
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural terpenoid glycosides with in vitro/vivo antithrombotic profiles from the leaves of Crataegus pinnatifida†
Pin-Yi Gaoab,
Ling-Zhi Li*a,
Ke-Chun Liuc,
Chen Sunc,
Xue Suna,
Ya-Nan Wua and
Shao-Jiang Song *a
*a
aKey Laboratory of Structure-Based Drug Design and Discovery, Ministry of Education, School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China. E-mail: songsj99@163.com; lilingzhijessie@163.com
bCollege of Pharmaceutical and Biotechnology Engineering, Institute of Functional Molecules, Shenyang University of Chemical Technology, Shenyang 110142, People's Republic of China
cBiology Institute of Shandong Academy of Sciences, Jinan, People's Republic of China
First published on 16th October 2017
Abstract
Two norditerpenoids (1–2) with unique carbon skeletons, four sesquiterpenoids (3–6) and nine nor-sesquiterpenoids (7–15) were isolated from the leaves of Crataegus pinnatifida and evaluated as possessing antithrombotic activities in vitro/vivo. Their structures with absolute configurations were determined via a combination of spectroscopic data, chemical methods, and quantum-chemical calculations (ECD, NMR, and OR data). Compound 3 showed an inhibitory effect on ADP induced platelet aggregation in vitro, which is mediated through the response to the specific receptor of P2Y12 by docking results. Compound 3 also clearly prolonged the time to form thrombocytes induced by FeCl3, in the caudal vessels of zebrafish.
Introduction
Abnormal thrombosis is a key factor in serious cardiovascular diseases (CVDs) that are among the leading killers around the world.1 The majority of cardiovascular diseases, including acute coronary syndrome, ischemic stroke and peripheral vascular diseases, are associated with thrombotic disorders.2 Platelet activation in atherosclerotic arteries is central to the development of arterial thrombosis. Therefore, the inhibition of platelet aggregation must occur to prevent a thrombotic event.3 Current antithrombotic discovery efforts target compounds that have improved therapeutic indices compared to the standard of care.4 This fact has stimulated great interest in the search for natural bioactive compounds (NBC) from herbal drugs or herbal medicinal preparations that are highly efficacious in thrombin reduction while having minimal or reduced bleeding liabilities.2,5 Research on novel bioactives and drugs with different mechanisms of action and increased efficacy, and low toxicity is highly desired. The leaves of Crataegus pinnatifida (hawthorn leaves) have been used to treat cardiovascular disease for several decades in Europe, China and the United States.6,7 An extract of hawthorn leaves, Yixintong medicinal preparations, is one of the most effective drugs available for blood-activating and stasis-dissolving.8,9 The flavonoids have been considered as the only effective component until terpenoids (monoterpenoid and sesquiterpenoid) with antithrombotic activity were obtained recently from the leaves of C. pinnatifida.10–13 As a new effective ingredient, the terpenoids exhibit potential antithrombotic activity both in vitro and in vivo.14In continuing the search for antithrombotic agents, two novel norditerpenoids with unique carbon skeletons were isolated as the first diterpenoids from the leaves of the title plant, together with two new sesquiterpenoids (3, 4) and 11 known ones (5–15). The absolute configurations of 1–4 were determined by producing of Rh2(OCOCF3)4 complexes or Mosher ester, and by comparison of ECD, NMR, and optical rotation (OR) data with the calculated values. In this report, the isolation, structure elucidation, and in vitro and in vivo antithrombotic evaluation of the isolates (Fig. 1) are described.
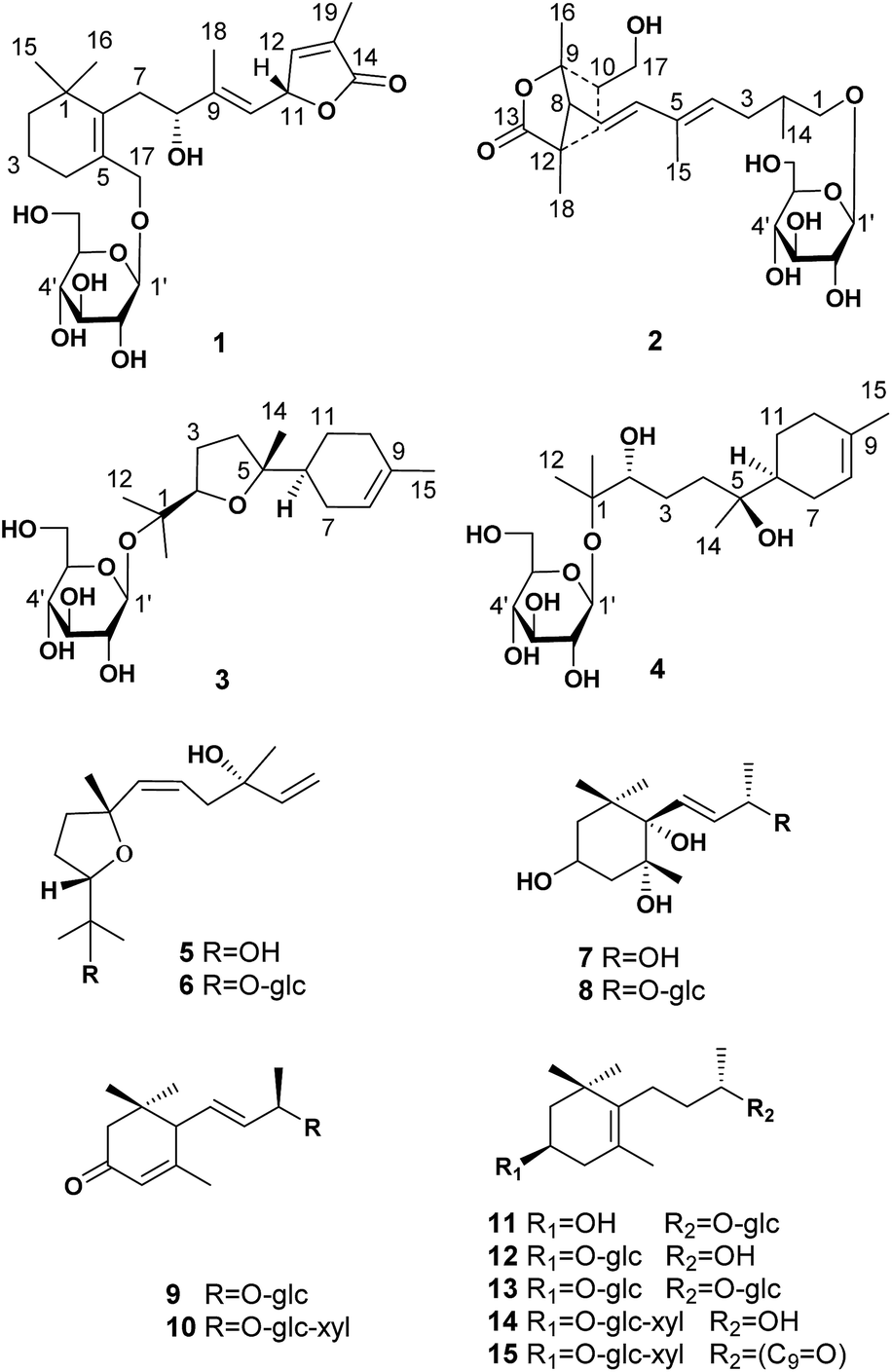 | ||
| Fig. 1 The structures of compounds 1–15. | ||
Results and discussion
The ethanol extract from the leaves of C. pinnatifida was separated and purified by macroporous resin, polyamide, silica gel, C18 reversed silica gel and semipreparative HPLC to afford two norditerpenoids (1–2), 13 sesquiterpenoids (3–15). The known compounds (5–15) were identified as shnyegenin B (5),12 shnyeside B (6),12 (3S,5R,6R,7E,9S)-megastiman-7-ene-3,5,6,9-tetrol (7),12 euodionosides D (8),13 (6R,9R)-3-oxo-α-ionol-9-O-β-D-glucopyranoside (9),13 (6S,7E,9R)-6,9-dihydroxy-4,7-megastiymadien-3-one-9-O-[β-D-xylopyranosy-β-D-glucopyranoside] (10),12 linarionoside A (11),13 linarionoside B (12),12 linarionoside C (13),12 3,9-dihydroxy-5-megastigmen-3-O-[β-D-xylopyranosy-β-D-glucopyranoside] (14),12 pinnatifidanoside C (15)14 by comparison of their spectroscopic data with literature data (ESI-E.2.†).Compound 1, a colorless oil, was assigned the molecular formula of C25H38O9 as determined by the HRESIMS ion at m/z 483.2582 [M + H]+ (calc. 483.2589). The 1H NMR data (Table 1) showed the presence of four tertiary methyls at δH 0.82 (3H, s), 0.88 (3H, s), 1.57 (3H, s) and 1.78 (3H, s), a pair of oxygenated methylene protons at δH 3.90 (1H, d, J = 10.8 Hz) and 4.48 (1H, d, J = 10.8 Hz), two oxygenated methine protons at δH 3.80 (1H, m) and 5.23 (1H, d, J = 9.6 Hz), and there were two olefinic H-atoms which appeared to be at two double bonds separately 5.49 (1H, d, J = 9.6 Hz) and 7.26 (1H, s). In addition, an anomeric proton signal appeared to be at 4.17 (1H, d, J = 7.5 Hz), while six oxygenated H-atoms at δH 2.90–3.60 indicated the presence of a sugar moiety. The 13C NMR spectrum (Table 1) exhibited 25 carbon resonances, from which, four methyls (δC 10.9, 14.7, 28.1, 28.2), four methylenes (δC 21.0, 31.0, 41.0, 45.2), a quaternary carbon (δC 36.5), three carbons bearing oxygen (δC 67.0, 70.7, 84.9), three double bond groups (δC 124.2, 128.6, 132.1, 138.6, 140.8, 151.7), one ester carbonyl group (δC 174.5), and a sugar group (δC 61.9, 70.8, 74.2, 77.6, 77.6 and 103.6) were identified, which suggested that compound 1 is a norditerpene glycoside. Further information about the 2D structure of 1 was obtained from an HMBC experiment. The key correlations of 1 (Fig. 2), δH 1.78 (19-Me)/δC 151.7 (C-12), 128.6 (C-13), 174.5 (C-14), δH 5.23 (H-11)/δC 124.2 (C-10), 151.7 (C-12), 128.6 (C-13) and δH 7.26 (H-12)/δC 84.9 (C-11), 128.6 (C-13), 174.5 (C-14), 10.9 (C-19), suggested the presence of an α, β unsaturated lactone. The other key correlations of 1 around δH 0.82 (15-Me), 0.88 (16-Me), 3.90 and 4.48 (17-CH2OH), 1.57 (18-Me) coupling with above part suggested the presence of the C19-norditerpenoid skeleton. In addition, the glycosidic site was established by a HMBC correlation from H-1′ (δH 4.17) and C-17 (δC 70.7). The geometry of the double bond at C-9 was assigned as E on the basis of the upfield shift of the methyl carbon signal in the 13C NMR, δC 14.7 for C-18,15 which was confirmed by NOE correlations with H-8 and H-10, as well as H-11 and H3-18 (Fig. 3).
| Position | 1a | 2a | ||||
|---|---|---|---|---|---|---|
| δC | Type | δH, mult. (J in Hz) | δC | Type | δH, mult. (J in Hz) | |
| a Measured at 150 MHz for 13C in DMSO-d6, and measured in 600 MHz for 1H in DMSO-d6, (s) singlet, (d) doublet, (m) multiplet. | ||||||
| 1 | 36.5 | C | 75.3 | CH2 | 3.65 m | |
| 2 | 41.0 | CH2 | a 1.30 m | 37.1 | CH | 1.52 m |
| b 1.40 m | ||||||
| 3 | 21.0 | CH2 | 1.54 m | 24.0 | CH2 | a 1.55 m |
| b 2.19 m | ||||||
| 4 | 31.0 | CH2 | a 2.04 m | 134.4 | CH | 5.54 m |
| b 2.30 m | ||||||
| 5 | 132.1 | C | 133.6 | C | ||
| 6 | 140.8 | C | 141.7 | CH | 6.28 d (15.3) | |
| 7 | 45.2 | CH2 | 2.89 m | 118.8 | CH | 5.54 m |
| 8 | 67.0 | CH | 3.80 m | 58.2 | CH | 2.48 m |
| 9 | 138.6 | C | 86.0 | C | ||
| 10 | 124.2 | CH | 5.49 d (9.6) | 38.6 | CH | 1.96 m |
| 11 | 84.9 | CH | 5.23 d (9.6) | 37.1 | CH2 | a 1.45 m |
| b 1.57 m | ||||||
| 12 | 151.7 | CH | 7.26 s | 46.7 | C | |
| 13 | 128.6 | C | 179.5 | C | ||
| 14 | 174.5 | C | 22.5 | CH3 | 1.16 d (6.0) | |
| 15 | 28.2 | CH3 | 0.82 s | 12.7 | CH3 | 1.75 s |
| 16 | 28.1 | CH3 | 0.88 s | 23.8 | CH3 | 1.25 s |
| 17 | 70.7 | CH2 | a 3.90 d (10.8) | 64.6 | CH2 | 3.51 m |
| b 4.48 d (10.8) | ||||||
| 18 | 14.7 | CH3 | 1.57 s | 19.5 | CH3 | 0.94 s |
| 19 | 10.9 | CH3 | 1.78 s | |||
| 1′ | 103.6 | CH | 4.17 d (7.5) | 103.4 | CH | 4.16 d (7.8) |
| 2′ | 74.2 | CH | 2.90 m | 74.3 | CH | 2.92 m |
| 3′ | 77.6 | CH | 3.11 m | 77.5 | CH | 3.12 m |
| 4′ | 70.8 | CH | 3.08 m | 70.8 | CH | 3.09 m |
| 5′ | 77.6 | CH | 3.10 m | 77.5 | CH | 3.10 m |
| 6′ | 61.9 | CH2 | a 3.33 m | 61.9 | CH2 | a 3.33 m |
| b 3.60 m | b 3.60 m | |||||
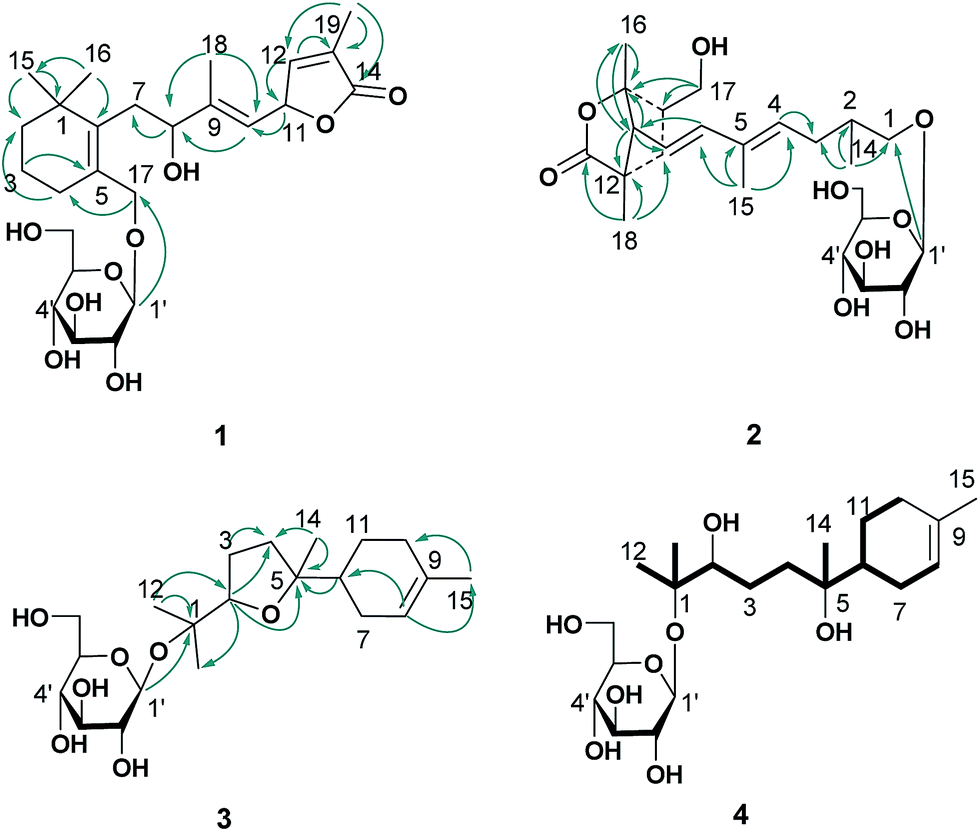 | ||
| Fig. 2 HMBC correlations (arrows) of 1–3, and 1H–1H COSY (bold lines) of 4. | ||
 | ||
| Fig. 3 Key NOESY correlations (H–H) of 1 and 3. | ||
The absolute configuration of the norditerpenoid genin (1′) obtained from acid hydrolysis of 1, was established by calculations of its ECD data,16,17 and the Bulkiness Rule for Rh2(OCOCF3)4 complexes of a secondary alcohol.18,19 A conformational search (MMFFs force field) for 1′a and its enantiomer 1′b led to 12 conformers, respectively, followed by geometric optimization of each one.
The optimized conformers subjected to ECD calculations in MeOH (CPCM) using the B3LYP functional and the 6-311+G(d) basis set for TDDFT. The final calculated ECD was obtained as the result of the Boltzmann-weighted average. Comparison of the experimental ECD curve 1′ and calculated ECD curves (Fig. 4) permitted the assignment of the absolute configuration of 1′ as 8R, 11R (ESI-Table 1†). In addition, the Rh2(OCOCF3)4-induced ECD experiment on the 8-OH of 1′ also confirmed the above result (negative E band at 340 nm in spectra Fig. S24†). The D-glucose was identified by acid hydrolysis of 1 then comparison with an authentic sample by an HPLC analysis using OR detector (ESI-Fig. 3†). Consequently, the structure of compound 1 was established and named norhawthornoid A.
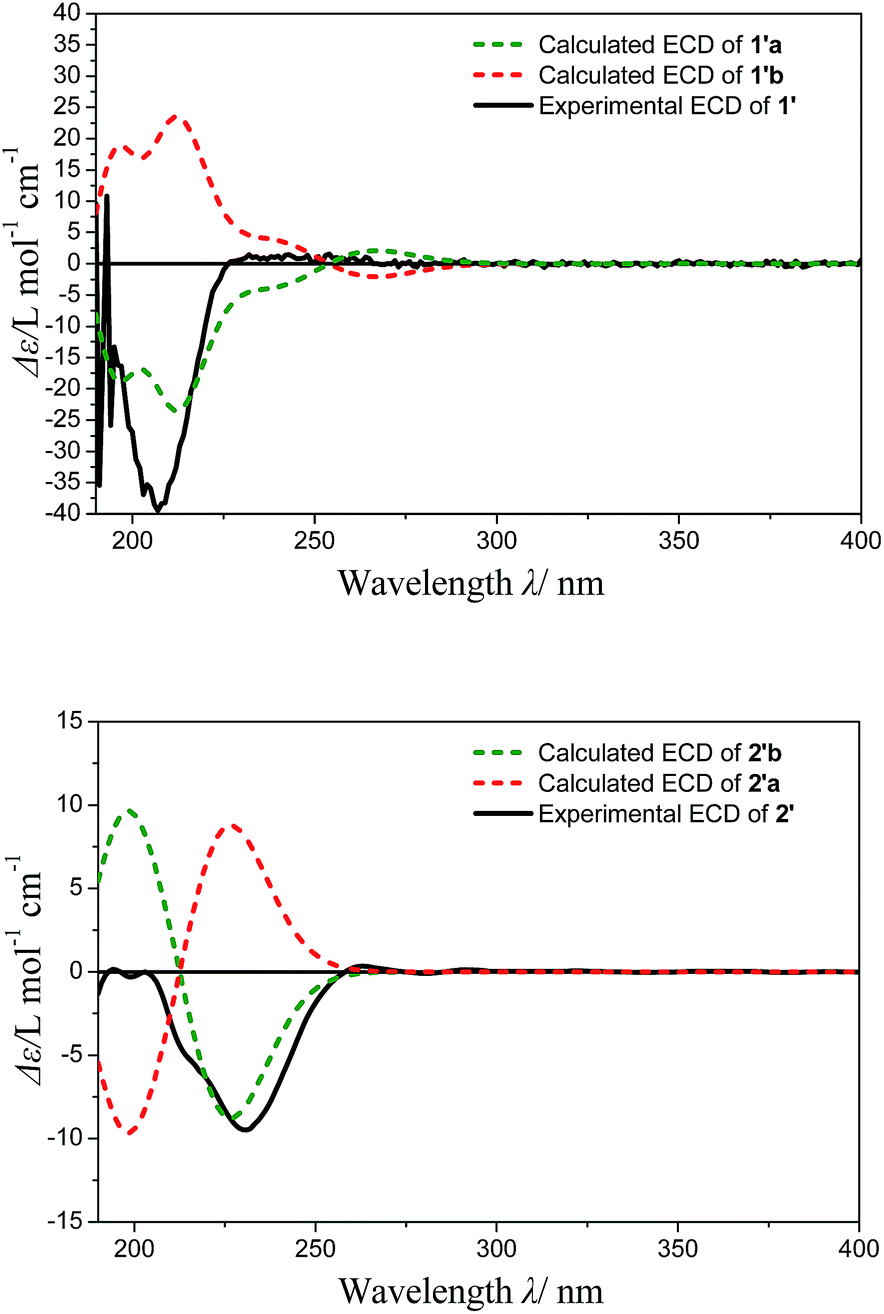 | ||
| Fig. 4 Experimental and calculated ECD for 1 and 2. | ||
Compound 2 was obtained as a colorless oil, and its HRESIMS exhibited a sodium adduct ion peak at m/z 471.2588 [M + H]+ (calc. 471.2589), corresponding to the molecular formula C24H38O9. In the 1H NMR spectrum (Table 1) of 2 there were resonances attributable to four methyls at δH 1.75 (3H, s), 1.25 (3H, s), 0.94 (3H, s) and 1.16 (3H, d, J = 6.0 Hz), two pair of oxygenated methylene protons at δH 3.51 (2H, m) and 3.65 (2H, m), three olefinic H-atoms at δH 6.28 (1H, d, J = 15.3 Hz), 5.54 (2H, m), an anomeric proton at δH 4.16 (1H, d, J = 7.8 Hz), and six oxygenated H-atoms at δH 2.92–3.60. The 13C NMR spectrum (Table 1) exhibited 24 carbon resonances, from which, four methyls (δC 12.7, 19.5, 22.5, 23.8), two methylenes (δC 24.0, 37.1), three methines (δC 37.1, 38.6, 58.2), a quaternary carbon (δC 46.7), four carbons bearing oxygen (δC 64.6, 75.3, 86.0), two double bond groups (δC 118.8, 133.6, 134.4, 141.7), one ester carbonyl group (δC 179.5), and a sugar group (δC 61.9, 70.8, 74.3, 77.5, 77.5 and 103.4) were identified, which suggested that compound 2 has a norditerpenoid skeleton. Analysis of the 1H-, 13C-NMR and HSQC data helped us to assign H- to their bonded C-atoms (Table 1), and further information about the 2D structure of 2 (Fig. 2) was obtained from the HMBC experiment. The presence of bicyclic [2.2.1] rigid rings of 2 was supported by the long-range HMBC interactions δH 1.25 (16-Me)/δC 58.2 (C-8), 86.0 (C-9), 38.6 (C-10); δH 3.51 (17-CH2OH)/δC 86.0 (C-9), 38.6 (C-10), 37.1 (C-11); δH 0.94 (18-Me)/δC 58.2 (C-8), 37.1 (C-11), 46.7 (C-12), 179.5 (C-13). The presence of the C9 side chain was established by other key correlations around δH 1.16 (14-Me), 1.75 (15-Me), 5.54 and 6.28 (olefinic H – 4, 6, 7). In addition, C-8 was assigned to the linkage site of above two moieties due to the key correlations between δH 2.48 (H-8) and δC 141.7 (C-6), 118.8 (C-7), 86.0 (C-9), 37.1 (C-11), 46.7 (C-12), 23.8 (C-16), 19.5 (C-18). The location of the sugar moiety was determined by the HMBC correlations between H-1′ (δH 4.16) and C-1 (δC 70.3) (Fig. 2). Furthermore, In the 1H NMR data, the vicinal coupling constant J(6,7) of 15.3 Hz suggested a trans relationship between the protons H-6 and H-7. The geometry of the double bond at C-4 was assigned as E on the basis of the upfield shift of the methyl carbon signal in the 13C NMR spectrum (δC 12.7 for C-15).15
The absolute configuration of the genin 2′ obtained from acid hydrolysis of 2, was established by comparison of the experimental and calculated ECD and NMR data.16,17 The experimental ECD curves of 2′ (Fig. 4) matched well with the theoretically calculated ECD data of 2′b using the TDDFT method at the B3LYP/6-311+G (d) level (ESI-Table 2†). Thus, the absolute configuration of 2′ was determined to 8R/9S/10R/12S. Furthermore, the configuration of C-2 was indicated by calculated NMR data. For this, our high-accuracy 13C-NMR calculation of the two possible stereoisomers (2R/8R/9S/10R/12S or 2S/8R/9S/10R/12S) indicated that the potential one is 2S/8R/9S/10R/12S (R2 = 0.9875, AveDev = 3.5) (ESI-Fig. 5†). Finally, the D configuration of the sugar moiety was determined by acid hydrolysis followed by an HPLC analysis with an authentic sample using an OR detector (ESI-Fig. 3†). On the basis of the above evidence, the structure of 2 was determined and named norhawthornoid B.
Compound 3, a colorless oil, showed the molecular formula of C21H36O7 from the HRESIMS ion at m/z 401.2536 [M + H]+ (calc. 401.2534). The 1H and 13C NMR data (Table 2), assigned by HSQC and HMBC spectra, showed signals of four tertiary methyls at δH 1.59 (3H, s), 1.16 (3H, s), 1.07 (3H, s), 1.02 (3H, s), an oxygenated methine proton at δH 3.82 (1H, m), an olefinic proton at δH 5.35 (1H, br s). Furthermore, an anomeric proton signal at 4.37 (1H, d, J = 7.8 Hz), and six oxygenated H-atoms at δH 2.86–3.62 indicated the presence of a sugar moiety. The 13C NMR spectrum (Table 2) exhibited 21 carbon resonances, from which, four methyls (δC 22.8, 24.0, 24.0, 24.0), five methylenes (δC 24.0, 27.1, 27.3, 27.3, 31.1), two methines (δC 43.7, 85.4), two quaternary carbons (δC 78.4, 85.2), a double bond group (δC 121.5, 133.9), and a sugar group (δC 61.9, 71.1, 74.4, 77.2, 77.8 and 98.0) were identified, which suggested that compound 3 is a sesquiterpenoid glycoside. In the HMBC spectrum (Fig. 2), the key correlations of 3 (Fig. 2), δH 1.07 (12-Me)/δC 78.4 (C-1), 85.4 (C-2), 24.0 (C-13); δH 1.16 (13-Me)/δC 78.4 (C-1), 85.4 (C-2), 22.8 (C-12); δH 1.02 (14-Me)/δC 27.3 (C-4), 85.2 (C-5), 43.7 (C-6), and δH 1.59 (15-Me)/δC 121.5 (C-8), 133.9 (C-9), 31.1 (C-10), suggested the presence of the bisabolane sesquiterpene skeleton. A long- range correlation between H-2 (δH 3.82) and C-5 (δC 85.2) suggested the presence of 2,5-ether moieties. In addition, the glycosidic site (C-1) was established unambiguously by an HMBC experiment, in which a long-range correlation between H-1′ (δH 4.37) and C-17 (δC 78.4) was observed (Fig. 2). The relative configuration of 3 was determined by the key NOE correlations between H-2/H-6, H-2/H2-7, which suggested that they were located on the same side of the tetrahydrofuran (THF) ring (C2–C5) (Fig. 3).
| Position | 3a | 4a | ||||
|---|---|---|---|---|---|---|
| δC | Type | δH, mult. (J in Hz) | δC | Type | δH, mult. (J in Hz) | |
| a Measured at 150 MHz for 13C in DMSO-d6, and measured in 600 MHz for 1H in DMSO-d6, (s) singlet, (d) doublet, (m) multiplet. | ||||||
| 1 | 78.4 | C | 77.4 | C | ||
| 2 | 85.4 | CH | 3.82 m | 80.2 | CH | 3.91 m |
| 3 | 24.0 | CH2 | 1.71 m | 21.2 | CH2 | 1.71 m |
| 4 | 27.3 | CH2 | 1.52 m | 38.2 | CH2 | 1.53 m |
| 5 | 85.2 | C | 72.5 | C | ||
| 6 | 43.7 | CH | 1.96 m | 42.3 | CH | 1.88 m |
| 7 | 27.1 | CH2 | 1.92 m | 24.5 | CH2 | 1.88 m |
| 8 | 121.5 | CH | 5.35 br s | 121.5 | CH | 5.34 br s |
| 9 | 133.9 | C | 133.6 | C | ||
| 10 | 31.1 | CH2 | 1.95 m | 31.1 | CH2 | 1.88 (2H, m) |
| 11 | 27.3 | CH2 | 1.65 m | 26.9 | CH2 | 1.67 (2H, m) |
| 12 | 22.8 | CH3 | 1.07 s | 23.7 | CH3 | 1.10 s |
| 13 | 24.0 | CH3 | 1.16 s | 23.8 | CH3 | 1.10 s |
| 14 | 24.0 | CH3 | 1.02 s | 23.8 | CH3 | 0.83 s |
| 15 | 24.0 | CH3 | 1.59 s | 23.5 | CH3 | 1.59 s |
| 1′ | 98.0 | CH | 4.37 d (7.8) | 97.5 | CH | 4.34 d (7.5) |
| 2′ | 74.4 | CH | 2.86 m | 74.0 | CH | 2.89 m |
| 3′ | 77.8 | CH | 3.12 m | 77.2 | CH | 3.11 m |
| 4′ | 71.1 | CH | 3.04 m | 70.6 | CH | 3.02 m |
| 5′ | 77.2 | CH | 3.06 m | 77.0 | CH | 3.04 m |
| 6′ | 61.9 | CH2 | a 3.30 br d (11.4) | 61.5 | CH2 | a 3.35 br d (10.8) |
| b 3.62 br d (11.4) | b 3.62 br d (10.8) | |||||
The absolute configuration of the sesquiterpene skeleton (3′) obtained by acid hydrolysis was established by comparison of the experimental and calculated OR.20 Due to the above NMR data, the configuration of 3′ was identified as one of the two isomers (2R, 5R, 6S or 2S, 5S, 6R). A conformational search using the MMFFs force field for the (2R, 5R, 6S)-stereoisomer and its enantiomer led to the identification of 11 conformers, followed by geometric optimization of each one. The optimized conformers were subjected to OR calculations in MeOH (CPCM) using the B3LYP functional and the 6-311+G(d) basis set for DFT. Final calculated ORs were obtained as the result of the Boltzmann-weighted average (ESI-Table 6†). From these results, the negative calculated ORs ([α]D20 −8.7) of the (2R, 5R, 6S)-stereoisomer was a better fit to the experimental OR value ([α]D20 −2.8, c 0.4, MeOH) than the positive one ([α]D20 +8.7). In addition, the D-glucosyl moiety was confirmed by acid hydrolysis of 3 and then comparison with an authentic sample. Consequently, the structure of compound 3 was given the trivial name pinnatifidanoside F.
Compound 4 was obtained as colorless oil and its molecular formula was determined as C21H38O8 by HRESIMS. In the 1H NMR spectrum (Table 2), the presence of four tertiary methyls at δH 1.59 (3H, s), 1.10 (6H, s), 0.83 (3H, s), a oxygenated methine proton at δH 3.91 (1H, m), a olefinic proton at δH 5.34 (1H, br s), were revealed, in addition, the anomeric proton signal at 4.34 (1H, d, J = 7.5 Hz) and six oxygenated H-atoms at δH 2.89–3.62 demonstrated the presence of a saccharide group. The 13C NMR spectrum (Table 2) exhibited 21 carbon resonances, from which, four methyls (δC 23.5, 23.7, 23.8, 23.8), five methylenes (δC 21.2, 24.5, 26.9, 31.1, 38.2), two methines (δC 42.3, 80.2), two quaternary carbons (δC 72.5, 77.4), a double bond group (δC 121.5, 133.6), and a sugar group (δC 61.5, 70.6, 74.0, 77.0, 77.2 and 97.5) were identified, which suggested that compound 4 has a sesquiterpenoid skeleton closed to the structure of 3. A careful comparison of the NMR spectroscopic data of 4 with that of 3 (Table 2) suggested that they have a closed sesquiterpenoid skeleton. Following analysis of their NMR data, the C2 and C5 atoms in 4 exhibiting −5.2 and −12.7 ppm deviations respectively indicated that the THF ring was replaced by a 2,5-dihydroxy moiety, which was confirmed by HRESIMS data (Fig. 1). The multi-method combination of Mosher's method and comparison of the experimental and calculated NMR and OR values21–23 was presented here for support in the study of the absolute configuration of the sesquiterpene genin (4′). Firstly, according to the MTPA shielding/deshielding effects of the two esters, the ΔδH (S–R) values (ESI-Fig. 4†) revealed a 2R configuration for 4′. Then, the configurations of C-5 and C-6 were indicated by calculated NMR and OR data. For this, our high-accuracy 13C-NMR calculation of the four possible stereoisomers (2R/5S/6S, 2R/5S/6R, 2R/5R/6S or 2R/5R/6R) indicated that the potential one is 2R/5S/6S (R2 = 0.9980 larger than others) (Fig. 5, ESI-Fig. 6 and 7†). Meanwhile, the OR value of the (2R/5S/6S)-stereoisomer predicted to be −21.8 at the DFT level was in agreement with the experimental OR value ([α]D20 −7.5, c 0.2, MeOH). Furthermore, HPLC analysis of the acidic hydrolysate of 4 showed that the sugar moiety was a D-glucose. Thus, the structure of compound 4 was named pinnatifidanoside G.
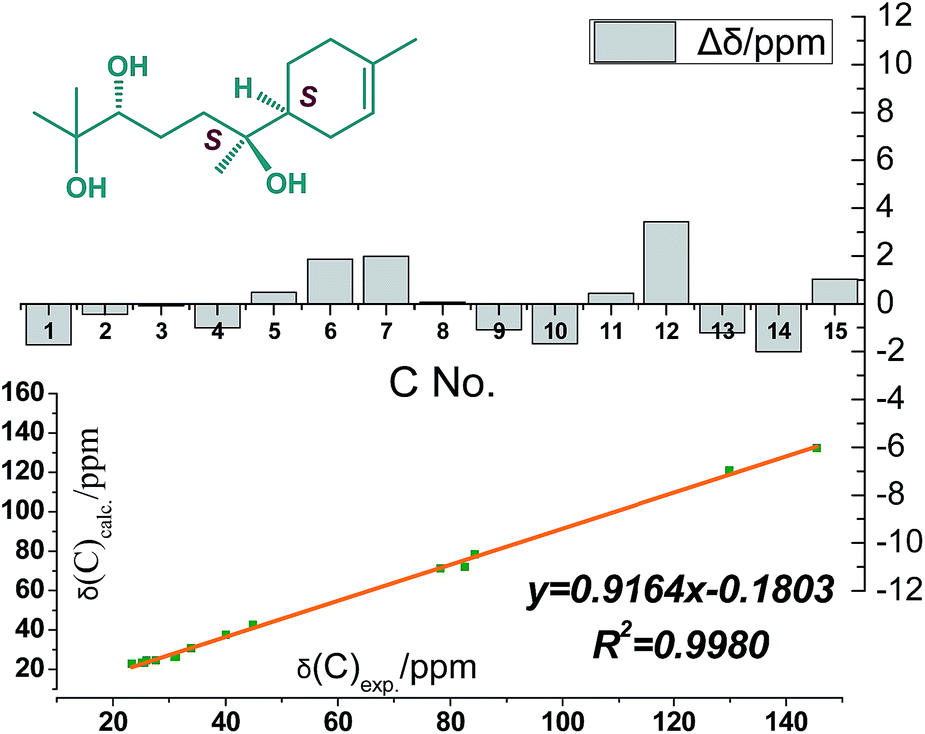 | ||
| Fig. 5 Experimental and calculated 13C chemical shifts of 4′a. | ||
The adenosine diphosphate (ADP), a granular content, act as agonists to activate more platelets and recruit them onto the subendothelial matrix, which contains cells, collagen, and von Willebrand factor, is exposed and tether to the site of injury. Our current study identified a key mechanism underlying antithrombotic effect of terpenoids from hawthorn leaves, showing that they blocked platelet activation by inhibiting the ADP pathway.14,24 In this study, all the isolates were tested in vitro for their antithrombotic effects by ADP induced platelet aggregation experiment.25 The inhibitory activity of individual compounds is summarized in Table 3. Compounds 1–4, 8, 11 and 15 showed potent anti-platelet activities. Compound 3 a sesquiterpenoid displayed the best inhibitory activity in particular, inhibiting platelet aggregation by 84.00 ± 4.00% at the final concentration of 0.25 mg mL−1 during the test interval. These results indicated that sesquiterpenoid inhibit platelet aggregations induced by ADP agonists, and thus it may have beneficial potential for the prevention of platelet-involved cardiovascular disease.
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ± SD, n = 3)
± SD, n = 3)
| Samples | Inhibition rate (%) | Samples | Inhibition rate (%) |
|---|---|---|---|
| 1 | 69.20 ± 6.35 | 9 | <50 |
| 2 | 74.00 ± 4.50 | 10 | <50 |
| 3 | 84.00 ± 4.00 | 11 | 68.25 ± 1.35 |
| 4 | 76.25 ± 7.25 | 12 | 64.55 ± 1.75 |
| 5 | <50 | 13 | <50 |
| 6 | <50 | 14 | 64.25 ± 2.35 |
| 7 | 54.05 ± 5.25 | 15 | 74.95 ± 10.45 |
| 8 | 76.95 ± 2.45 | ||
| Aspirin | 82.65 ± 4.65 |
The P2 family of receptors mediates the platelet response to ADP, and mammalian platelets express three ADP subtypes receptors, namely, P2Y1, P2Y12, and P2X1.24,26 Among them, the P2Y12 receptor (P2Y12R), a member of the P2Y purinergic GPCR family stimulated by ADP, is one of the most important clinical drug targets for inhibition of platelet aggregation.26 Clopidogrel inhibits platelet activation through irreversible binding to the P2Y12 ADP receptor on platelet membrane. Therefore, compound 3, the most potent inhibitor of the ADP-induced platelet aggregation, was investigated as an antagonist of the direct-acting P2Y12 receptor by molecular modeling studies (Fig. 6). Also AZD1283 (the co-crystal ligand from crystal protein PDB code 4PXZ) was used as a positive control.26 These investigations indicate that (a) the 3′-OH and 1-O- of 3 were in close proximity to the catalytic residues Tyr 109 (2.97 Å) and Arg 256 (5.01 Å) and underwent hydrogen bonding interactions. Also, the same hydrogen bonding interactions were oriented between AZD1283 and Tyr 109 and Arg 256 (distance 3.29 and 3.18 Å) (Fig. 7) (b) both AZD1283 and compound 3 were oriented in an aromatic pocket consisting of Tyr 109, Phe 252, Arg 256, and Lys 280. In addition, Tyr 105, Asn 191, Gln 195, Tyr 259, and Leu 276 are also considered as key active residues interacting with the natural ligand. In addition, the results of the ADP-induced platelet aggregation experiment indicated that these isolates may block platelet activation by inhibiting P2Y12R.
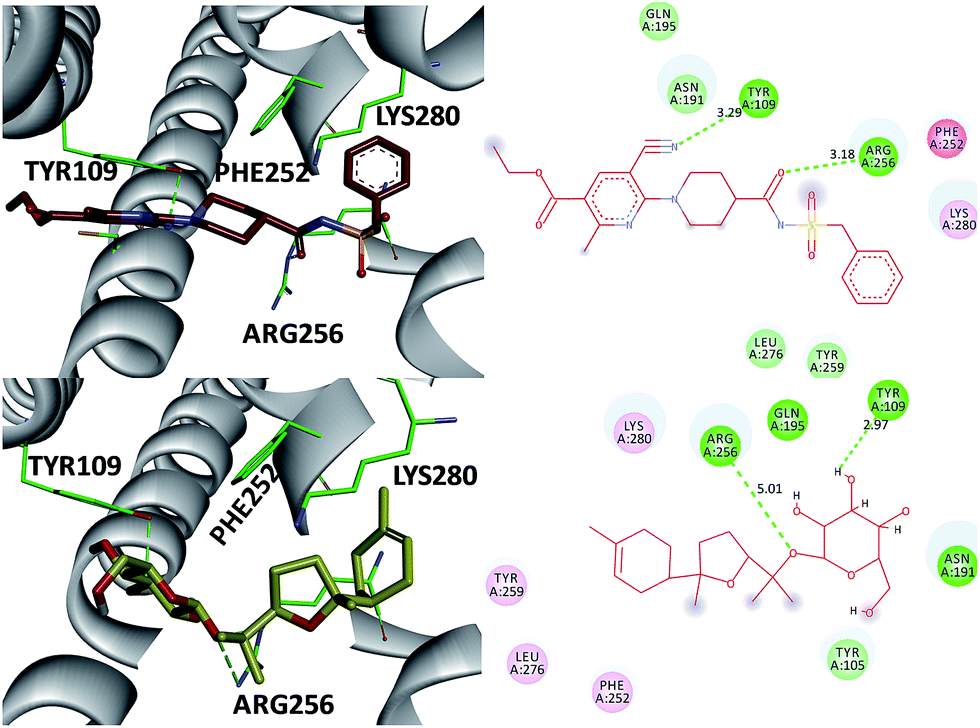 | ||
| Fig. 6 Amino acids interacting with compound 3 and AZD1283. Hydrogen bonds are shown as green dots. | ||
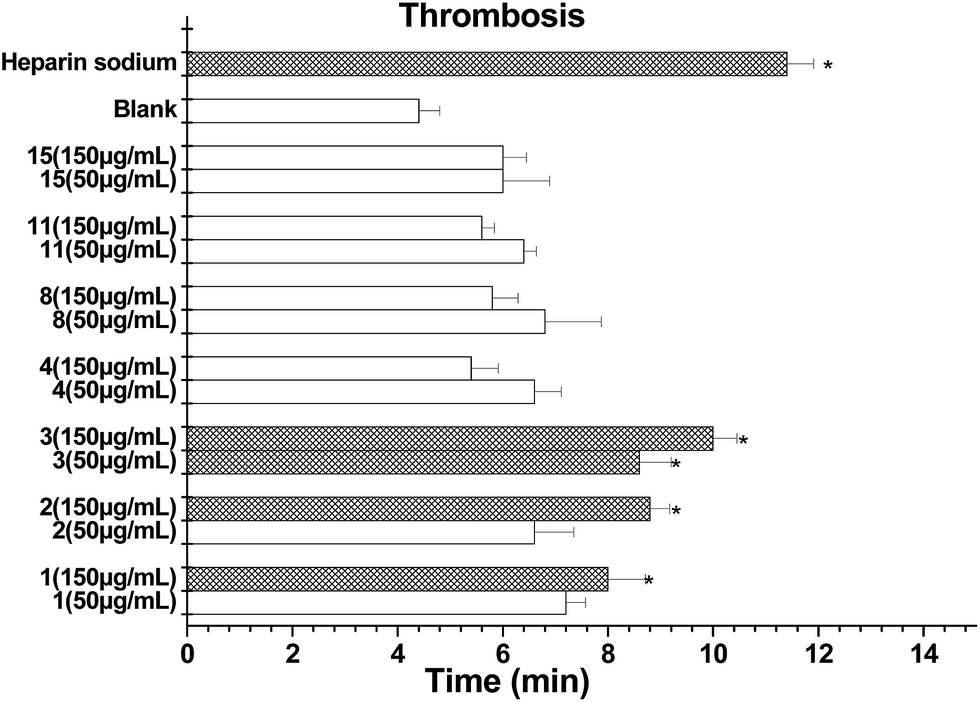 | ||
| Fig. 7 Antithrombotic activity of compounds 1–4, 8, 11 and 15 on zebrafish. | ||
More recently, the genetic screening of natural products has been developed for an FeCl3-induced thrombosis model in zebrafish in vivo, which overcomes the limitations of the mouse model.27,28 Prior to investigating the antithrombotic effect, we determined the viability of zebrafish embryos pretreated with increasing doses of isolates (50 and 150 μg mL−1) for 48 h. The results obtained showed that compounds 1–4, 8, 11 and 15 did not influence the embryo viability in cultured zebrafishes up to 150 μg mL−1. Next, different doses of each compound were investigated using this thrombosis model. Compounds 1–3 clearly prolonged the time to form a thrombus in the tested zebrafish (Fig. 7). Among them, the thrombosis time of 3 was around or over 2.2-fold that of the model control and was the most potential.
Experimental
General experimental procedures
Optical rotations were obtained using an Autopol IV automatic polarimeter (Rudolph Research Analytical). UV spectra were recorded on a Shimadzu double-beam 210A spectrometer. The ECD spectra were measured with a BioLogic MOS-450 spectropolarimeter. NMR spectra were recorded on a Bruker ARX-400 and 600 spectrometer with TMS as internal standard in dimethyl sulfoxide-d6 (DMSO-d6). Mass spectra were determined on a HRESI-MS: MicroTOF spectrometer (Bruker Daltonics, CA). TLC silica gel (GF254), silica gel (200–300 mesh) and polyamide (200–400 mesh) were purchased from Qingdao Marine Chemical Co., China. Reversed-phase C18 silica gel (ODS 70–80 μm Merck, Germany). Solvents were of industrial purity and distilled prior to use.Plant material and animals
The leaves of Crataegus pinnatifida (Rosaceae) were gathered from Liaoning in November 2015, and authenticated by pharmacognosy Prof. Lu Jin-Cai. A voucher specimen (HY151108) is kept in the Nature Products Laboratory of Shenyang Pharmaceutical University, Shenyang, China.Sprague–Dawley (SD) rats (8 weeks, 220–240 g) were obtained from Liao Ning Chang Sheng Biotechnology Co., Ltd. They were housed in a conventional animal facility with free access to food and water in a temperature and relative humidity controlled environment under a 12 h light/dark schedule. Adult zebrafish (AB) were obtained from Biology Institute of Shandong Academy of Sciences (Jinan, China), and fish were maintained with a 14/10 h light/dark cycle. Zebrafish were fed with Tetramin granulated food and live brine shrimps (Artemia nauplii). Embryos were obtained from natural spawning, which was induced in the next morning after mating, and were placed in the embryos medium (5.0 mM NaCl, 0.17 mM KCl, 0.4 mM CaCl2, 0.16 mM MgSO4) under 28 °C, to feed them for three days post fertilization (dpf). All animal procedures were carried out according to the Regulations of Experimental Animal Administration issued by State Committee of Science and Technology of China, and approved by the institutional ethical committee (IEC) of Shenyang Pharmaceutical University.
Extraction and isolation
Exhaustive extraction of C. pinnatifida leaves (15 kg) with 50% ethanol (total 270 L) followed by reflux two times, which was prepared according to Pharmacopoeia of People's Republic of China (Yixintong medicinal preparations in page 396–397). The concentrated crude extract (2.0 kg) was separated by macroporous resin (20 kg) with EtOH/H2O (v/v = 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 10:90, 60:40, 95:5). The 60% ethanol fraction (280 g) was further separated by polyamide (2 kg) column chromatography (EtOH/H2O 20:70, 40:60, 60:40, 95:5) to provide 4 fractions (Frs. a–d). Fr. a (26 g) was subjected to silica gel (200 g) chromatography eluting with a CH2Cl2–EtOH (v/v = 30:1, 15:1, 10:1, 8:1) and further purified by ODS (50 g) column chromatography with EtOH:H2O (3:4) to yield compounds 8 (12 mg), 10 (23 mg), 13 (23 mg), 14 (9 mg) and 15 (15 mg) which were further purified by preparative HPLC. And the same method was employed to obtain compounds 3 (27 mg), 4 (11 mg) and 6 (22 mg), 9 (16 mg), 11 (53 mg) and 12 (45 mg) from the next fraction (Frs. b, 35 g). Compounds 1 (9 mg), 2 (12 mg), 5 (28 mg) and 7 (19 mg) from Frs. c, (23 g).
ε) 208 (2.21) nm; ECD (MeOH) λ (Δε) 209 (−39.50) nm; see Table 1 for 1H NMR, 13C NMR; HRESIMS m/z: 483.2582 [M + H]+ (calcd C25H39O9 483.2589).ε) 228 (2.21) nm; ECD (MeOH) λ (Δε) 230 (−9.80) nm; see Table 1 for 1H NMR, 13C NMR; HRESIMS m/z: 471.2588 [M + H]+ (calcd C24H38O9 471.2589).
:100, 10:90, 60:40, 95:5). The 60% ethanol fraction (280 g) was further separated by polyamide (2 kg) column chromatography (EtOH/H2O 20:70, 40:60, 60:40, 95:5) to provide 4 fractions (Frs. a–d). Fr. a (26 g) was subjected to silica gel (200 g) chromatography eluting with a CH2Cl2–EtOH (v/v = 30:1, 15:1, 10:1, 8:1) and further purified by ODS (50 g) column chromatography with EtOH:H2O (3:4) to yield compounds 8 (12 mg), 10 (23 mg), 13 (23 mg), 14 (9 mg) and 15 (15 mg) which were further purified by preparative HPLC. And the same method was employed to obtain compounds 3 (27 mg), 4 (11 mg) and 6 (22 mg), 9 (16 mg), 11 (53 mg) and 12 (45 mg) from the next fraction (Frs. b, 35 g). Compounds 1 (9 mg), 2 (12 mg), 5 (28 mg) and 7 (19 mg) from Frs. c, (23 g).
ε) 208 (2.21) nm; ECD (MeOH) λ (Δε) 209 (−39.50) nm; see Table 1 for 1H NMR, 13C NMR; HRESIMS m/z: 483.2582 [M + H]+ (calcd C25H39O9 483.2589).ε) 228 (2.21) nm; ECD (MeOH) λ (Δε) 230 (−9.80) nm; see Table 1 for 1H NMR, 13C NMR; HRESIMS m/z: 471.2588 [M + H]+ (calcd C24H38O9 471.2589).Characteristic data of compounds 5–15 see ESI-E.2.†
Acid hydrolysis of 1–4
Compounds 1–4 (each 2.0 mg) were individually hydrolyzed by 6% HCl (1.0 mL) under reflux for 2 h. The reaction mixtures were extracted with CH2Cl2, and the organic layers evaporated under reduced pressure to yield 1′ (0.2 mg), 2′ (0.5 mg), 3′ (0.7 mg), and 4′ (0.8 mg). D-Glucose was obtained from the aqueous layers of 1–4 and identified by HPLC confirmation (Shodex Asahipak NH2P-50 4E, 250 × 4.6 mm no. N1490155; flow rate 1.0 mL min−1; eluent CH3CN/H2O, 3:1) with the authentic sample using optical rotation detector (Jasco OR-4090).
Preparation of the (R)- and (S)-MTPA esters of 4′
To a solution of 4′ (1.0 mg) in dry pyridine (200 μL) was added (S)-MTPA chloride (20 μL). The mixture was allowed to stand under N2 at room temperature for 2 h. The solution was concentrated under vacuum to afford a residue, which was subjected to semipreparative HPLC with an RP-C18 column afforded the (S)-MTPA esters of 4′. The (R)-MTPA ester of 4′ was prepared with (R)-MTPA chloride and was purified in the same manner. 4′S: from 1.0 mg of 4′ was obtained 0.3 mg of (S)-MTPA ester. 4′R: from 1.0 mg of 4′ was obtained 0.2 mg of (R)-MTPA ester.Rh2(OCOCF3)4-induced ECD experiment of 1′ and 5
Compound 1′ and 5 (0.5 mg) were dissolved in a dry solution of [Rh2(OCOCF3)4] complex (1.0 mg) in CDCl3 (700 μL). The first ECD spectrum was recorded immediately after mixing, and its time evolution was monitored until stationary (10–15 min after mixing). The inherent ECD was subtracted. The observed sign of the E band at about 350 nm in the induced ECD spectrum was correlated to the absolute configuration of the C-8 of 1′ (secondary alcohol moiety), as well as the C-3 of 5 (tertiary alcohol moiety).29Computational methods
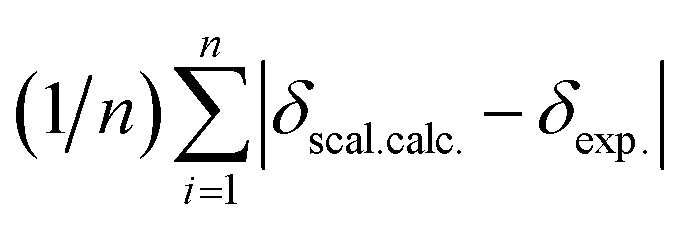 .
.Evaluation of antiplatelet activity in rat PRP
Evaluation of the antiplatelet aggregation activities of pure compounds 1–15, positive control (aspirin) and blank (normal saline) were performed using an antiplatelet aggregation method.25 SD rats weighing 200–240 g were lightly anesthetized with diethyl ether. A volume of 8–10 mL of blood was collected from the abdominal aorta into a syringe containing 3.8% sodium citrate. The ratio of blood to 3.8% sodium citrate was adjusted to 1:9 v/v and then centrifuged at 150g for 10 min to obtain platelet-rich plasma (PRP) used for the aggregation study. PRP (2 × 108 platelets per mL) was prewarmed at 37 °C for 2 min. The isolates 1–15 dissolved respectively in 5% dimethylsulfoxide at a concentration of 6.25 mg mL−1 (10 μL) were added to the diluted PRP (230 μL) before the addition of the platelet aggregation agent. PRP aggregation was induced by 10 μL ADP (62.5 μM). The final concentration of all samples was 0.25 mg mL−1. In positive and blank control experiments, normal saline and aspirin were added instead of the isolates. From each series of experiments, the inhibitors were tested in at least three times, and the extent of antiplatelet aggregation was measured by the percentage inhibition of the control value. The results obtained are shown in Table 3, ESI-Fig. 8.†
Antithrombotic assay using a zebrafish system
The arachidonic acid (FeCl3) was used to induce larval zebrafish thrombus as model group. Three dpf (days post fertilization) AB-type zebrafish embryos were placed into 24 well plates with ten ones per well. Then the embryos were exposed to select positive control (20 U mL−1 heparin sodium) and test sample solutions at final concentrations (50, 150 μg mL−1) in 0.2% DMSO for 48 h until five dpf. The negative control group was the solvent group, which not only represented the back ground value of the test group, but also made sure that the cardiac red blood cells of larval zebra fish were normal. The larval zebrafish were treatment with 60 μg mL−1 of FeCl3, and the time was recorded until a thrombus was observed in the venous system. Meanwhile, it also needed to be observed if there was the presence of embryonic death or deformity. The thrombosis time of the heparin sodium was normalised to 100% and the results were expressed as a percentage of the control (Fig. 7).Molecular modeling (docking) studies
Docking of AZD1283 and compound 3 into the P2Y12 was performed with Molegro virtual docker (Molegro). The X-ray crystal structure of the P2Y receptor P2Y12 (PDB code 4PXZ) was obtained from the RCSB Protein Data Bank. The co-crystal ligand AZD1283 was extracted to define the binding site in docking. The structure of compound 3 was constructed and energy minimized for 1000 iterations reaching a convergence of 0.01 kcal mol−1 Å−1. The docking experiments on P2Y12 were carried out by superimposing the minimized ligand into the receptor. Ten runs were performed and five poses returned. All the other docking parameters were set as default. The pose for which the biotin entity of AZD1283 and 3 binds to P2Y12 receptor in a similar way as observed in the original ligand–enzyme complex was exported and examined with Discovery Studio Visualizer 4.0 (Accelrys).Conclusions
In summary, our current study describes the discovery of two uncommon norditerpenoids (1–2) and 13 sesquiterpenoids (3–15). The compounds 1 and 2 represent the first examples of diterpenoids obtained from Crataegus genus. The new compounds 1–3 exhibited exceptionally potent antithrombotic activities in vitro/vivo. The most potent one, pinnatifidanoside F (3), is about 2.2-fold more potent compared with the model control in the zebrafish model for thrombosis (in vivo). It is interesting to study the kinds of antithrombotic ingredients present in the title plant. Particularly, these terpenoids could play an important role on their own or in good synergy with flavonoids. This need to be examined in future research studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Project of National Natural Science Foundation of P. R. China (81302661), Innovation Team (LT2015027) of Liaoning of P. R. China, and Foundation (L2015419) from the Project of Education Department of Liaoning province of P. R. China are gratefully acknowledged. The authors thank Molegro ApS for kindly providing us with a free evaluation of their software packages. The authors also thank Li W. and Sha Y. of Shenyang Pharmaceutical University for recording NMR spectra.Notes and references
- N. L. Smith, L. B. Harrington, M. Blondon, K. L. Wiggins, J. S. Floyd, C. M. Sitlani, B. McKnight, E. B. Larson, F. R. Rosendaal, S. R. Heckbert and B. M. Psaty, J. Thromb. Haemostasis, 2016, 7, 1384 CrossRef PubMed.
- E. Fuentes and I. Palomo, J. Funct. Foods, 2014, 6, 73 CrossRef CAS.
- Y. N. Shi, Y. M. Shi, L. Yang, X. C. Li, Z. H. Zhao, Y. Qu, H. T. Zhu, D. Wang, R. R. Cheng, C. R. Yang, M. Xu and Y. J. Zhang, J. Ethnopharmacol., 2015, 162, 87 CrossRef CAS PubMed.
- J. X. Qiao, T. C. Wang, S. Hiebert, C. H. Hu, W. A. Schumacher, S. A. Spronk, C. G. Clark, Y. Han, J. Hua, L. A. Price, H. Shen, S. A. Chacko, G. Everlof, J. S. Bostwick, T. E. Steinbacher, Y. X. Li, C. S. Huang, D. A. Seiffert, R. Rehfuss, R. R. Wexler and P. Y. S. Lam, ChemMedChem, 2014, 9, 2327 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed.
- R. Guo, M. H. Pittler and E. Ernst, Cochrane Database Syst. Rev., 2008, 1, 1 Search PubMed.
- P. Z. Liu, H. K. Kallio, D. G. Lü, C. S. Zhou, S. Ou and B. J. Yang, J. Agric. Food Chem., 2010, 58, 1012 CrossRef CAS PubMed.
- M. Luo, J. Y. Hu, Z. Y. Song, J. Jiao, F. S. Mu, X. Ruan, Q. Y. Gai, Q. Qiao, Y. G. Zu and Y. J. Fu, RSC Adv., 2015, 5, 67532 RSC.
- H. F. Pan, X. Wang, Y. L. Du, Y. R. Li, S. N. Zhao and W. D. Pan, Anal. Methods, 2014, 6, 7475 RSC.
- B. M. Fraga, Nat. Prod. Rep., 2012, 29, 1334 RSC.
- J. E. Edwards, P. N. Brown, N. Talent, T. A. Dickinson and P. R. Shipley, Phytochemistry, 2012, 79, 5 CrossRef CAS PubMed.
- S. J. Song, L. Z. Li, P. Y. Gao, Y. Peng, J. Y. Yang and C. F. Wu, Food Chem., 2011, 129, 933 CrossRef CAS PubMed.
- P. Y. Gao, L. Z. Li, Y. Peng, F. F. Li, C. Niu, X. X. Huang, M. Ming and S. J. Song, Biochem. Syst. Ecol., 2010, 38, 988 CrossRef CAS.
- L. Z. Li, P. Y. Gao, S. J. Song, Y. Q. Yuan, C. T. Liu, X. X. Huang and Q. B. Liu, J. Funct. Foods, 2015, 12, 237 CrossRef CAS.
- J. H. Shin, Y. W. Seo and K. W. Cho, J. Org. Chem., 1999, 64, 1853 CrossRef CAS PubMed.
- Y. Tang, Y. B. Xue, G. Du, J. P. Wang, J. J. Liu, B. Sun, X. N. Li, G. M. Yao, Z. W. Luo and Y. H. Zhang, Angew. Chem., Int. Ed., 2016, 55, 4069 CrossRef CAS PubMed.
- G. Q. Zhan, J. J. Liu, J. F. Zhou, B. Sun, H. J. Akber Aisa and G. M. Yao, Eur. J. Med. Chem., 2017, 127, 771 CrossRef CAS PubMed.
- J. Frelek and W. J. Szczepek, Tetrahedron: Asymmetry, 1999, 10, 1507 CrossRef CAS.
- M. T. Liu, S. Lin, M. L. Gan, M. H. Chen, L. Li, S. J. Wang, J. C. Zi, X. N. Fan, Y. Liu, Y. K. Si, Y. C. Yang, X. G. Chen and J. G. Shi, Org. Lett., 2012, 14, 1004 CrossRef CAS PubMed.
- L. W. Tian, Y. J. Feng, T. D. Tran, Y. Shimizu, T. Pfeifer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2015, 78, 1756 CrossRef CAS PubMed.
- D. P. Curran, Q. S. Zhang, H. J. Lu and V. Gudipati, J. Am. Chem. Soc., 2006, 128, 9943 CrossRef CAS PubMed.
- M. Diaz, J. Jaballas, J. Arias, H. Lee and T. Onak, J. Am. Chem. Soc., 1996, 118, 4405 CrossRef CAS.
- G. Barone, D. Duca, A. Silvestri, L. Gomez-Paloma, R. Riccio and G. Bifulco, Chem.–Eur. J., 2002, 8, 3240 CrossRef CAS.
- P. Jagadeeswaran, M. Gregory, K. Day, M. Cykowski and B. Thattaliyath, J. Thromb. Haemostasis, 2005, 3, 46 CrossRef CAS PubMed.
- G. H. Tang, G. H. Jiang, S. Z. Wang and L. F. Zheng, Chin. J. Pharmacol. Toxicol., 2001, 15, 317 CAS.
- J. Zhang, K. Zhang, Z. G. Gao, S. Paoletta, D. Zhang, G. W. Han, T. Li, L. Ma, W. Zhang, C. E. Muller, H. Yang, H. Jiang, V. Cherezov, V. Katritch, K. A. Jacobson, R. C. Stevens, B. Wu and Q. Zhao, Nature, 2014, 509, 119 CrossRef CAS PubMed.
- K. D. Kurz, B. W. Main and G. E. Sandusky, Thromb. Res., 1990, 60, 269 CrossRef CAS PubMed.
- M. R. Lang, G. Gihr, M. P. Gawaz and I. I. Müller, J. Thromb. Haemostasis, 2010, 8, 1159 CrossRef CAS PubMed.
- L. Xiong, C. G. Zhu, Y. R. Li, Y. Tian, S. Lin, S. P. Yuan, J. F. Hu, Q. Hou, N. H. Chen, Y. H. Yang and J. P. Shi, J. Nat. Prod., 2011, 74, 1188 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., C. T. Wallingford, 2010 Search PubMed.
- G. Barone, L. Gomez-Paloma, D. Duca, A. Silvestri, R. Riccio and G. Bifulco, Chem.–Eur. J., 2002, 8, 3233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, HRESIMS, ECD and Rh2(OCOCF3)4-ICD spectra of compounds; details of calculated and experimental ECD, 13C NMR and ORs, MTPA esters, acid hydrolysis and in vitro and in vivo antithrombotic assays. See DOI: 10.1039/c7ra10768d |
| This journal is © The Royal Society of Chemistry 2017 |