
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of methoxetamine, its metabolites and deuterium labelled analog as analytical standards and their HPLC and chiral capillary electrophoresis separation†
B. Jurasek ab,
M. Himlc,
R. Jurokbc,
K. Hajkovabde,
A. Vobinuskovad,
P. Rezankad and
M. Kuchar*abe
ab,
M. Himlc,
R. Jurokbc,
K. Hajkovabde,
A. Vobinuskovad,
P. Rezankad and
M. Kuchar*abe
aDepartment of Chemistry of Natural Compounds UCT Prague, Technická 5, Prague, 166 28, Czech Republic. E-mail: kuchara@vscht.cz
bForensic Laboratory of Biologically Active Substances UCT Prague, Technická 5, Prague, 166 28, Czech Republic
cDepartment of Organic Chemistry UCT Prague, Technická 5, Prague, 166 28, Czech Republic
dDepartment of Analytical Chemistry UCT Prague, Technická 5, Prague, 166 28, Czech Republic
eNational Institute of Mental Health CZ, Topolová 748, Klecany, 250 67, Czech Republic
First published on 18th December 2017
Abstract
Methoxetamine, a designer drug marketed as a replacement for the dissociative anaesthetic ketamine, has been associated with significant numbers of hospital related intoxications and deaths in Europe. The fast and user-friendly identification and quantification of methoxetamine and its metabolites is a key factor for successful treatment of intoxication. Therefore, we suggested a convenient preparation method which was used for the synthesis of methoxetamine, seven methoxetamine metabolites and a deuterium labelled derivative as analytical standards. Methoxetamine and normethoxetamine were used as starting materials for the preparation of O-demethylated and N-dealkylated metabolites. The multistep synthesis starts from commercially available compounds and offers good yields. Our prepared analytical standards were used for the confirmation of the suggested structure of methoxetamine metabolites in rat urine by LCMS. Capillary electrophoresis was used for the chiral separation of MXE and its metabolites using β-cyclodextrin, carboxymethylated β-cyclodextrin, and sulphated β-cyclodextrin as chiral selectors at various concentrations. Chiral separation was successful for four analytes. A mixture of MXE and its metabolites was subsequently analyzed under optimal conditions, i.e. when using 15 mmol L−1 β-cyclodextrin in 50 mmol L−1 phosphate buffer, pH 2.5. In this case, chiral separation was achieved for three analytes and all analytes were separated from each other.
Introduction
According to the annual drug reports published by the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), a new class of psychoactive substances, known as ‘legal highs’, ‘herbal incense’, ‘designer drugs’ or ‘research chemicals’ has emerged on the drug market in the last decade. The new psychoactive substances (NPSs) include a wide range of products, from natural plant-originating substances to semisynthetic and synthetic compounds.1,2 In recent years, the use of new psychoactive substances has grown dramatically. The growing popularity of these compounds is based on availability and apparent ‘legality’ since many NPSs are yet to be covered by legislative controls pertaining to narcotic and psychotropic substances.3–5 In 2016, 66 NPS were notified to the EU Early Warning System operating under EMCDDA. The total number of monitored substances increased to more than 620.4 The NPS market is rapidly growing and its legislative regulation is very difficult. In addition to the large number of NPS notified every year, there is a very limited supply (often complete absence) of analytical standards that are important for research, clinical and forensic practice. EMCDDA divides NPS into several classes.6 Dissociative anaesthetics are an interesting, however dangerous, group of NPS. The dissociative anaesthetics include compounds with an aryl moiety connected to the cyclohexylamine (arylcyclohexylamines). Arylcyclohexylamines are represented by ketamine and its analogues (Fig. 1).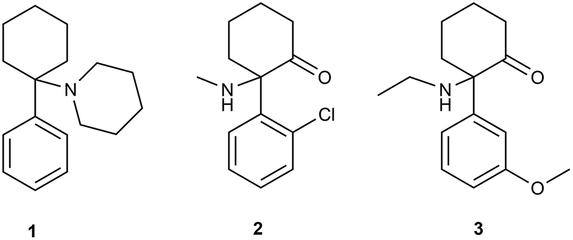 | ||
| Fig. 1 Phencyclidine (1), ketamine (2), methoxetamine (3). | ||
Ketamine is a well-known anaesthetic with a good safety profile, which is used particularly in anaesthesia, and as co-analgesics in pain management. Moreover, ketamine is suggested as a novel treatment for major depressive disorder and bipolar depression.7,8 On the other hand, ketamine (2), phencyclidine (PCP, 1) and methoxetamine (MXE, 3) have an abuse potential.9–11 While ketamine (2) has been registered for pharmaceutical usage, many less well characterized arylcyclohexylamines have appeared on the black market as ‘research chemicals’ and can be obtained from internet retailers.11,12
Methoxetamine (3) is a structural analogue of ketamine. MXE was extensively advertised for recreational use as a ‘legal’ and ‘bladder friendly’ alternative to ketamine with the main effects of hallucinations, depersonalization and dissociation of the physical body.13–16 MXE has a sub-micromolar affinity for the glutamate N-methyl-D-aspartate (NMDA) receptor (pKi = 6.59), which is comparable to that of ketamine (pKi = 6.18), and binds to the phencyclidine site on the NMDA receptor.17 Very recently, we have published that methoxetamine behaves as a standard dissociative anaesthetic more potent than its analogue ketamine, which in turn might increase the risk of overdose, loss of consciousness and potentially fatal complications. MXE was found to be generally anxiogenic and psychotomimetic, which, in humans is likely to produce unpleasant experiences, and negative psychological sequelae.10
Data on the prevalence of use of methoxetamine (3) is limited, but reports of acute intoxications including fatalities have recently appeared.18–20 Methoxetamine is associated with 120 non-fatal intoxications and 22 deaths (reported through Early warning system (EWS) by European Member States), however only 55 of these cases have been analytically confirmed.15,21 Although many related publications have focused on intoxication with methoxetamine, complete synthesis have not been published yet. Only a brief study of methoxetamine analysis reported the possibility of methoxetamine synthesis analogously to ketamine.22
Toxicological detection is necessary as a proof of intoxication or abuse of a certain compound. Urine analysis is one of the most common detection methods in forensic toxicology. Urine samples are usually analysed because of longer detectability, however the knowledge about the metabolism of the investigated compound is often essential, especially, if the substance is completely metabolised and only metabolites are detectable in the urine samples. Furthermore, knowledge about metabolism is a prerequisite for the risk assessment concerning pharmacokinetic interactions caused, e.g. by cytochrome P450 (CYP) enzymes. The first structures of methoxetamine metabolites have been suggested by Meyer et al. and Menzies et al.23,24 They used full-scan GCMS (EI) technique, but this determination has some limitations. Therefore, we synthetized standards of methoxetamine metabolites for the confirmation of their suggested structures and for the pharmacokinetic and metabolism studies. To support the pharmacokinetic and metabolism studies of methoxetamine, stable isotopically labelled standards are an essential tool. Deuterium-labelled internal standards are particularly effective for mass spectrometry analyses and quantification. Many studies have been reported for the preparation of deuterium-labelled controlled drugs as internal standards for the use in GCMS and LCMS analysis.25,26
Therefore, our aim was to develop a convenient method for the synthesis of methoxetamine, its metabolites and deuterium-labelled derivative as analytical standards. All prepared high purity compounds were used for pharmacokinetics.10,27
Chiral analysis of drugs and their metabolites can provide very useful information about metabolic pathways in living organisms because it can be expected that metabolic pathways can be different for each enantiomer. Thus enantiomers may have different pharmacological effects. For this reason we used capillary electrophoresis (CE) as chiral separation method for enantioseparation of MXE and its metabolites using cyclodextrin and their derivatives as chiral additives to the background electrolyte (BGE).
Results and discussion
Synthesis of methoxetamine and its metabolites
The synthetic route to obtain N-dealkylated methoxetamine metabolites is outlined in Fig. 2. Following the synthetic route, treating commercially available 3-methoxybromobenzene (4) with cyclohexanone and activated magnesium under the Grignard protocol afforded intermediate 5,28 which under acidic conditions eliminated water to cyclohexene intermediate 629 in high yield 90%. Following epoxidation underwent smoothly in biphases system CH2Cl2/H2O without phase transfer catalyst to obtain the epoxide 7.30 Regioselective opening of the epoxide with HBr (aq) gave unstable bromohydrine, which was directly oxidized with Jones reagent to afford compound 8 in 64% overall yield. Nucleophilic substitution of 8 with NaN3 was followed by almost quantitative azide 9 reduction using catalytic hydrogenation. The metabolite normethoxetamine 10 was treated with NaBH4 affording dihydro-normethoxetamine 11 in average yield 65%. The synthesis of metabolite dihydro-O-desmethyl-normethoxetamine 13 started from normethoxetamine 10 through intermediate O-desmethyl-normethoxetamine 12 obtained by methoxy group cleavage with HBr (aq) reflux followed by reduction with NaBH4 in high 85% overall yield. O-Desmethyl-normethoxetamine (12) was selectively protected with di-tert-butyldicarbonate and then methylated with CD3I. Final acid-promoted deprotection provided deuterium labelled metabolite 14.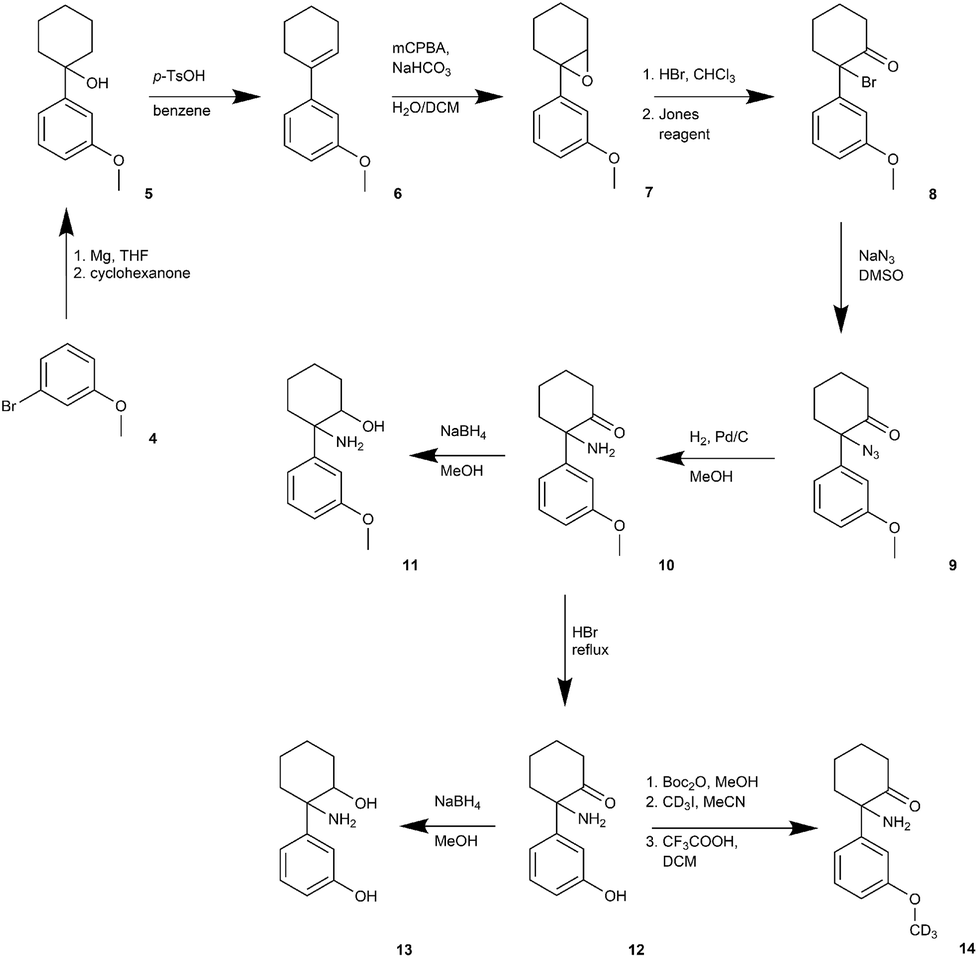 | ||
| Fig. 2 Synthesis of N-dealkylated metabolites 10, 11, 13 and deuterium labelled metabolite 14. | ||
A synthetic route for the preparation of O-demethylated metabolites (Fig. 3) was led through the parent compound methoxetamine (3). We followed the methoxetamine synthesis described by Hays.22 Briefly, 3-methoxybenzonitrile 15 underwent Grignard reaction to form 3-methoxyphenyl cyclopentyl ketone (16).31 Further bromination led to alpha-bromo ketone 17,32 which was treated with ethylamine to form Schiff's base and final heating provided methoxetamine (3).22 Metabolite dihydro-O-desmethyl-methoxetamine (21) was obtained by methoxy group cleavage with HBr (aq) under reflux followed by reduction with NaBH4 in high overall yield (65%). Direct reduction of methoxetamine with NaBH4 afforded dihydromethoxetamine metabolite 19 in high yield (96%).
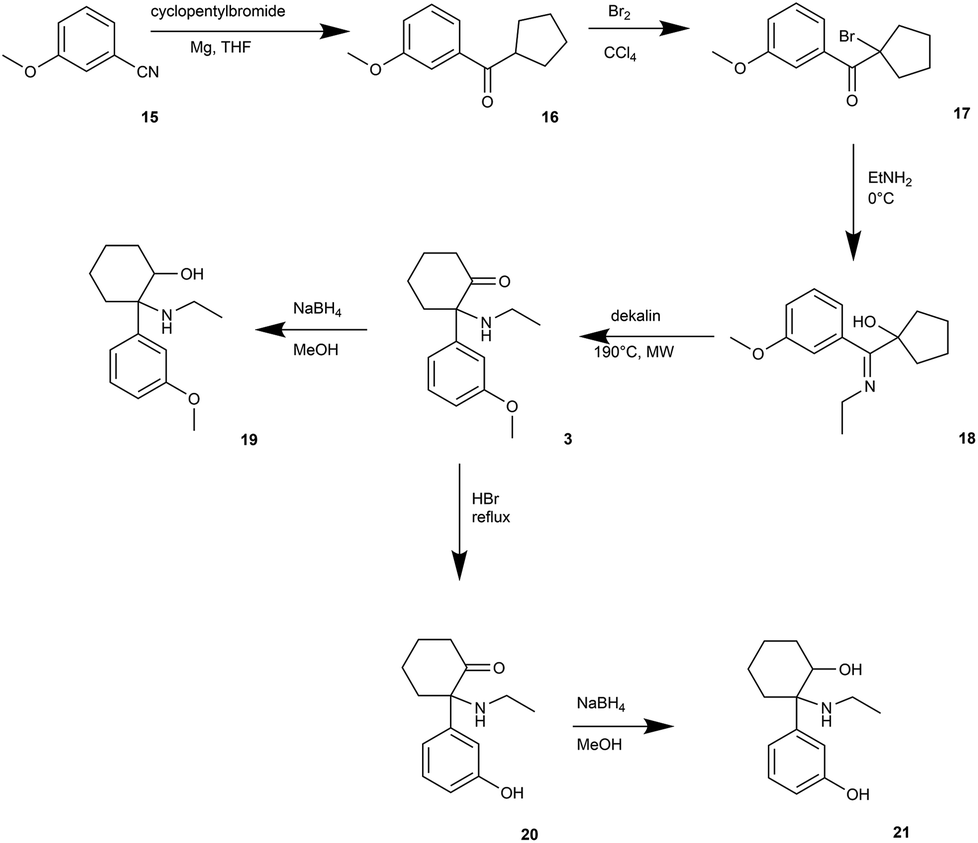 | ||
| Fig. 3 Synthesis of methoxetamine (3) and N-demethylated metabolites 19, 20, 21. | ||
LC-MS/MS analysis of prepared standards
A sample preparation technique with an LC-MS/MS detection method using synthesized standards was developed and extensively evaluated before it was used for the real samples of pharmacokinetic study. Full description of this method and collected data is shown in our previous work.10,27The aim of the chromatography was to separate compounds with similar m/z so that there was no misidentification because of isobar occurrence. There were just two co-elutions within the analysis; the first one was the common co-elution of 10 and its deuterium-labelled analog 14 and the second one was caused by similar retention of 3 and 11, which differ in m/z. The first stage of separation was accomplished successfully and the second stage was to create sensitive multiple reaction monitoring method (MRM) for MS/MS acquisition. Source conditions and mass transitions were optimized for each analyte. Mixtures of analyzed compounds were measured and evaluated on different concentration levels, and subsequently several different matrices were used (listed in Fig. 4.).10,27
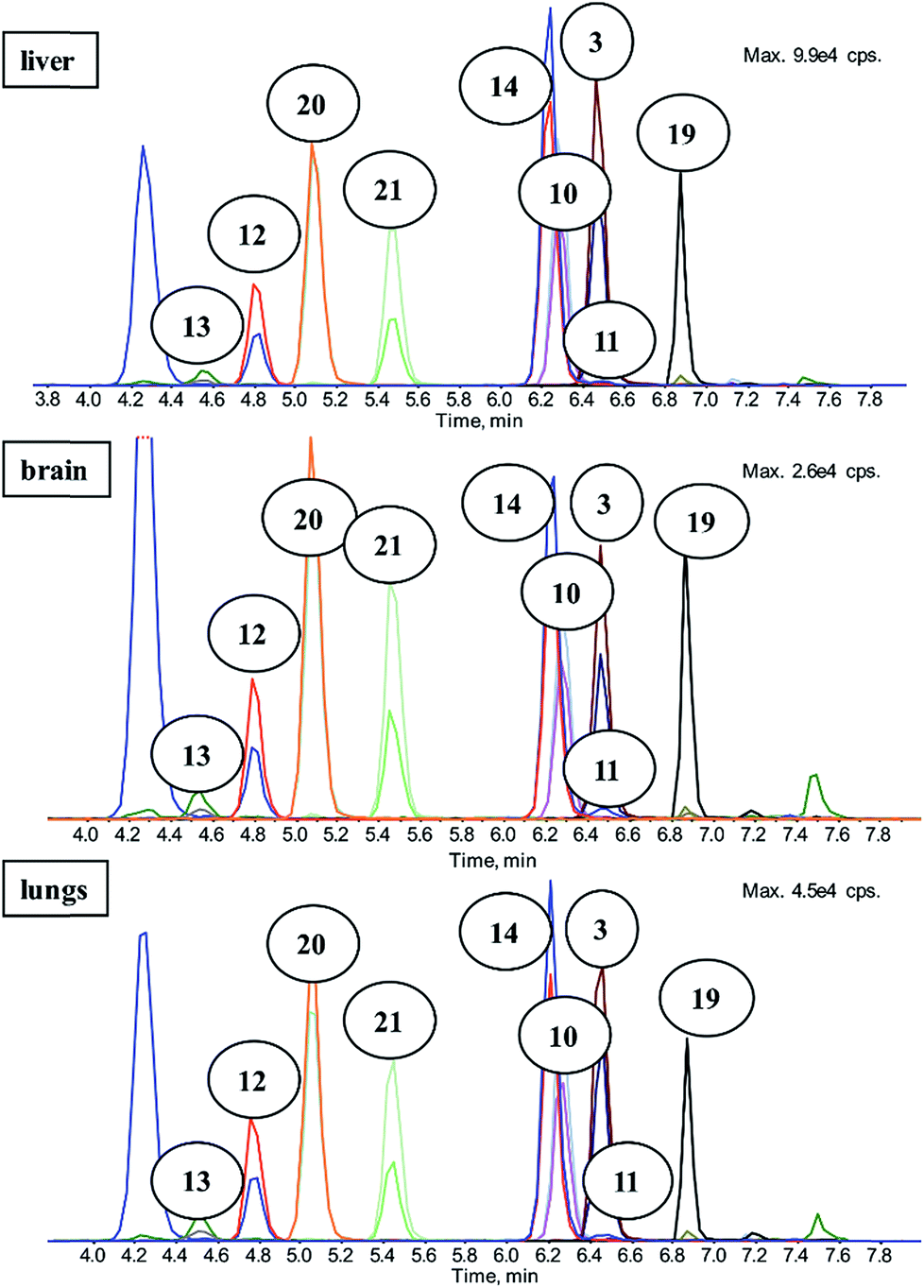 | ||
| Fig. 4 Chromatograms of matrix-matched calibration on 50 ng mL−1 (approx. 0.2 μmol L−1) concentration level. Different tissues – liver, brain, and lung – were enriched by standard compounds and prepared in the same way as the real samples. | ||
However, chiral separation of the analytes was not pursued in the aforementioned publications. In addition, there is no information on the metabolic pathways for particular enantiomers. Thus, it is important to develop not only chromatographic separation method, but, in particular, a method that can be used to differentiate between the enantiomers of a particular drug or its metabolite.
CE analysis of prepared standards
Our CE study dealt with a suitable chiral selector, its concentration, and pH of BGE for the enantiomers separations of studied MXE and its metabolites. Three selected cyclodextrins were tested at all above mentioned conditions, i.e. at 4 different concentration levels and 6 different pH. For β-cyclodextrin (BCD) that is often used due to low cost, its chiral separation was achieved for compound 3 (R = 0.8) and metabolites 11 (R = 0.6) and 20 (R = 1.1). The second tested cyclodextrin was carboxymethylated β-cyclodextrin (CMBCD) that is often used for separation of compounds containing basic nitrogen group. In this case, compound 3 was partially enantioseparated (R = 1.0) and metabolites 10, 11, and 20 were fully enantioseparated (R > 1.5) (Fig. 5). Using sulphated β-cyclodextrin (SBCD) did not lead to the better resolution, although the same previously mentioned four compounds were also at least partially separated (0.5 < R < 1.5). To our best knowledge, we achieved enantioseparations of these compounds by CE for the first time.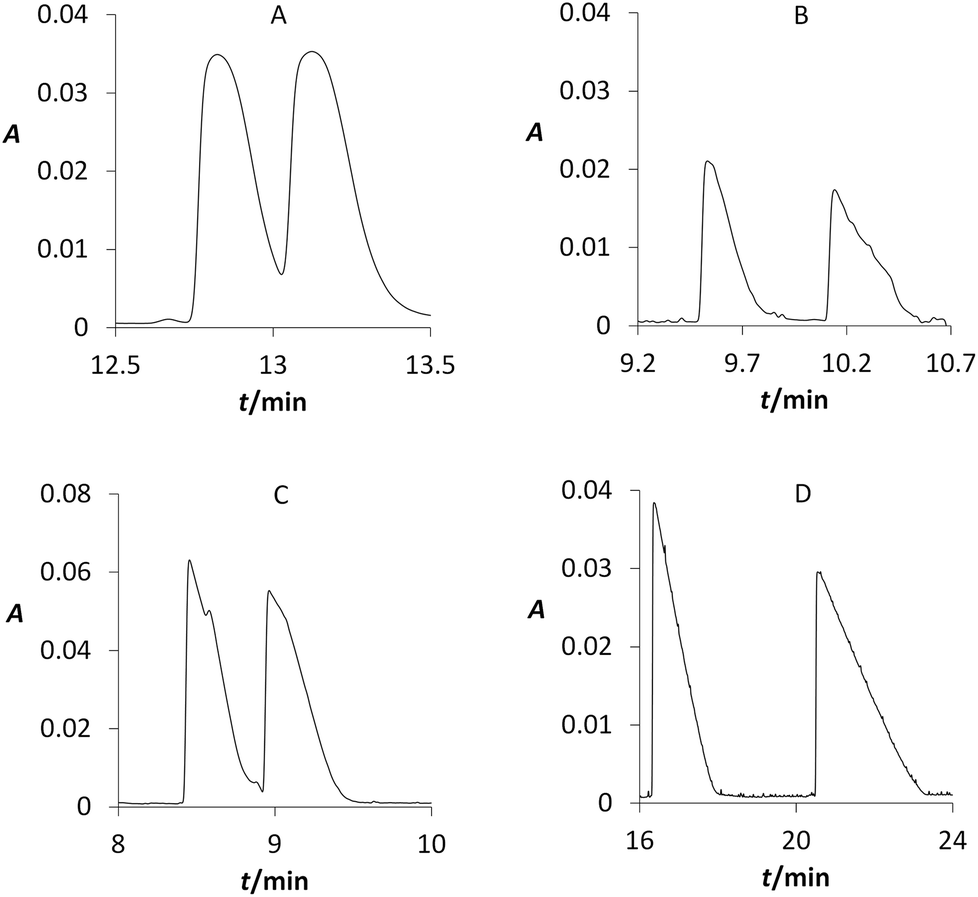 | ||
| Fig. 5 Electropherograms of analyzed compounds; dependences of absorbance at 207 nm on electromigration time. Separated analytes (BGE): (A) 3 (10 mmol L−1 CMBCD, 50 mmol L−1 phosphate buffer, pH 2.5); (B) 10 (10 mmol L−1 CMBCD, 50 mmol L−1 formic buffer, pH 4.0); (C) 11 (10 mmol L−1 CMBCD, 50 mmol L−1 acetic buffer, pH 5.0); (D) 20 (7 mmol L−1 CMBCD, 50 mmol L−1 formic buffer, pH 4.0); fused-silica capillary: od/id = 375/75 μm, total/effective length 58.5/50.0 cm; voltage 20 kV; temperature 25 °C. | ||
Because in the living organisms above-mentioned compounds will occur simultaneously, the second part of our CE study focused on the separation of mixtures of MXE and its metabolites. The measurement was carried out under optimal conditions that were chosen from experiments at above-mentioned conditions, i.e. 3 types of cyclodextrins, 4 different concentration levels and 6 different pH. The optimal BGE consists of 50 mmol L−1 phosphate buffer (pH 2.5) with 15 mmol L−1 BCD as a chiral additive.
To identify individual signals in the electropherogram as depicted in Fig. 6, new mixtures were prepared with various concentrations of studied chiral compounds. These mixtures were analysed under the identical conditions as the original mixture and the assignment of the signals was carried out using the peak areas and UV-vis spectra of individual analysed compound. As can be seen in Fig. 6, six analytes were separated from each other and the half of them was partially enantioseparated.
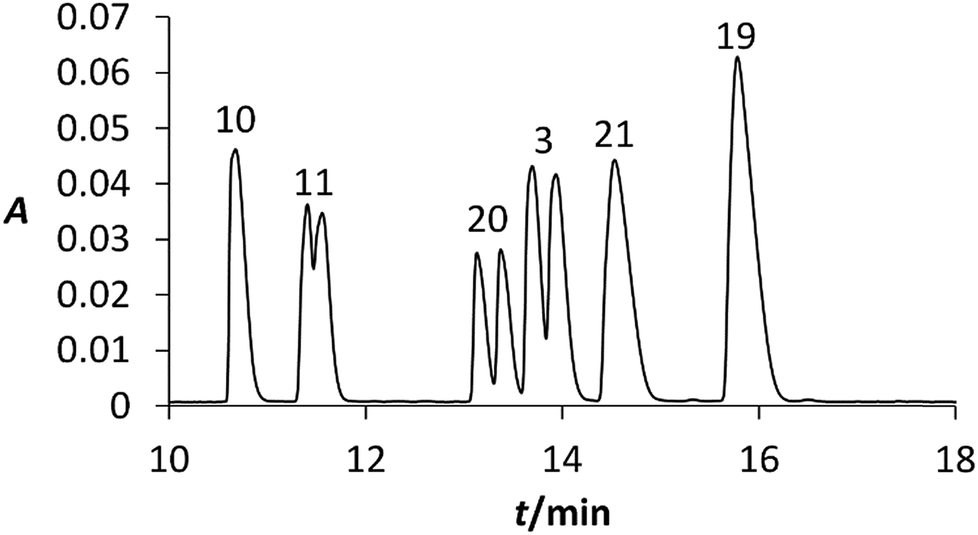 | ||
| Fig. 6 Electropherogram of analyzed mixture of MXE and its metabolites; dependences of absorbance at 207 nm on electromigration time; BGE: 15 mmol L−1 BCD, 50 mmol L−1 sodium phosphate, pH 2.5; fused-silica capillary: od/id = 375/75 μm, total/effective length 58.5/50.0 cm; voltage 20 kV; temperature 25 °C. | ||
Conclusions
In conclusion, we established an efficient synthetic route towards normethoxetamine-d3 (14) and seven methoxetamine metabolites, namely, dihydro-O-desmethyl-normethoxetamine (13), O-desmethyl-normethoxetamine (12), O-desmethylmethoxetamine (20), dihydro-O-desmethylmethoxetamine (21), normethoxetamine (10), dihydro-normethoxetamine (11), dihydromethoxetamine (19).A sample preparation technique with LC-MS/MS method using synthesized standards was developed and extensively evaluated before it was used on the real samples of pharmacokinetic study. Mixtures of analyzed compounds were measured and evaluated on different concentration levels, and subsequently several different matrices were also used. Full description of this method and collected data is shown in our previous work.10,27
We studied the influence of the type and concentration of CDs on the enantioselectivity of methoxetamine and its derivatives by chiral CE. The results of the analyses (carried out in 50 mmol L−1 buffer at pH 2.5 to 5.0 with the addition of 4 to 15 mmol L−1 BCD, CMBCD or SBCD) demonstrate a significant dependence of enantioseparations on the conditions studied. Four compounds (3, 10, 11, and 20) were successfully enantioseparated.
Experimental section
Chemicals and materials
Hydrochloric acid (30%) (Suprapur, Merck, Germany), 1 mol L−1 sodium hydroxide (Tripur, Merck), ortho-phosphoric acid (50%), β-cyclodextrin (BCD), carboxymethylated β-cyclodextrin (CMBCD) and sulphated β-cyclodextrin (SBCD) (all Sigma-Aldrich, Czech Republic). Diethyl ether solution of hydrochloride was prepared from commercial diethyl ether, commercial concentrated hydrochloric acid and calcium chloride as drying agents. Solvents were acquired from commercial sources and were used after distillation. Other commercial reagents were used without further purification. The reactions were monitored with the aid of thin-layer chromatography (TLC Silica gel 60 F254) and pre-coated reverse phase gel plates (TLC RP-18 F254). Visualization was carried out with UV light. Chromatography refers to flash column chromatography and it was carried out with the indicated solvents on silica gel (particle size 0.040–0.060 mm) or reverse phase column (Redisep RF Gold C18). All operations involving air-sensitive reagents were performed under an inert atmosphere of dry argon using syringe and cannula techniques, oven-dried glassware, and freshly distilled and dried solvents. Thermal rearrangement were carried out in Microwave reactor (Biotage Initiator Classic) melting points were determined using a melting point apparatus (PGH Rundfunk–Fernsehen), and they are uncorrected. NMR spectra were recorded in CDCl3 or CD3OD at room temperature on a Varian Gemini 300 MHz (1H NMR 300 MHz, 13C NMR 75 MHz) or an Agilent 400 MR DDR2 (1H NMR 400 MHz, 13C NMR 100 MHz). The spectra were referenced to residual solvent protons in the 1H NMR spectra (7.26 ppm for CDCl3 and 4.84 and 3.31 ppm for CD3OD) and to solvent carbons in the 13C NMR spectra (77.0 ppm for CDCl3 and 49.05 ppm for CD3OD). Infrared (IR) spectra were measured on FTIR Nicolet 6700 (Thermo-Nicolet) coupled with ATR cell GladiATR (Pike). High-resolution MS (HRMS) were run either by the ESI ionization or atmospheric pressure chemical ionization (APCI) mode. High resolution mass spectra (HRMS) were recorded using a Thermo Scientific LTQ Orbitrap Velos.CE separations were performed with an Agilent CE instrument (Agilent 3D HPCE, Germany) equipped with UV-vis diode-array detector. Bare fused silica capillary of 375/75 μm od/id and 58.5/50 cm total/effective length obtained from Polymicro Technologies (AZ, USA) was used.
LC conditions
LC-MS/MS was performed using an Agilent 1200 HPLC series (Agilent Technologies, Germany) in tandem with API 3200 MS system (AB Sciex, Canada). ZORBAX Eclipse Plus C18 (50 × 2.1 mm, 5 μm) with a pre-column was used for the chromatographic separation with a gradient of 0.1% (v/v) formic acid in deionized water (mobile phase A) and 0.1% (v/v) formic acid in methanol (mobile phase B) at flow rate of 250 μL min−1. The gradient set-up was: 0–4 min from 90% A to 60% A; 4–5 min to 0% A; 5–5.5 min at 0% A; 5.5–5.7 min back to 90% A, and equilibration at the same level to 10 min. The valve arrangement was: 3.1–7.6 min to the MS source. The MS conditions were following: electrospray ionization (ESI) in positive mode, ionization voltage of 5500 V, source temperature of 500 °C and gas flow rates-curtain gas 15, GS1 and GS2 50 arbitrary units. Data were acquired in multiple reaction monitoring mode (MRM) using Analyst 1.4.2 (AB Sciex, Canada).27CE conditions
Individual analyzed compounds were dissolved in water at 10 mmol L−1 concentration. For the CE experiments the analyte solutions were further diluted with water to the final concentration 1 mmol L−1. For the determination of migration order of particular compounds the mixture composed of analyzed compounds at different concentration was used. The background electrolyte consisted of 50 mmol L−1 buffers at pH 2.5, 3.0 (50 mmol L−1 orthophosphoric acid adjusted to appropriate pH with 1 mol L−1 NaOH), 3.5, 4.0 (50 mmol L−1 formic acid adjusted to appropriate pH with 1 mol L−1 NaOH), 4.5, 5.0 (50 mmol L−1 acetic acid adjusted to appropriate pH with 1 mol L−1 NaOH), and various concentrations of the studied CDs (4, 7, 10, 15 mmol L−1 for BCD, CMBCD, and SBCD). A new fused silica capillary was first rinsed with 1 mol L−1 NaOH for 30 min, then with H2O for 30 min. Between the runs the capillary was rinsed at 99.4 kPa first with 0.1 mol L−1 NaOH for 2 min then with H2O also for 2 min, and finally with running buffer again for 2 min (for the capillary washing a different buffer solution than for the subsequent analysis was used). The analytes were injected hydrodynamically by pressure of 1.5 kPa for 5 s. All the separations were performed at 20 kV (anode at the injection capillary end) with a voltage ramp time of 12 s. Detection was carried out at 207 nm and the capillary was thermostated at 25 °C during the analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by a specific university research (project MSMT No. 20-SVV/2017) and by the Ministry of Interior of the Czech Republic (project VG20172020056).Notes and references
- European Monitoring Centre for Drugs and Drug Addiction and Europol, EU drug markets report – a strategic analysis, Portugal and Haag, 2013 Search PubMed.
- UNODC Early warning advisory on new psychoactive substances, What are NPS?, https://www.unodc.org/LSS/Page/NPS, accessed 1 January 2017.
- L. Hondebrink, J. J. Nugteren-van Lonkhuyzen, D. Van Der Gouwe and T. M. Brunt, Drug Alcohol Depend., 2015, 147, 109–115 CrossRef PubMed.
- European Monitoring Centre for Drugs and Drug Addiction and Europol, European Drug Report 2017: Trends and Developments, Luxembourg, 2017 Search PubMed.
- European Monitoring Centre for Drugs and Drug Addiction, European Drug Report 2016, Luxembourg, 2016 Search PubMed.
- European Monitoring Centre for Drugs and Drug Addiction and Europol, EU drug markets report: In-depth analysis, Luxembourg, 2016 Search PubMed.
- E. E. Lee, M. P. Della Selva, A. Liu and S. Himelhoch, Gen. Hosp. Psychiatr., 2015, 37, 178–184 CrossRef PubMed.
- S. Sassano-Higgins, D. Baron, G. Juarez, N. Esmaili and M. Gold, Depression Anxiety, 2016, 33, 718–727 CrossRef CAS PubMed.
- C. J. Botanas, J. B. de la Peña, I. J. dela Peña, R. Tampus, R. Yoon, H. J. Kim, Y. S. Lee, C. G. Jang and J. H. Cheong, Pharmacol., Biochem. Behav., 2015, 133, 31–36 CrossRef CAS PubMed.
- R. R. Horsley, E. Lhotkova, K. Hajkova, B. Jurasek, M. Kuchar and T. Palenicek, Brain Res. Bull., 2016, 126, 102–110 CrossRef CAS PubMed.
- J. B. Zawilska, Toxicol. Lett., 2014, 230, 402–407 CrossRef CAS PubMed.
- H. Morris and J. Wallach, Drug Test. Anal., 2014, 6, 614–632 CrossRef CAS PubMed.
- C. Ornella, S. Fabrizio, S. Pierluigi, F. Susanne, A. Sulaf, S. Jacqueline, C. John and T. Giuseppina, Hum. Psychopharmacol., 2012, 27, 145–149 CrossRef PubMed.
- P. I. Dargan, H. C. Tang, W. Liang, D. M. Wood and D. T. Yew, Clin. Toxicol., 2014, 52, 176–180 CrossRef CAS PubMed.
- European Monitoring Centre for Drugs and Drug Addiction, Methoxetamine – Report on the risk assessment of 2-(3-methoxyphenyl)-2-(ethylamino)cyclohexanone (methoxetamine) in the framework of the Council Decision on new psychoactive substances, Luxembourg, 2014 Search PubMed.
- B. Jurásek and M. Kuchař, Drugs & Forensics Bulletin, 2016, 1, 27–31 Search PubMed.
- B. L. Roth, S. Gibbons, W. Arunotayanun, X.-P. Huang, V. Setola, R. Treble and L. Iversen, PLoS One, 2013, 8, 1–5 Search PubMed.
- K. E. Hofer, B. Grager, D. M. Müller, C. Rauber-Lüthy, H. Kupferschmidt, K. M. Rentsch, A. Ceschi, D. M. Muller, C. Rauber-Luthy, H. Kupferschmidt, K. M. Rentsch and A. Ceschi, Ann. Emerg. Med., 2012, 60, 97–99 CrossRef PubMed.
- D. M. Wood, S. Davies, M. Puchnarewicz, A. Johnston and P. I. Dargan, Eur. J. Clin. Pharmacol., 2012, 68, 853–856 CrossRef CAS PubMed.
- M. Wikstrom, G. Thelander, M. Dahlgren and R. Kronstrand, J. Anal. Toxicol., 2013, 37, 43–46 CrossRef PubMed.
- M. T. Zanda, P. Fadda, C. Chiamulera, W. Fratta and L. Fattore, Behav. Pharmacol., 2016, 27, 489–496 CrossRef CAS PubMed.
- P. A. Hays, J. F. Casale and A. L. Berrier, Microgram J., 2012, 9, 3–17 CAS.
- M. R. Meyer, M. Bach, J. Welter, M. Bovens, A. Turcant and H. H. Maurer, Anal. Bioanal. Chem., 2013, 405, 6307–6321 CrossRef CAS PubMed.
- E. L. Menzies, S. C. Hudson, P. I. Dargan, M. C. Parkin, D. M. Wood and A. T. Kicman, Drug Test. Anal., 2014, 6, 506–515 CrossRef CAS PubMed.
- R. S. Sulake, C. Chen, H.-R. Lin and A.-C. Lua, Bioorg. Med. Chem. Lett., 2011, 21, 5719–5721 CrossRef CAS PubMed.
- A. L. Patton, K. A. Seely, S. Pulla, N. J. Rusch, C. L. Moran, W. E. Fantegrossi, L. D. Knight, J. M. Marraffa, P. D. Kennedy, L. P. James, G. W. Endres and J. H. Moran, Anal. Chem., 2014, 86, 1760–1766 CrossRef CAS PubMed.
- K. Hajkova, B. Jurasek, D. Sykora, T. Palenicek, P. Miksatkova and M. Kuchar, Anal. Bioanal. Chem., 2016, 408, 1171–1181 CrossRef CAS PubMed.
- J. Wallach, G. De Paoli, A. Adejare and S. D. Brandt, Drug Test. Anal., 2014, 6, 633–650 CrossRef CAS PubMed.
- D. Huang, H. Wang, F. Xue and Y. Shi, J. Org. Chem., 2011, 76, 7269–7274 CrossRef CAS PubMed.
- A. C. White and G. Bradley, US Pat., US4387097 A1, 1983.
- X. Cheng, R. Goddard, G. Buth and B. List, Angew. Chem., Int. Ed., 2008, 47, 5079–5081 CrossRef CAS PubMed.
- E. Benchikh, P. Fitzgerald, P. Lowry and I. McConnell, US Pat., US2017/3308, 2017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10893a |
| This journal is © The Royal Society of Chemistry 2017 |