1 is attributed to the stretching mode of the N–H group.54 Thus, the FTIR results confirm the bond formation of boron nitride.
Raman spectroscopy was further employed to explore the phase purity of the as-prepared BN nanosheets. As shown in Fig. 1(c), in the Raman spectrum of the synthesized h-BN sample a broad peak at ca. 1375 cm−1 is observed, which is ascribed to the high-frequency E2g vibrational mode of h-BN.55 The bulk BN powder displays a Raman shift at 1367 cm−1 as an intrinsic E2g vibration.56 It has been reported that crystal domain size, stack layer ordering, amorphization, elemental doping and porosity could lead to such shifting and broadening of the Raman peak compared to bulk BN.57 Furthermore, highly porous BN displays the Raman shift at a higher frequency and broader peaks compared to bulk BN.58 Therefore, the appearance of the wide peak for the h-BN sample prepared in this work is also attributed to the existence of rather weak interactions between atomic layers leading to a disordered porous structure. This Raman finding is also later confirmed by the FESEM and BET results.
The optical properties of the BN sample were investigated via UV-vis spectroscopy. As shown in Fig. 1(d), the UV-vis absorption spectrum of the BN nanosheets dispersed in ethanol exhibits a relatively broad band with a strong absorption at about 210 nm, which corresponds to a band gap of ∼5.92 eV and is quite close to the reported value for BN (∼6.15 eV).59 Besides, there is also an absorption tail at the lower energy side of the absorption peak, which indicates that high density structural defects exist in the BN nanosheets.59
The morphology and structure of the as-prepared BN sample were investigated via SEM. Fig. 2 shows the SEM images of the BN nanostructure at different magnifications. The SEM images show that the obtained particles are sheet-like in shape with a thickness of 40–50 nm. As shown in Fig. 2(a)–(d), the as-prepared h-BN sample presents the layered structure of a graphene-analogue material. According to the SEM images, the sample consists of fine loosely aggregated plate-like particles with lengths in the range of 0.5 to 3.5 μm. The high magnification images in Fig. 2(e) and (f) indicate that the surface of the sheets is rough and compact with porous features, and they are composed of BN fine nanoplates and nanoparticles with thicknesses or sizes of 20–30 nm, which is consistent with the above-calculated value from the Scherrer equation. Furthermore, the SEM images reveal that the BN nanosheets have many pores and holes with diameters ranging from 20 to 100 nm, which are created by gas bubbles during the synthetic process.
 | ||
| Fig. 2 Different magnification SEM images of the h-BN nanosheets. | ||
The size and microstructure of the as-prepared BN particles were further investigated via TEM, as shown in Fig. 3. The TEM sample was prepared by dispersing BN powder in ethanol under ultrasonic vibration. From the TEM images in Fig. 3(a)–(d), clearly the morphology of the particles is plate-like in shape with a thickness of 20–30 nm, which indicates that the as-prepared h-BN has a graphene-like structure. As evident from the images, the morphology of the BN sample from the TEM images agrees with the SEM results.
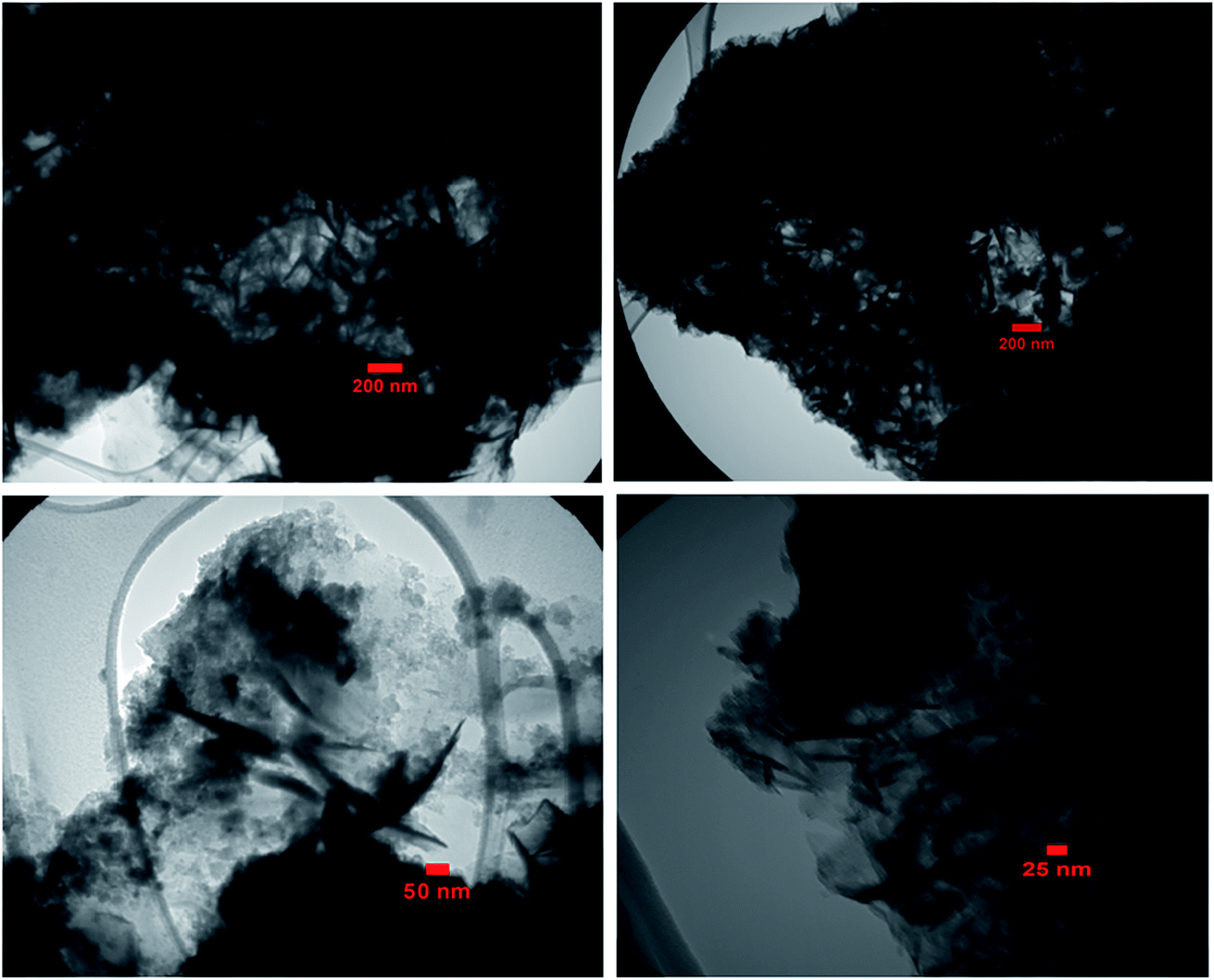 | ||
| Fig. 3 TEM images of the h-BN nanosheets. | ||
The chemical composition of the BN nanosheets was also investigated via energy dispersive X-ray (EDX) spectroscopy. Fig. 4 shows the EDX spectrum of the as-prepared product. In the EDX spectrum, strong peaks of boron and nitrogen with the atomic ratio of B:N of 50.74:49.26 were observed. This finding indicates that the nanosheets are composed of boron and nitrogen atoms with an atomic ratio of 1.03:1, which is in good agreement with the chemical stoichiometric ratio of BN. Slightly rich boron may result from the surface hydrolysis during purification which could result in a small loss in nitrogen. The existence of the gold (Au) peak at ca. 2.2 keV is ascribed to the coating of the gold film before SEM observation. The inset in Fig. 4 shows the elemental mappings of the product. In these images, a uniform distribution of B and N elements is observed.
 | ||
| Fig. 4 EDX analysis of the h-BN nanosheets. | ||
Further evidence for the formation of BN was obtained from the XPS spectra of the sample, as shown in Fig. 5. They show that the sample surface consists of nitrogen, boron, carbon and oxygen, with binding energies of N 1s, B 1s, C 1s and O 1s at 398.7, 191.5, 284.40 and 532.40 eV, respectively (Fig. 5(a)). The C 1s and O 1s peaks indicate that a small amount of impurity elements exists such as C and O due to the adsorption of CO2, H2O and O2 on the surface of the sample. The B 1s peak at 191.5 eV (Fig. 5(b)) and the N 1s peak at 398.7 eV (Fig. 5(c)) indicate BN, which is in good agreement with that in the literature.58 The quantification of B 1s and N 1s peaks confirms that the atomic ratio of B:N is 1.05:1, which closely agrees with the stoichiometric composition of BN.
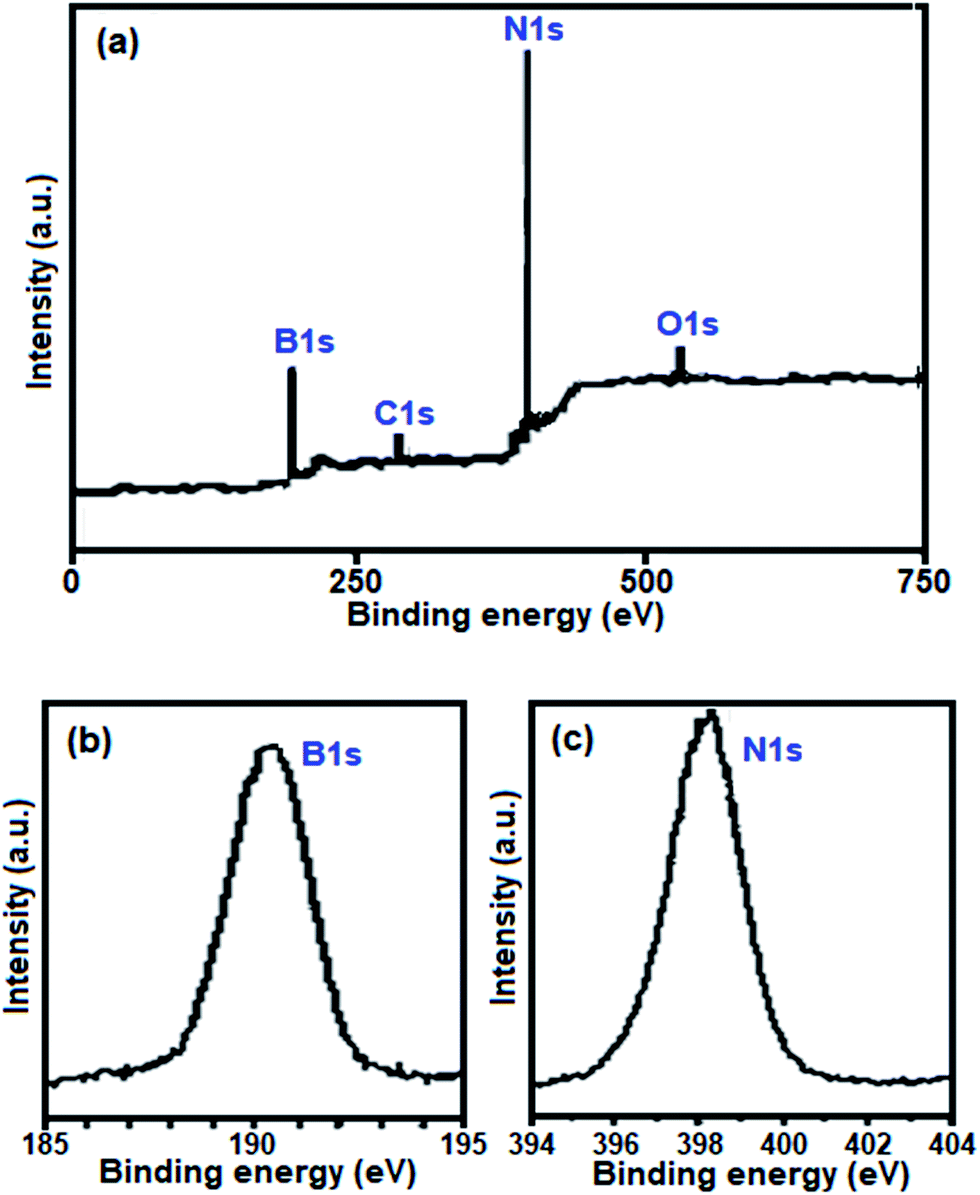 | ||
| Fig. 5 (a) Full range survey XPS spectrum of the as-prepared BN nanosheets. High-resolution XPS spectra of (b) B 1s and (c) N 1s. | ||
The surface area and porous structures of the BN nanosheets were measured based on nitrogen adsorption–desorption. As shown in Fig. 6(a), the isotherm shape could be categorized as type IV with a distinct H3 hysteresis loop at high relative pressures according to the IUPAC classification, which indicates the presence of a mesoporous structure.60 The BET specific surface area obtained from the isotherms is 177.2 m2 g−1. It should be noted that the surface area is much higher than that of commercial BN powder (25 m2 g−1).61 The large surface area is possibly caused by escaping gas for instance HCl, H2O and NH3 formed from the decomposition of the precursors. The total pore volume is 0.686 cm3 g−1. The pore size of the sample is 2.305 nm, which is estimated using the Barrett–Joyner–Halenda (BJH) method from the adsorption and desorption branches of the N2 isotherms (Fig. 6(b)). Owing to their large specific surface area, mesoporous nature, small pore diameter and high structural defect density, the BN nanosheets are expected to be an excellent adsorbent.
 | ||
| Fig. 6 (a) Nitrogen adsorption–desorption isotherm and (b) pore size distribution of BN nanosheets. | ||
Surface charge is one of the important parameter that controls the type of adsorbate to be absorbed. The zeta potential reveals the type and value of charge on the dispersed particles. For the as-obtained h-BN nanosheets, the zeta potential value was evaluated to be −52.5 mV in aqueous suspension and at natural pH. The negative value of the zeta potential demonstrates the presence of a negative charge on the surface of the product sample.
Finally, the preparation of BN by the present method was compared with some reported methods in recent years.62–75 From the Table 1, it is clear that with respect to reaction conditions and time, the present method is more suitable and superior. Most of the reported methods required high temperature and/or pressure, complicated expensive instruments, expensive and/or sensitive starting materials, long reaction times, inert atmosphere and strict operating conditions. Our method has several advantages, such as low cost, high yield, lack of organic solvents, substrates or vacuum systems and especially a lack of inert atmosphere. For instant, the borax and NH4Cl used in this work are easily available, non-toxic and very cheap sources of boron and nitrogen elements. The above advantages make this MW-assisted synthesis process a very attractive approach for the rapid and large-scale production of BN in the future.
3.2. Dye adsorption studies
Boron nitride (BN) nanosheets, so-called ‘white graphene’, consist of a few layers of hexagonal BN planes and have unique properties compared with graphene. In addition, the polarity of the B–N bond and high surface area of h-BN-related nanostructures provide good adsorption properties for various substances ranging from organic pollutants to hydrogen.76,77 Since BN is composed of light elements, it is a promising adsorbent with a high uptake gravimetric capacity. Its high chemical and thermal stability also suggest that recycling could be easily achieved. Nanostructured BN is therefore an ideal candidate as an absorbent material.To evaluate the adsorption capability of the as-prepared h-BN nanomaterial for the removal of organic dyes from contaminated water, three organic pollutants including cationic MB (molecule size: 1.38 nm × 0.64 nm × 0.21 nm), cationic RhB (molecule size: 1.56 nm × 1.35 nm × 0.42 nm) and anionic MO (molecule size: 1.54 nm × 0.48 nm × 0.28 nm) were selected as models, considering that both the charge and size of organic dyes are the most important factors controlling the adsorption process. The adsorption process was monitored using the characteristic absorption peaks of the dyes at 664, 553 and 463 nm for MB, RhB and MO, respectively. A decrease in the intensity of the characteristic peaks with adsorption time indicates a decrease in the amount of dye in the solution. The time dependent UV-vis absorption spectra of the dyes in the presence of the BN nanomaterial are shown in Fig. 7(a)–(c). The digital images and UV-vis spectroscopic results show that the cationic MB and RhB dyes are almost removed from aqueous solution in 0.5 min. As it can be seen in Fig. 7(c), the characteristic absorbance band of MO decreased slightly after 4 min with negligible fading of the orange color. The results show that the BN nanomaterial is a poor absorbent for anionic MO dye from aqueous solution. In comparison, in Fig. 7(d), the adsorption rate of the MB and RhB solutions quickly reached 98% and 78%, respectively, in the first 0.5 min while the removal efficiency of MO dye is less than 10% even after 4 min. Obviously, BN has different adsorption abilities towards different organic dyes. The different effects on the removal of dyes is related to the structure and charge of the dye molecules and the structure of the adsorbent material. The presence of a negative charge on the surface of the BN nanomaterial, as depicted by its zeta potential, is consistent with the results of adsorption of differently charged dye molecules. Therefore, the BN nanosheets demonstrate good adsorption properties toward the cationic dye molecules MB and RhB. However, the removal percent of RhB dye is lower than that of MB dye, which is probably due to the large volume of RhB molecules, which causes steric hindrance with the active adsorption sites on the hybrid, and its weak π–π interactions with the BN nanosheets. On the other hand, the appearance of electrostatic repulsion between the negatively charged BN nanomaterial and anionic MO dye molecule results in less adsorption compared with cationic dye molecules. Therefore, the adsorption of dyes is found to be very specific towards the charge present on the surface of the adsorbate. The ultrafast removal rate in the initial 0.5 min is attributed to the rapid diffusion and strong electrostatic interaction of the cationic dyes to the external surfaces of the BN nanosheets. Therefore, the as-prepared BN nanosheets can be categorized as an ultrafast adsorbent which would be highly useful for the quick and significant removal of harmful compounds.
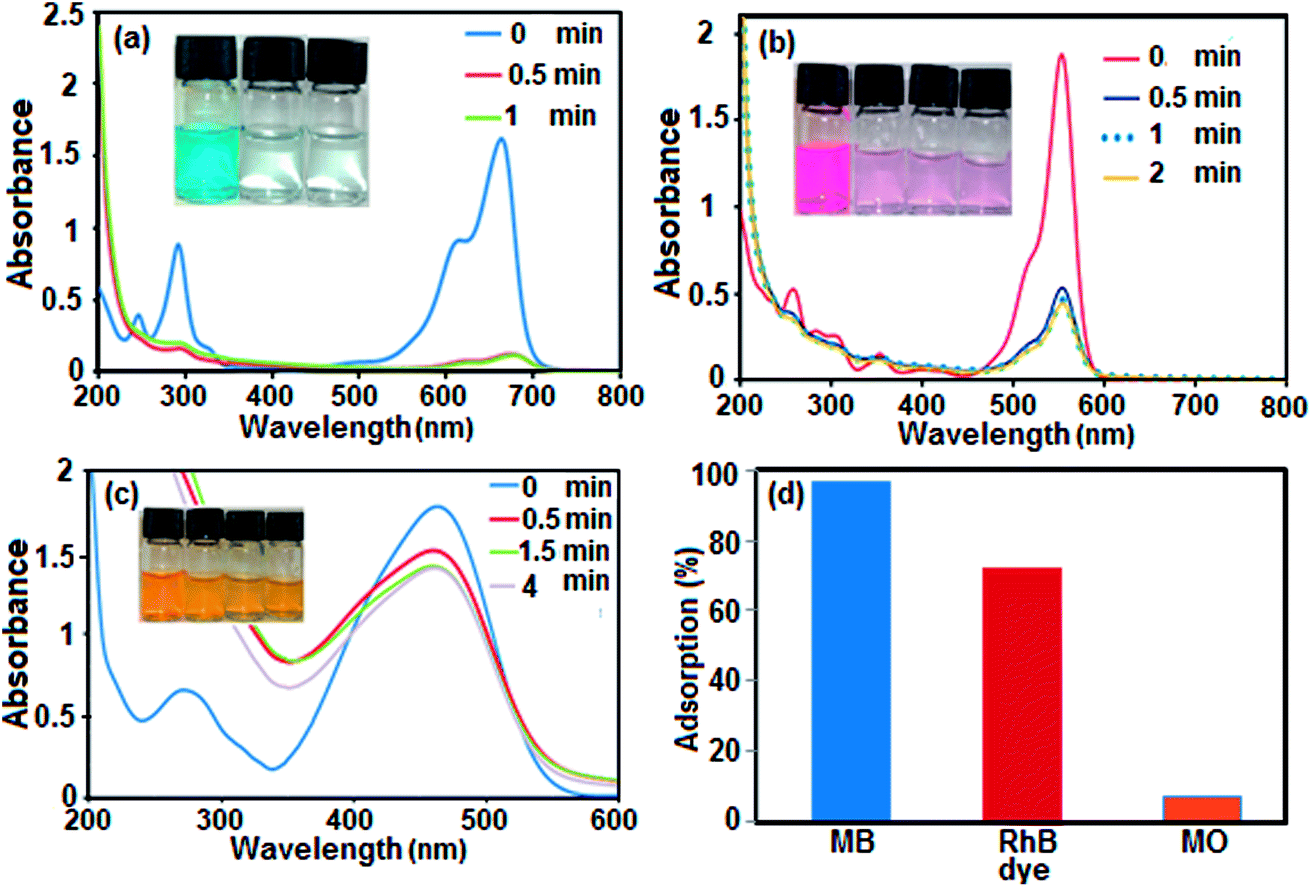 | ||
| Fig. 7 UV-vis spectral changes of dye aqueous solutions over BN nanosheets at different time intervals: (a) MB dye, (b) RhB dye and (c) MO dye. (d) Comparison of adsorption efficiency (%) of dyes. Conditions: [dye] = 25 mg L−1 in 50 mL aqueous solution and mBN = 60 mg/50 mL at room temperature. The insets show digital photos of the organic dyes in aqueous solution during the adsorption process. | ||
Selective adsorption and separation of a specific dye is more attractive and challenging in the process of dye-wastewater treatment. In this study, in view of the large uptake capacity of the BN nanosheets for MB and RhB dye, it can be anticipated that the BN nanosheets may also have outstanding adsorption and separation behavior in the treatment of dye mixtures. The selective uptake of dyes was tested using dye mixtures and the process was monitored by UV-vis spectroscopy. Since MB and MO have similar molecule sizes, the preferable uptake of cationic MB from the MB/MO mixture (Fig. 8(a)) may be assigned to the existence of a negative charge on the BN nanosheets. For further confirmation, cationic RhB was selected to mix with anionic MO and the results reveal that RhB dye was also preferably adsorbed on the BN nanomaterial from this binary dye mixture, as illustrated in Fig. 8(b). To further validate this point, a ternary mixture of MB, RhB and MO dye was investigated. As exhibited in Fig. 8(c), the representative bands of MB and RhB all disappeared quickly in the mixed dyes and only the characteristic absorption peaks of MO were left, which suggests that the BN nanosheets could selectively capture cationic dyes when utilized in corresponding ternary mixtures. The same conclusion is displayed in the insets of Fig. 8(a)–(c), where only the orange color of MO dye can be seen in the final solutions of mixed dyes after the adsorption process. This can be attributed to the negative charge of the dye molecule, which repels each other between MO and the BN nanosheets. This further indicates that the adsorption process occurs mainly due to the electrostatic interactions between the BN nanosheets and dye molecules. Meanwhile, the aromatic backbone further strengthens the connection between the adsorbent (BN nanosheet) and adsorbates (MB and RhB dyes) via π–π stacking interaction.78 Due to the synergistic effect of electrostatic interactions and π–π stacking interactions the BN nanosheets possess selective adsorption ability towards some cationic aromatic dyes.78 Thus, the as-prepared BN nanomaterial is an environmentally friendly active adsorbent for the removal of different cationic organic pollutants.
 | ||
| Fig. 8 Selective absorption capacity of BN toward mixed dyes of (a) MB/MO, (b) MO/RhB and (c) MB/RhB/MO. Conditions: C0(MB) = C0(MO) = C0(RhB) = 25 mg L−1, V = 50 mL, mBN = 60 mg at room temperature for time = 2 min. The insets show digital photos of the organic dyes in aqueous solutions during the adsorption process. | ||
The effect of adsorbent dosage on the removal efficiency for MB and RhB dye was investigated. The adsorbent dosages were varied from 15 to 30 mg and then to 60 mg at the initial dye concentration of 25 mg L−1 and pH of about 6 for both dyes. The removal efficiencies of both dyes with different amounts of adsorbent are shown in Fig. 9. The results indicate an increase in removal efficiency with an increase in adsorbent dosage, which can be explained on the basis of the increase in the availability of adsorption sites on the surface of the adsorbent. With the adsorbent dose of 60 mg, both dyes (MB and RhB) showed high removal (%) in the short time of 0.5 min.
 | ||
| Fig. 9 Effect of adsorbent dosage on the removal of MB and RhB dyes. Conditions: [dye] = 25 mg L−1, 50 mL, mBN = 60 mg at room temperature for time = 0.5 min. | ||
The as-prepared BN nanosheets exhibited the adsorption capacity of 472.4 mg g−1 when 180 mg of BN nanosheets was soaked in 1 L of 500 mg L−1 aqueous MB solution for 1 h. Table 2 lists a comparison of the adsorption capacity for MB adsorption on various types of adsorbents. It can be seen that the adsorption capacity of the BN nanosheets used in this work is larger than that of other adsorbents reported in the literature.79–95 Based on the above observations and the characteristics of the BN nanosheets, we infer that the large specific surface area, mesoporous nature, high porosity and high structural defect density of the BN nanosheets contribute to its enhanced adsorption capacity. Also, noncovalent interactions such as π–π stacking interactions between lamellar BN nanosheets and aromatic rings also makes an important contribution to its adsorption capacity. Thus, this material is a promising excellent adsorbent for the treatment of toxic organic pollutants in dye-wastewater.
| Entry | Adsorbent | Adsorption capacity (mg g−1) | Ref. |
|---|---|---|---|
| 1 | Activated carbon | 135 | 79 |
| 2 | Clay | 6.3 | 80 |
| 3 | Zeolite | 10.86 | 81 |
| 4 | Graphene/Fe3O4 | 33.66 | 82 |
| 5 | Carbon nanotubes | 35.4 | 83 |
| 6 | Peanut husk | 72.13 | 84 |
| 7 | MWCNTs/Fe2O3 | 42.3 | 85 |
| 8 | CoFe2O4/MWCNT | 14.3 | 86 |
| 9 | Chitosan/montmorillonite | 95.24 | 87 |
| 10 | Chitosan/Fe3O4 | 45.1 | 88 |
| 11 | Polyurethane foam | 23.03 | 89 |
| 12 | WO3 nanosheets | 33.33 | 90 |
| 13 | MOF-235 | 187 | 91 |
| 14 | Graphene | 153.85 | 92 |
| 15 | H6P2W18O62/MOF-5 | 51.81 | 93 |
| 16 | Graphene oxide (GO) | 144.92 | 94 |
| 17 | Polyaniline nanotubes | 4.8 | 95 |
| 18 | BN nanosheets | 472.4 | This work |
The stability and reusability of adsorbents are important standards for their practical application. To verify whether the composite material is stable and can be recycled during adsorption experiments, cycle tests using the BN nanosheets for the removal of MB were conducted. After each cycle, the adsorbent was completely separated by filtration. Subsequently, fast release of the adsorbed MB was achieved by thoroughly washing the adsorbent with a dilute solution of NaCl and ethanol three times. Then, the desorbed adsorbent was added to 50 mL 25 mg L−1 MB solution under stirring. As described in Fig. 10, the BN nanomaterial showed almost identical rapid adsorption of MB, and after four cycles, the regenerated adsorbent was still able to remove 92% MB from the aqueous solution. Thus, we conclude that the BN nanomaterial is reusable during adsorption experiments.
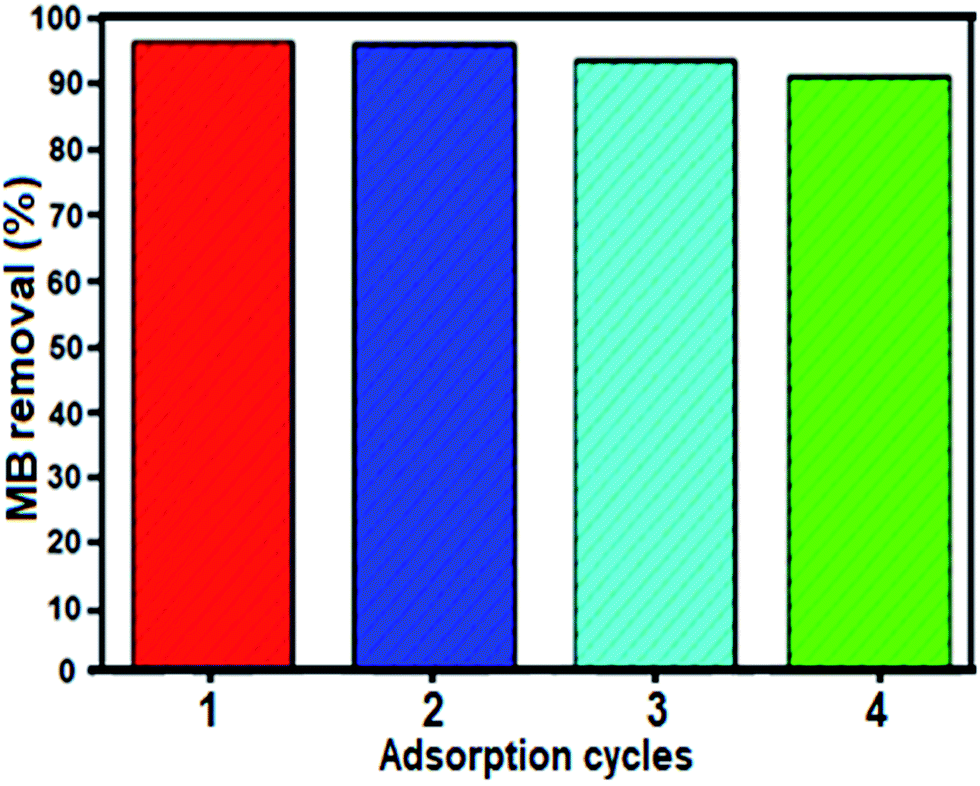 | ||
| Fig. 10 Recyclability of the BN nanosheets in the removal of MB dye. Conditions: [dye] = 25 mg L−1, 50 mL, mBN = 60 mg at room temperature for time = 0.5 min. | ||
Now we further discuss the stability of this material. As shown in Fig. 11(a) and (b), the XRD pattern and FTIR spectrum of the recovered adsorbent after the fourth run are consistent with that of the fresh BN sample (see Fig. 1(a) and b). These observations confirm that the structure of the BN nanosheets is stable under the reaction conditions and is not affected by the reactants. The morphology of the recycled adsorbent particles was also analyzed. Fig. 11(c) and (d) show the SEM images of the adsorbent after four cycles. It can be observed that the recovered adsorbent almost retained its initial size and morphology (see Fig. 2). Considering the abovementioned experiment results, we conclude that the structure of the compound remains intact, which further confirms its excellent stability and recyclability.
 | ||
| Fig. 11 (a) XRD pattern, (b) FT-IR spectrum, and (c) and (d) SEM images of the recovered BN nanomaterial after the fourth run. | ||
4. Conclusion
In summary, we have successfully synthesized graphene-like h-BN nanosheets by reacting borax and NH4Cl under MWI in 10 min without any solvent or surfactant. The as-prepared h-BN nanosheets were used as a novel adsorbent for the removal of dye in aqueous solution. They exhibit the maximum adsorption capacity of 472.4 mg g−1 for MB dye. More importantly, they present excellent ultrafast adsorption characteristics for cationic organic dyes. Only 30 seconds is enough to achieve adsorption equilibrium. Interestingly, they not only exhibit a rapid adsorption rate and high uptake capacity towards cationic dyes (MB and RhB), but also exhibit fast selective separation and recovery from mixed dyes (MB + MO, RhB + MO, and MB + RhB + MO). This difference in the adsorption capacity of BN nanosheets for organic dyes is attributed to the synergistic effect of electrostatic interactions, π–π stacking between adsorbent and adsorbate, and chemical structure of dye molecules. Thus, the h-BN nanosheets present powerful potential in separating dye mixtures in aqueous solution. Furthermore, their high adsorption capacity and ultrafast adsorption characteristics make them a potentially attractive adsorbent in water purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported in part by the Lorestan University Research Council and Iran Nanotechnology Initiative Council (INIC).References
- T. A. Saleh and V. K. Gupta, Environ. Sci. Pollut. Res., 2012, 19, 1224–1228 CrossRef CAS PubMed.
- Y. Wang, M. Yao, Y. Chen, Y. Zuo, X. Zhang and L. Cui, J. Alloys Compd., 2015, 627, 7–12 CrossRef CAS.
- A. S. Özcan, B. Erdem and A. Özcan, J. Colloid Interface Sci., 2004, 280, 44–54 CrossRef PubMed.
- T. A. Khan, E. A. Khan and S. Jahan, Appl. Clay Sci., 2015, 107, 70–77 CrossRef CAS.
- V. K. Gupta, R. Jain, A. Nayak, S. Agarwal and M. Shrivastava, Mater. Sci. Eng., C, 2011, 31, 1062–1067 CrossRef CAS.
- E. V. Datskevich, R. V. Prikhod'ko, I. V. Stolyarova, A. V. Lozovskii and V. V. Goncharuk, Russ. J. Appl. Chem., 2010, 83, 1785–1793 CrossRef CAS.
- Z. Chen, J. Fu, M. Wang, X. Wang, J. Zhang and Q. Xu, Appl. Surf. Sci., 2014, 289, 495–501 CrossRef CAS.
- Y. Li, Q. Du, T. Liu, J. Sun, Y. Wang, S. Wu, Z. Wang, Y. Xia and L. Xia, Carbohydr. Polym., 2013, 95, 501–507 CrossRef CAS PubMed.
- C. A. P. Almeida, N. A. Debacher, A. J. Downs, L. Cottet and C. A. D. Mello, J. Colloid Interface Sci., 2009, 332, 46–53 CrossRef CAS PubMed.
- A. T. Paulino, M. R. Guilherme, A. V. Reis, G. M. Campese, E. C. Muniz and J. Nozaki, J. Colloid Interface Sci., 2006, 301, 55–62 CrossRef CAS PubMed.
- S. Karthikeyan, V. K. Gupta, R. Boopathy, A. Titus and G. Sekaran, J. Mol. Liq., 2012, 173, 153–163 CrossRef CAS.
- Y. Liu, C. Luo, J. Sun, H. Li, Z. Sun and S. Yan, J. Mater. Chem. A, 2015, 3, 5674–5682 CAS.
- T. A. Khan and M. Nazir, Environ. Prog. Sustainable Energy, 2015, 34, 1444–1454 CrossRef CAS.
- T. A. Saleh and V. K. Gupta, Adv. Colloid Interface Sci., 2014, 211, 93–101 CrossRef CAS PubMed.
- T. A. Khan, M. Nazir and E. A. Khan, Toxicol. Environ. Chem., 2013, 95, 919–931 CrossRef CAS.
- J. Chen, X. Wang, X. Liu, J. Huang and Z. Xie, J. Coastal Res., 2015, 73, 1–3 CrossRef CAS.
- M. Auta and B. H. Hameed, Environ. Eng. Manage. J., 2015, 14, 955–963 CAS.
- S. X. Yang, L. Y. Wang, X. D. Zhang, W. J. Yang and G. L. Song, Chem. Eng. J., 2015, 275, 315–321 CrossRef CAS.
- M. Visa, C. Bogatu and A. Duta, J. Hazard. Mater., 2015, 289, 244–256 CrossRef CAS PubMed.
- M. Vakili, M. Rafatullah, B. Salamatinia, A. Z. Abdullah, M. H. Ibrahim, K. B. Tan, Z. Gholami and P. Amouzgar, Carbohydr. Polym., 2014, 113, 115–130 CrossRef CAS PubMed.
- E. Alvarez-Ayuso, A. Garcia-Sanchez and X. Querol, Water Res., 2003, 37, 4855–4862 CrossRef CAS PubMed.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5856 CrossRef CAS PubMed.
- M. Liu, C. Chen, J. Hu, X. Wu and X. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CAS.
- J. W. Zhang, J. H. Luo, P. M. Wang, B. Ding, Y. C. Huang, Z. L. Zhao, J. Zhang and Y. G. Wei, Inorg. Chem., 2015, 54, 2551–2559 CrossRef CAS PubMed.
- Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2015, 283, 329–339 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- H. Zhang, Y. J. Chen, J. H. Ma, H. X. Tong, J. Yang, D. W. Ni, H. M. Hu and F. Q. Zheng, J. Alloys Compd., 2011, 509, 6616–6620 CrossRef CAS.
- F. Iskandar, S. G. Kim, A. B. D. Nandiyanto, Y. Kaihatsu, T. Ogi and K. Okuyama, J. Alloys Compd., 2009, 471, 166–171 CrossRef CAS.
- I. V. Povstugar, A. N. Streletskii, D. G. Permenov, I. V. Kolbanev and S. N. Mudretsova, J. Alloys Compd., 2009, 483, 298–301 CrossRef CAS.
- X. L. Meng, N. Lun, Y. X. Qi, H. L. Zhu, F. D. Han, L. W. Yin, R. H. Fan, Y. J. Bai and J. Q. Bi, J. Solid State Chem., 2011, 184, 859–862 CrossRef CAS.
- C. Tan, X. Cao, X. J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- M. Li, L. Xu, L. Yang, Z. Bai and Y. Qian, Diamond Relat. Mater., 2009, 18, 1421–1425 CrossRef CAS.
- G. L. Wood, J. F. Janik, E. A. Pruss, D. Dreissig, W. J. Kroenke, T. Habereder, H. Nöth and R. T. Paine, Chem. Mater., 2006, 18, 1434–1442 CrossRef CAS.
- C. Tang, Y. Bando, Y. Huang, C. Zhi and D. Golberg, Adv. Funct. Mater., 2008, 18, 3653–3661 CrossRef CAS.
- L. Lin, Z. Li, Y. Zheng and K. Wei, J. Am. Ceram. Soc., 2009, 92, 1347–1349 CrossRef CAS.
- S. J. Yoon and A. Jha, J. Mater. Sci., 1995, 30, 607–614 CrossRef CAS.
- J. L. Huang, C.-H. Pan and D.-F. Lii, Surf. Coat. Technol., 1999, 122, 166–175 CrossRef.
- R. R. Rye, T. T. Borek, D. A. Lindquist and R. T. Paine, J. Am. Ceram. Soc., 1990, 73, 1409–1412 CrossRef CAS.
- L. Wang, L. Shen, X. Xu, L. Xu and Y. Qian, RSC Adv., 2012, 2, 10689–10693 RSC.
- M. Zheng, Y. Liu, Y. Gu and Z. Xu, Sci. China, Ser. B, 2008, 51, 205–210 CrossRef CAS.
- L. Laversenne, Š. Miljanić, P. Miele, C. Goutaudier and B. Bonnetot, Mater. Sci. Forum, 2007, 555, 355–362 CrossRef CAS.
- C. C. Hwang and S. L. Chung, J. Mater. Res., 1998, 13, 680–686 CrossRef CAS.
- I. P. Borovinskaya, Inorg. Mater., 2003, 39, 588–593 CrossRef CAS.
- I. P. Borovinskaya, V. A. Bunin and A. G. Merzhanov, Mendeleev Commun., 1997, 7, 47–48 CrossRef.
- X. Yang, L. Qin, L. Wang, R. Ding, L. Shi and B. Lv, Chem. Eng. J., 2018, 333, 191–199 CrossRef CAS.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed and references cited therein.
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed.
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef CAS PubMed.
- L. H. Li, J. Cervenka, K. Watanabe, T. Taniguchi and Y. Chen, ACS Nano, 2014, 8, 1457–1462 CrossRef CAS PubMed.
- M. Du, Y. Wu and X. Hao, CrystEngComm, 2013, 15, 1782–1786 RSC.
- N. Meyer, M. Devillers and S. Hermans, Catal. Today, 2015, 241, 200–207 CrossRef CAS.
- G. R. Bhimanapati, D. Kozuch and J. A. Robinson, Nanoscale, 2014, 6, 11671–11675 RSC.
- X. Zeng, L. Ye, S. Yu, H. Li, R. Sun, J. Xu and C. P. Wong, Nanoscale, 2015, 8, 6774–6781 RSC.
- C. Huang, W. Ye, Q. Liu and X. Qiu, ACS Appl. Mater. Interfaces, 2014, 6, 14469–14476 CAS.
- R. Arenal, A. C. Ferrari, S. Reich, L. Wirtz, J. Y. Mevellec, S. Lefrant, A. Rubio and A. Loiseau, Nano Lett., 2006, 6, 1812–1816 CrossRef CAS PubMed.
- Y. Kubota, K. Watanabe, O. Tsuda and T. Taniguchi, Science, 2007, 317, 932–934 CrossRef CAS PubMed.
- R. V. Gorbachev, I. Riaz, R. R. Nair, R. Jalil, L. Britnell, B. D. Belle, E. W. Hill, K. S. Novoselov, K. Watanabe, T. Taniguchi, A. K. Geim and P. Blake, Small, 2011, 7, 465–468 CrossRef CAS PubMed.
- C. Huang, C. Chen, X. Ye, W. Ye, J. Hu, C. Xu and X. Qiu, J. Mater. Chem. A, 2013, 1, 12192–12197 CAS.
- J. S. Lauret, R. Arenal, F. Ducastelle and A. Loiseau, Phys. Rev. Lett., 2005, 94, 037405 CrossRef CAS PubMed.
- S. J. Teng, J. N. Wang and X. X. Wang, J. Mater. Chem., 2011, 21, 5443–5450 RSC.
- F. M. Gao, L. Hou, G. F. Sun, H. Y. Gou and M. Tian, Cryst. Growth Des., 2007, 7, 535–540 Search PubMed.
- M. Kakiage, T. Shoji and H. Kobayashi, J. Ceram. Soc. Jpn., 2016, 124, 13–17 CrossRef CAS.
- G. Lian, X. Zhang, H. Si, J. Wang, D. Cui and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12773–12778 CAS.
- L. Wang, L. Shen, X. Xu, L. Xu and Y. Qian, RSC Adv., 2012, 2, 10689–10693 RSC.
- C. Xiong and W. Tu, Eur. J. Inorg. Chem., 2014, 19, 3010–3015 CrossRef.
- T. Lu, L. Wang, Y. Jiang, Q. liu and C. Huang, J. Mater. Chem. B, 2016, 4, 6103–6110 RSC.
- J. Xiong, W. Zhu, H. Li, W. Ding, Y. Chao, P. Wu, S. Xun, M. Zhang and H. Li, Green Chem., 2015, 17, 1647–1656 RSC.
- W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4(1777), 1–7 Search PubMed.
- D. Liu, W. Lei, S. Qin and Y. Chen, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- A. Nag, K. Raidongia, K. P. S. S. Hembram, R. Datta, U. V. Waghmare and C. N. R. Rao, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS PubMed.
- M. Zheng, H. Dong, Y. Xiao, S. Liu, H. Hu, Y. Liang, L. Sun and Y. Liu, RSC Adv., 2016, 6, 45402–45409 RSC.
- J. Ma, J. Li, G. Li, Y. Tian, J. Zhang, J. Wu, J. Zheng, H. Zhuang and T. Pan, Mater. Res. Bull., 2007, 42, 982–988 CrossRef CAS.
- J. Duan, R. Xue, Y. Xu and C. Sun, Mater. Lett., 2008, 62, 3355–3357 CrossRef CAS.
- Y. Zhang, L. Xu, B. Tang and Z. Li, Catal. Sci. Technol., 2013, 3, 222–229 CAS.
- X. L. Meng, N. Lun, Y. Q. Qi, J. Q. Bi, Y. X. Qi, H. L. Zhu, F. D. Han, Y. J. Bai, L. W. Yin and R. H. Fan, Eur. J. Inorg. Chem., 2010, 20, 3174–3178 CrossRef.
- X. Zhang, G. Lian, S. J. Zhang, D. L. Cui and Q. L. Wang, CrystEngComm, 2012, 4, 4670–4676 RSC.
- D. Portehault, C. Giordano, C. Gervais, I. Senkovska, S. Kaskel, C. Sanchez and M. Antonietti, Adv. Funct. Mater., 2010, 20, 1827–1833 CrossRef CAS.
- X. Zhang, G. Lian, S. Zhang, D. Cui and Q. Wang, CrystEngComm, 2012, 14, 4670–4676 RSC.
- R. Li, X. Q. Ren, J. S. Zhao, X. Feng, X. Jiang, X. X. Fan, Z. G. Lin, X. G. Li, C. G. Hu and B. Wang, J. Mater. Chem. A, 2014, 2, 2168–2173 CAS.
- Q. Zhou, Q. Gao, W. Luo, C. Yan, Z. Ji and P. Duan, Colloids Surf., A, 2015, 470, 248–257 CrossRef CAS.
- C. D. Woolard, J. Strong and C. R. Erasmus, Appl. Geochem., 2002, 17, 1159–1164 CrossRef CAS.
- Y. Yao, S. Miao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS.
- Y. J. Yao, F. F. Xu, M. Chen, Z. X. Xu and Z. W. Zhu, Bioresour. Technol., 2010, 101, 3040–3046 CrossRef CAS PubMed.
- J. Song, W. Zou, Y. Bian, F. Su and R. Han, Desalination, 2011, 265, 119–125 CrossRef CAS.
- S. Qu, F. Huang, S. Yu, G. Chen and J. Kong, J. Hazard. Mater., 2008, 160, 643–647 CrossRef CAS PubMed.
- A. A. Farghali, M. Bahgat, W. M. A. El Rouby and M. H. Khedr, J. Solution Chem., 2012, 41, 2209–2225 CrossRef CAS.
- J. Chang, J. Ma, Q. Ma, D. Zhang, N. Qiao, M. Hu and H. Ma, Appl. Clay Sci., 2016, 119, 132–140 CrossRef CAS.
- D. W. Cho, B. H. Jeon, C. M. Chon, F. W. Schwartz, Y. Jeong and H. Song, J. Ind. Eng. Chem., 2015, 28, 60–66 CrossRef CAS.
- E. E. Baldez, N. F. Robaina and R. J. Cassella, J. Hazard. Mater., 2008, 159, 580–586 CrossRef CAS PubMed.
- V. Vadivelan and K. V. Kumar, J. Colloid Interface Sci., 2005, 286, 90–100 CrossRef CAS PubMed.
- E. Haque, J. W. Jun and S. H. Jhung, J. Hazard. Mater., 2011, 185, 507–511 CrossRef CAS PubMed.
- T. Liu, Y. Li, Q. Du, J. Sun, Y. Jiao, G. Yang, Z. Wang, Y. Xia, W. Zhang and K. Wang, Colloids Surf., B, 2012, 90, 197–203 CrossRef CAS PubMed.
- X. Liu, W. Gong, J. Luo, C. Zou, Y. Yang and S. Yang, Appl. Surf. Sci., 2016, 362, 517–524 CrossRef CAS.
- Y. Li, Q. Du, T. Liu, J. Sun, Y. Wang, S. Wu, Z. Wang, Y. Xia and L. Xia, Carbohydr. Polym., 2013, 95, 501–507 CrossRef CAS PubMed.
- M. M. Ayad and A. A. EI-Nasr, J. Phys. Chem. C, 2010, 114, 14377–14383 CAS.
| This journal is © The Royal Society of Chemistry 2017 |