
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aldol condensation of acetic acid with formaldehyde to acrylic acid over Cs(Ce, Nd) VPO/SiO2 catalyst
Aili Wang a,
Jing Hua,
Hengbo Yin*a,
Zhipeng Lua,
Wuping Xuea,
Lingqin Shena and
Shuxin Liub
a,
Jing Hua,
Hengbo Yin*a,
Zhipeng Lua,
Wuping Xuea,
Lingqin Shena and
Shuxin Liub
aFaculty of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: yin@ujs.edu.cn; Tel: +86-511-88787591
bSchool of Chemistry and Chemical Engineering, Mianyang Normal University, Mianyang 621000, China
First published on 16th October 2017
Abstract
Cs, Ce, and Nd cation-modified VPO/SiO2 catalysts were prepared by a deposition method. The VPO/SiO2 catalyst was composed of VOPO4 and (VO)2P2O7 phases. When Cs, Ce, and Nd cations were added in the VPO/SiO2 catalyst, Cs4P2O7, CePO4, and NdPO4 were formed and V5+/V4+ ratio increased, respectively. The basicities of the metallic cation-modified catalysts increased while their acidities exhibited maximum values with the increase in the ratios of metallic cation to vanadium. The metallic cation-modified VPO/SiO2 catalysts exhibited higher catalytic activities for the aldol condensation reaction of acetic acid with formaldehyde to acrylic acid than the VPO/SiO2 catalyst. The high acidity and basicity of the metallic cation-modified VPO/SiO2 catalysts favored the formation of acrylic acid.
1. Introduction
Acrylic acid is widely used as an important raw material in the chemical industry for the production of superabsorbent polymers, dispersants, paints, adhesives, textile auxiliaries, and oilfield chemicals by virtue of its unsaturated bond and carboxylic acid group.1–5 The two-step catalytic oxidation of propylene to acrylic acid is a commonly used technique in commercial production of acrylic acid.6 However, propylene, as the feedstock for acrylic acid production, is non-sustainable and high cost because petroleum is a non-renewable resource and is gradually exhausted.Considering both the environment and the economy, more and more attention has been paid to renewable and economic acrylic acid production methods,3–5,7 such as, lactic acid dehydration,2,8 glycerol deoxydehydration–oxidation,9 and aldol condensation of acetic acid with formaldehyde.3,4,7,10 Production of acrylic acid from renewable resource is limited by the high price of lactic acid and the low acrylic acid yield in glycerol deoxydehydration–oxidation. However, the production of acetic acid in China was around 5000 kilotons per year in 2015 and acetic acid has been oversupplied in Chinese market. The aldol condensation of acetic acid with formaldehyde to acrylic acid is a potential production technology using coal-derivative acetic acid and formaldehyde as the feeds.
It was reported that supported alkali and alkaline earth metal hydroxides exhibited good catalytic activity for the aldol condensation reaction between acetic acid (methyl propionate and methyl acetate) and formaldehyde. Vitcha et al.11 firstly reported the synthesis of acrylic acid by the aldol condensation reaction between acetic acid and formaldehyde over alkali and alkaline earth metal aluminosilicate catalysts at the reaction temperatures of 275–385 °C. With the high acetic acid/formaldehyde ratios of 8–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the total reactant feed of 0.11 to 0.14 mol Lcat−1 h−1, acrylic acid yields of 80–100% were obtained. The low space-time yield probably limits the commercial application of these base catalysts. Ai12 reported that silica gel-supported cesium hydroxide catalyst gave a high methyl methacrylate selectivity of 86.5% at the formaldehyde conversion of 77.3% when methyl propionate and formaldehyde were used as the stating materials with their feed rates of 2.29 and 0.458 mmol gcat−1 h−1, respectively, at the reaction temperature of 360 °C. Yan et al.13 reported that when vapor phase condensation of methyl acetate with formaldehyde (mole ratio, 1:2) was catalyzed by SBA-15-supported cesium catalyst at 350–420 °C and a total reactant feed rate of 3.6 mL gcat−1 h−1, the selectivity of methyl acrylate and the conversion of methyl acetate were around 95% and 48%, respectively. They suggested that the high catalytic activity was mainly due to the suitable strength of weak acid–base properties of the catalyst.
:1 and the total reactant feed of 0.11 to 0.14 mol Lcat−1 h−1, acrylic acid yields of 80–100% were obtained. The low space-time yield probably limits the commercial application of these base catalysts. Ai12 reported that silica gel-supported cesium hydroxide catalyst gave a high methyl methacrylate selectivity of 86.5% at the formaldehyde conversion of 77.3% when methyl propionate and formaldehyde were used as the stating materials with their feed rates of 2.29 and 0.458 mmol gcat−1 h−1, respectively, at the reaction temperature of 360 °C. Yan et al.13 reported that when vapor phase condensation of methyl acetate with formaldehyde (mole ratio, 1:2) was catalyzed by SBA-15-supported cesium catalyst at 350–420 °C and a total reactant feed rate of 3.6 mL gcat−1 h−1, the selectivity of methyl acrylate and the conversion of methyl acetate were around 95% and 48%, respectively. They suggested that the high catalytic activity was mainly due to the suitable strength of weak acid–base properties of the catalyst.
Vanadium phosphorus oxide (VPO) catalyst also exhibited good catalytic activity for the aldol condensation of acetic acid with formaldehyde to acrylic acid. Ai14 reported that when trioxane was used as the formaldehyde source, the yield of acrylic acid of ca. 98% was obtained, based on the charged formaldehyde with the acetic acid/formaldehyde mole ratio of 2.5. In the case of formalin as the formaldehyde source, the yield of ca. 75% was obtained. The main side-reaction is the decomposition of acetic acid to form acetone and CO2. Feng et al.15 found that when the aldol condensation of methyl acetate or acetic acid with formaldehyde was catalyzed by VPO catalyst at 360 °C, the total formation rate of acrylic acid and methyl acrylate was ca. 1.188 mmol gcat−1 h−1 at the formaldehyde conversion of 85%. The VOPO4 and (VO)2P2O7 were considered as the active components in the VPO catalyst for the aldol condensation reactions.15,16 In our previously work,17 we found that when the VPO/SBA-15 catalyst with the P/V mole ratio of 2:1 catalyzed the aldol condensation of acetic acid with formaldehyde at 330–370 °C, the acrylic acid selectivities were 90.8–70.2% at the formaldehyde conversions of 14.3–68.7%. The maximum formation rate of acrylic acid was 5.5 mmol gcat−1 h−1 and the acid/base properties of the supported VPO catalysts played important roles in the aldol condensation reaction.
For the aldol condensation of acetic acid with formaldehyde to acrylic acid, the supported VPO catalyst exhibited higher formation rate of acrylic acid as compared to the alkali hydroxide and bulk VPO catalysts.11,15,17 Both surface acidity and basicity of catalyst obviously affected the formation of acrylic acid.17 On the other hand, the alkali and alkaline earth metal hydroxide catalysts exhibited high selectivity to acrylic acid or methyl acrylate in the aldol condensation reactions. Therefore, to obtain high catalytic activity for the aldol condensation reaction, the use of metallic action to adjust the surface acidity/basicity of the supported VPO catalyst is practical.
In the selective oxidation of n-butane to maleic anhydride, it was found that metallic cations could stabilize the catalyst performance, affect the surface acidity of catalyst, and modify the structure of VPO catalyst, giving high catalytic activity.18–24 However, to the best of our knowledge, the aldol condensation of acetic acid with formaldehyde to acrylic acid catalyzed by metallic cation-modified VPO catalyst has not been reported so far. The structural effect of metallic cation-modified VPO catalyst on the aldol condensation reaction is worth of investigation.
In our present work, the aldol condensation of acetic acid with trioxane (as the source of formaldehyde) to acrylic acid catalyzed by cesium-, cerium-, and neodymium-modified SiO2-supported vanadium phosphorus oxide (VPO/SiO2) catalysts was investigated. The structures and acid/base properties of the resultant metallic cation-modified VPO/SiO2 catalysts were analyzed. The presence of metallic cations in the VPO/SiO2 catalysts significantly improved the yield and the formation rate of acrylic acid. The effect of the structures and acid/base properties of these catalysts on their catalytic activities was discussed.
2. Experimental
2.1 Materials
V2O5 (99%), phosphoric acid (≥85%), iso-butyl alcohol, benzyl alcohol, acetone, acetic acid (≥99%), trioxane (>99%), acrylic acid, methyl acrylate, cesium carbonate, ammonium cerium nitrate, and neodymium nitrate hexahydrate were of reagent grade and were purchased from Shanghai Sinopharm Co. Ltd. Silica aerogel (SiO2) was purchased from Jiangsu Haoneng Chemical Co. Ltd. The chemicals were used as received without further purification.2.2 Preparation of catalyst
Vanadium-phosphorous oxide (VPO) was prepared according to the reported organic methods.25–27 The procedures were briefly illustrated as follows. 1.53 g of V2O5 was refluxed in a mixture of iso-butyl/benzyl alcohol (60 mL:60 mL) for 5.5 h, and then 4.26 g of phosphoric acid (85%) was added dropwise under refluxing for 5.5 h. A light blue suspension was obtained and cooled down to 50 °C. Given amounts of cesium carbonate, ammonium cerium nitrate, and neodymium nitrate hexahydrate salts were dissolved in 40 mL iso-butyl alcohol, respectively. The salt solutions were added into the VPO suspension, respectively. And then 8 g of SiO2 aerogel as a support was added into the suspension and stirred rapidly for 3 h. After cooling the reaction mixture to room temperature, the metallic cation-modified SiO2-supported VPO catalysts were filtrated and washed with iso-butanol and acetone, respectively. The samples were dried at 120 °C for 24 h and then calcined at 500 °C for 4.5 h under atmospheric condition. The as-prepared catalysts were denoted Mn-VPO/SiO2, where, M and n indicate the metallic cation and the molar ratio of metallic cation to vanadium, respectively. The catalysts were extruded as pellets at 10 MPa and then crushed into particles with the sizes in a range of 0.45–0.8 mm. The compositions of the catalysts are listed in Table 1.
| Catalysts | M/P/V mole ratio | BET surface areas (m2 g−1) | BJH average pore diameters (nm) | CO2 desorption peak (°C) | NH3 desorption peak (°C) | Total base quantities (μmol CO2 per gcat) | Total acid quantities (μmol NH3 per gcat) | Catalyst components |
|---|---|---|---|---|---|---|---|---|
| VPO | 2.2/1 | — | — | — | — | — | — | VOPO4, (VO)2P2O7 |
| VPO/SiO2 | 2.2/1 | 115.2 | 5.6 | 705 | 187 | 28.4 | 1294.8 | VOPO4, (VO)2P2O7 |
| Cs0.05-VPO/SiO2 | 0.05/2.2/1 | 104.1 | 5.0 | 680 | 189 | 102.8 | 1385.3 | VOPO4, (VO)2P2O7, Cs4P2O7 |
| Cs0.1-VPO/SiO2 | 0.1/2.2/1 | 86.0 | 5.0 | 686 | 177 | 145.4 | 1843.1 | |
| Cs0.2-VPO/SiO2 | 0.2/2.2/1 | 80.1 | 5.0 | 679 | 168 | 152.6 | 1245.1 | |
| Cs0.3-VPO/SiO2 | 0.3/2.2/1 | 70.9 | 4.6 | 667 | 175 | 203.6 | 825.9 | |
| Ce0.05-VPO/SiO2 | 0.05/2.2/1 | 108.6 | 5.2 | 677 | 188 | 107.2 | 1471.5 | (VO)2P2O7, CePO4 |
| Ce0.1-VPO/SiO2 | 0.1/2.2/1 | 99.8 | 5.0 | 713 | 184 | 182.2 | 1937.2 | |
| Cs0.2-VPO/SiO2 | 0.2/2.2/1 | 86.4 | 5.0 | 718 | 181 | 194.7 | 1348.0 | |
| Ce0.3-VPO/SiO2 | 0.3/2.2/1 | 80.6 | 4.8 | 692 | 189 | 254.4 | 1177.5 | |
| Nd0.05-VPO/SiO2 | 0.05/2.2/1 | 113.6 | 5.4 | 680 | 177 | 106.4 | 2455.3 | VOPO4, αII-VOPO4, NdPO4 |
| Nd0.1-VPO/SiO2 | 0.1/2.2/1 | 103.9 | 5.0 | 694 | 184 | 143.5 | 1842.3 | |
| Nd0.2-VPO/SiO2 | 0.2/2.2/1 | 93.6 | 5.0 | 701 | 174 | 191.3 | 1180.8 | |
| Nd0.3-VPO/SiO2 | 0.3/2.2/1 | 88.7 | 5.0 | 697 | 185 | 197.4 | 1064.1 |
2.3 Characterization
Nitrogen adsorption/desorption isotherms of the catalysts were measured at −196 °C with a NOVA 2000e physical adsorption apparatus. Prior to the measurement, all the catalysts were degassed at 120 °C for 4 h under vacuum. The specific surface areas and average pore sizes of the catalysts were calculated according to the BET and BIH methods, respectively.X-ray powder diffraction (XRD) was conducted at room temperature on a D8 super speed Bruke-AEX Company diffractometer with Cu Kα radiation (λ = 1.54056 Å). The scanning rate was at 2° min−1 (2θ).
X-ray photoelectron spectra (XPS) were performed on an XSAM800 spectrometer (Kratos Company) using Al Kα radiation (1486.6 eV). The binding energies were referred to the C 1s signal (284.6 eV).
CO2 temperature-programmed desorption (CO2-TPD) was performed to measure the basicity of catalyst while the acidity was measured using NH3 temperature-programmed desorption (NH3-TPD). 0.1 g of catalyst was heated at 400 °C for 0.5 h and then the temperature was reduced to room temperature in a helium flow (30 mL min−1). The pre-treated samples were exposed in a CO2 or a NH3 stream for 0.5 h at room temperature. And then the samples were purged with helium at 100 °C for 1 h to remove the physically adsorbed CO2 and NH3. CO2-TPD and NH3-TPD were monitored with a thermal conductivity detector (TCD) from 100 to 800 °C at a heating rate of 10 °C min−1. The base and acid amounts of the samples were quantitatively calculated according to the CO2 and NH3 desorption peak areas, respectively.
2.4 Catalytic test
The aldol condensation of acetic acid with formaldehyde was carried out in a fixed bed tubular stainless steel reactor operating at atmospheric pressure. 3 g of catalyst was charged in the reactor. The reaction temperatures were in a range of 320–400 °C. Trioxane ((HCHO)3) was used as a source of formaldehyde because it can be decomposed to formaldehyde in an acid solution. Trioxane was dissolved in acetic acid with an acetic acid/formaldehyde mole ratio of 3:1. The feed stream was pumped into an evaporator at 300 °C at the fixed rate of 8.6 g h−1 (0.165 mol h−1). Nitrogen was fed into the reactor through the evaporator as a carrier gas at a flow rate of 30 mL min−1.
The reaction mixture was collected in a cold trap at different reaction temperatures after reacting for 1 h and analyzed on a gas chromatograph with a flame ion detector (FID) and a HT-WAX capillary column. The unreacted formaldehyde was analyzed by the iodometry method. CO2 was analyzed on a gas chromatograph with a TCD and a TDX-01 packed column. n-Butanol was used as an internal standard for component quantification. The formaldehyde conversion (X) and the selectivities of acrylic acid (SAA) and methyl acrylate (SMA) based on formaldehyde were calculated according to the following equations, respectively.
| X = (n0 − nt)/n0 × 100% | (1) |
| SAA = nAA/(n0 − nt) × 100% | (2) |
| SMA = 2nMA/(n0 − nt) × 100% | (3) |
The yields of acetone (YA) and CO2 (YCO2) based on acetic acid were calculated according to the following equations, respectively.
| YA = nA/nAC × 100% | (4) |
| YCO2 = nCO2/nAC × 100% | (5) |
3. Results and discussion
3.1 XRD analysis
Fig. 1 shows the XRD patterns of the VPO, VPO/SiO2, and Cs (Ce and Nd) cation-modified VPO/SiO2 catalysts. For the bulk VPO catalyst, the diffraction peaks at 2θ = 18.5, 23.0, 28.4, 29.9, 43.2, 49.5, and 58.9° indicated the presence of (VO)2P2O7 (JCPDS, 34-1381) while the peaks at 2θ = 18.4, 21.2, and 28.1° confirmed the presence of VOPO4 (JCPDS, 37-0809).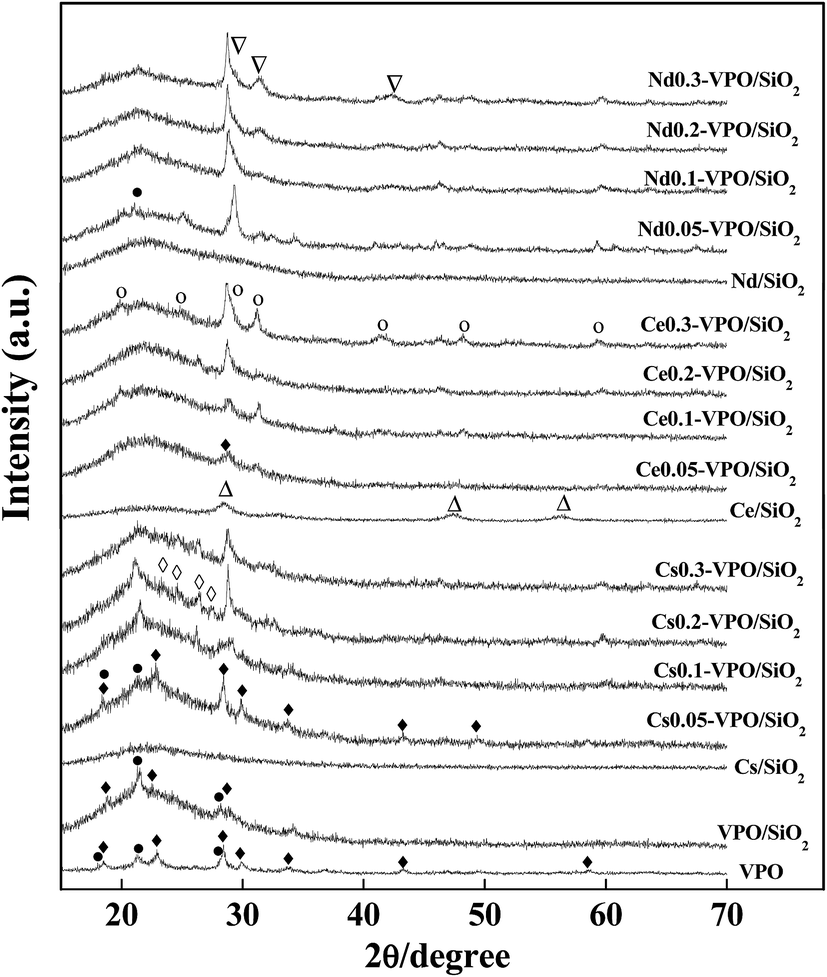 | ||
| Fig. 1 XRD patterns of the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2. (♦) (VO)2P2O7 (JCPDS, 34-1381); (●) VOPO4 (JCPDS, 37-0809); (◊) Cs4P2O7 (JCPDS, 38-0001); (Δ) CeO2 (JCPDS, 34-0394); (○) CePO4 (JCPDS, 34-1380); (□) αII-VOPO4 (JCPDS, 34-1247); (∇) NdPO4 (JCPDS, 04-0644). | ||
The XRD spectra of the VPO/SiO2 catalyst show that the characteristic diffraction peaks of (VO)2P2O7 appeared at 18.8, 22.6, and 28.9° and the diffraction peaks of VOPO4 appeared at 21.5 and 28.2°. After loading VPO on silica aerogel, the diffraction peak intensities of (VO)2P2O7 and VOPO4 decreased, indicating that the dispersibility of VPO component was improved. As compared to the XRD spectra of the bulk VPO catalyst, the peak shifts of the silica-supported VPO catalyst indicated that there was an interaction between VPO and silica support.
When VPO/SiO2 catalyst was modified with Cs cation, the diffraction peaks of (VO)2P2O7 at 18.4, 22.8, 28.4, 29.9, 33.8, 43.2, and 49.4° and the diffraction peaks of VOPO4 at 18.4, 21.3, and 28.4° were observed. With increasing the Cs/V ratio to 0.2, four weak peaks at 23.7, 24.7, 26.5, and 27.6° were observed, indicating the formation of Cs4P2O7 (JCPDS, 38-0001).
When VPO/SiO2 catalyst was modified with Ce cation, a characteristic diffraction peak of (VO)2P2O7 at 28.7° was observed. There was no diffraction peak of VOPO4 observed. With increasing the Ce/V ratio to 0.3, the characteristic diffraction peaks of CePO4 (JCPDS, 34-1380) were clearly observed, appearing at 20.0, 24.8, 29.1(shoulder), 31.2, 41.5, 48.4, and 59.4°, respectively.
When VPO/SiO2 catalyst was modified with Nd cation, the characteristic diffraction peak of VOPO4 (JCPDS, 37-0809) was observed at 21.2°. The characteristic diffraction peaks of αII-VOPO4 (JCPDS, 34-1247) at 20.2, 24.9, and 29.3° were also observed. There was no diffraction peak of (VO)2P2O7 observed. With increasing the Nd/V ratio to 0.3, the characteristic diffraction peaks of NdPO4 (JCPDS, 04-0644) were clearly observed, appearing at 29.5(shoulder), 31.6, and 42.4°, respectively.
When Cs, Ce, and Nd cations were loaded on silica aerogel support, the diffraction peaks of Ce0.2/SiO2 catalyst appeared at 28.6, 33.1, 47.5, and 56.3°, respectively, indicating that crystal CeO2 was formed on SiO2 surface. There were no diffraction peaks of cesium and neodymium oxides detected over the Cs0.2/SiO2 and Nd0.2/SiO2 catalysts. Cesium and neodymium oxides could be well dispersed on SiO2 surface. However, when Cs, Ce, and Nd cations were added in VPO/SiO2 catalyst, the compositions of VPO components were changed to some extent. The Cs, Ce, and Nd cations reacted with phosphorous component to form their phosphate salts, respectively.
3.2 XPS analysis
The XPS spectra of the VPO/SiO2, Cs0.2-VPO/SiO2, Ce0.2-VPO/SiO2, Nd0.2-VPO/SiO2, and used Cs0.2-VPO/SiO2 catalysts are shown in Fig. 2–4. The binding energies of components are listed in Table 2.| Catalysts | Binding energies (eV) | Atomic P/V ratio | V5+/V4+ relative amount | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P 2p | Si 2p | O 1s | V 2p3/2 | Cs 3d | Ce 3d | Nd 3d | ||||||
| V5+ | V4+ | 3d3/2 | 3d5/2 | 3d3/2 | 3d5/2 | 3d5/2 | ||||||
| VPO/SiO2 | 134.3 | 103.4 | 532.9 | 518.6 | 517.0 | — | — | — | — | — | 6.15 | 0.99 |
| Cs0.2-VPO/SiO2 | 134.5 | 103.4 | 532.8 | 518.5 | 516.9 | 738.4 | 724.3 | — | — | — | 7.16 | 1.34 |
| Ce0.2-VPO/SiO2 | 134.6 | 103.4 | 533.0 | 518.8 | 517.2 | — | — | 904.1 | 885.7 | — | 7.93 | 2.17 |
| Nd0.2-VPO/SiO2 | 134.8 | 103.4 | 533.0 | 518.8 | 517.2 | — | — | — | — | 982.4 | 5.68 | 1.50 |
| Cs0.2-VPO/SiO2 used | 134.6 | 103.4 | 532.8 | 518.5 | 517.0 | 737.6 | 723.8 | — | — | — | 4.30 | 0.09 |
The XPS spectra show that when Cs, Ce, and Nd cations were added in VPO/SiO2 catalyst, the binding energies of P 2p3/2 shifted to high values as compared with that in the VPO/SiO2 catalyst (Fig. 2). The O 1s binding energies of the Ce0.2-VPO/SiO2 and Nd0.2-VPO/SiO2 catalysts slightly shifted to high values while the O 1s binding energies of the fresh Cs0.2-VPO/SiO2 and used Cs0.2-VPO/SiO2 catalysts slightly shifted to low values. It could be explained as that the formation of phosphate salts changed the chemical states of phosphorous and oxygen. The binding energies of Si 2p for all the catalysts kept constant at 103.4 eV, indicating that silica support probably had a weak interaction with the supported component.
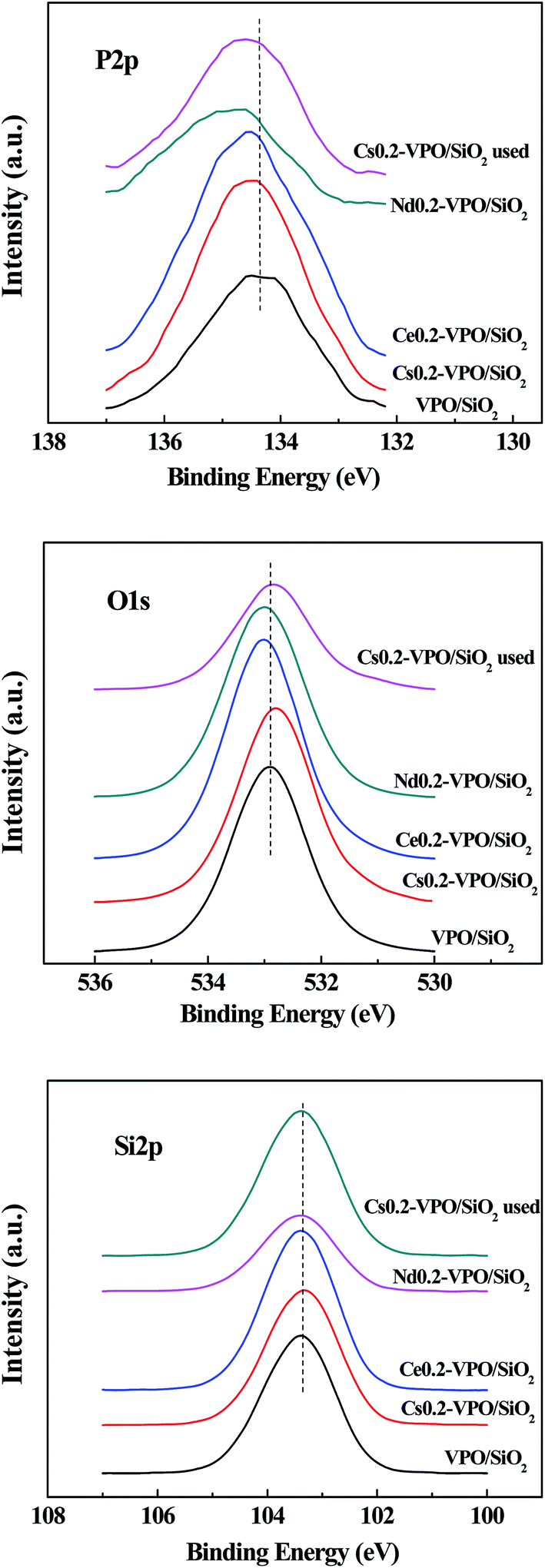 | ||
| Fig. 2 XPS spectra of P 2p, O 1s, and Si 2p of the VPO/SiO2, Cs0.2-VPO/SiO2, Nd0.2-VPO/SiO2, Ce0.2-VPO/SiO2, and used Cs0.2-VPO/SiO2 catalysts. | ||
The Cs 3d3/2 and Cs 3d5/2 binding energies of the fresh and used Cs0.2-VPO/SiO2 catalysts were 738.4, 724.3; 737.6, and 723.8 eV, respectively (Fig. 3). It was reported that the Cs 3d5/2 binding energy of Cs4P2O7 was 723.6 eV, referring to the residual C 1s of 284.6 eV.28 The difference between the Cs 3d5/2 binding energies in fresh Cs0.2-VPO/SiO2 and pure Cs4P2O7 indicated that there was an interaction between the Cs4P2O7 and the VPO oxides in the Cs0.2-VPO/SiO2 catalyst. The difference between the Cs 3d5/2 binding energies in both fresh and used Cs0.2-VPO/SiO2 catalysts revealed that the catalytic reaction also affected the chemical state of Cs cation.
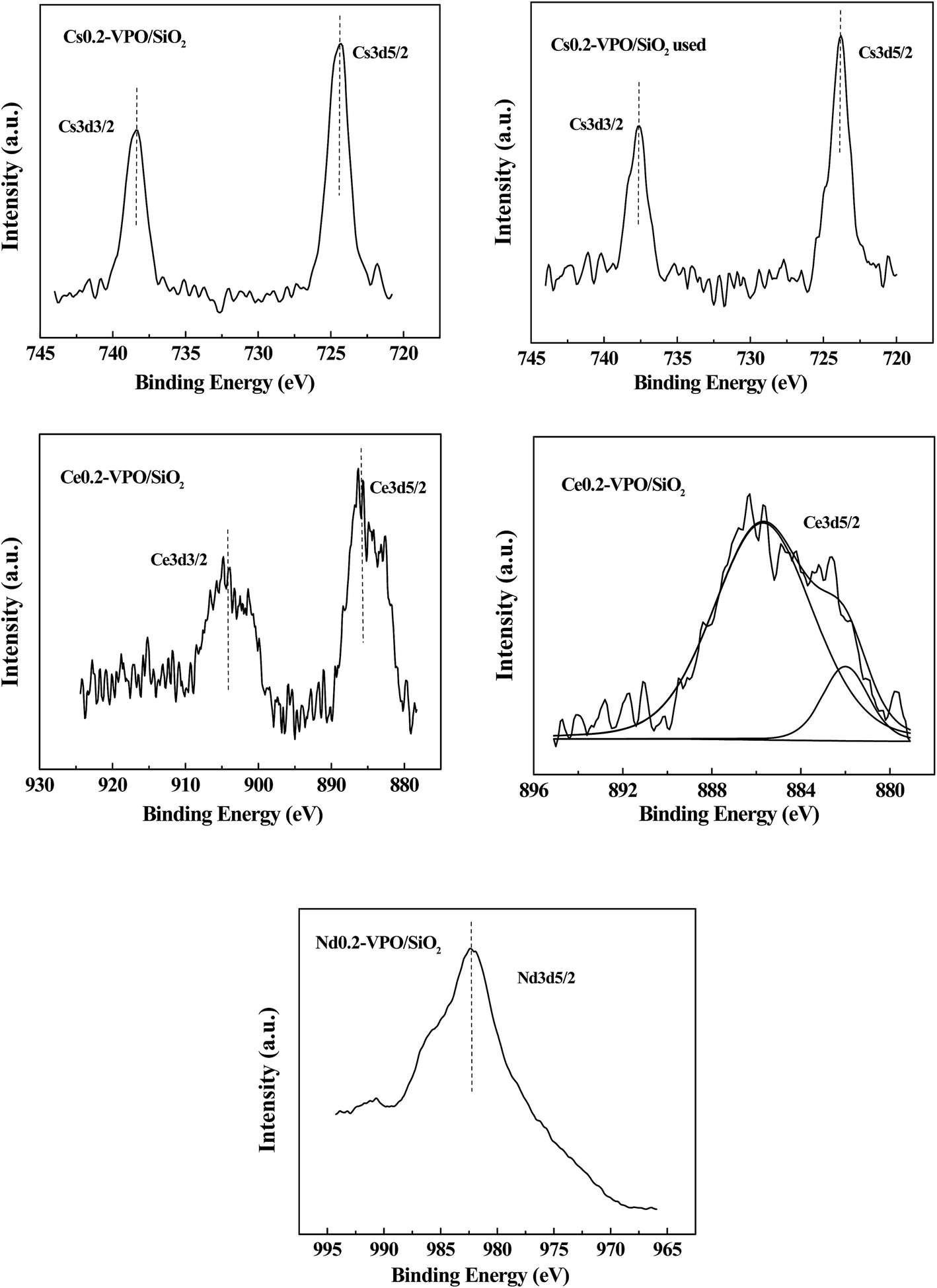 | ||
| Fig. 3 XPS spectra of Cs 3d, Ce 3d, and Nd 3d of the Cs0.2-VPO/SiO2, used Cs0.2-VPO/SiO2, Nd0.2-VPO/SiO2, and Ce0.2-VPO/SiO2 catalysts. | ||
The Ce 3d spectra of the Ce0.2-VPO/SiO2 catalyst did not exhibit a peak near 915 eV assigned to the Ce4+ cation.29 Cerium in Ce0.2-VPO/SiO2 catalyst was in the 3+ oxidation state. The Ce 3d3/2 and Ce 3d5/2 peaks had satellites, which were probably attributed to the existence of different final states.29 The Ce 3d5/2 peak can be separated by curve fitting with the software XPS PEAK 4.1 (Fig. 3). The spacing between the main and satellite peaks in the Ce 3d5/2 curve was 3.7 eV, being the same as that calculated by Pemba-Mabiala et al.29 The Ce 3d5/2 binding energy was 885.7 eV, which was in agreement with the value of Ce 3d5/2 (885.6 eV) in CePO4.29 The formation of CePO4 in the Ce0.2-VPO/SiO2 catalyst was certified by XPS analysis, which was consistent with the XRD analysis.
The Nd 3d5/2 spectrum of the Nd0.2-VPO/SiO2 catalyst shows that the Nd 3d5/2 binding energy was 982.4 eV (Fig. 3). It was reported that the Nd 3d5/2 binding energies of Nd3+ in Nd2O3 (ref. 30 and 31) and neodymium phosphate32 were 983.1, 983.3, and 982.8 eV, respectively. The binding energy of Nd 3d5/2 in the Nd0.2-VPO/SiO2 catalyst was close to that of Nd3+ cation, indicating the Nd element was present in 3+ oxidation state.
The profiles of V 2p3/2 were asymmetric, suggesting the co-presence of V5+/V4+ cations (Fig. 4). Deconvolution of the V 2p3/2 spectra of the representative catalysts based on a Gaussian signal was carried out according to the method reported in literatures.33–37 The V 2p3/2 binding energies for V5+ and V4+ cations were around 518.6 and 517.0 eV, respectively (Table 2). It was found that the atomic ratios of P/V and V5+/V4+ were obviously affected by the presence of metallic cations, indicating that the surface compositions of the catalyst active components were changed by the addition of metallic cations. After taking part in the reaction, the atomic ratio of V5+/V4+ of the used Cs0.2-VPO/SiO2 catalyst obviously decreased as compared with the fresh one, indicating that V5+/V4+ cation pairs probably took part in the catalytic reaction.
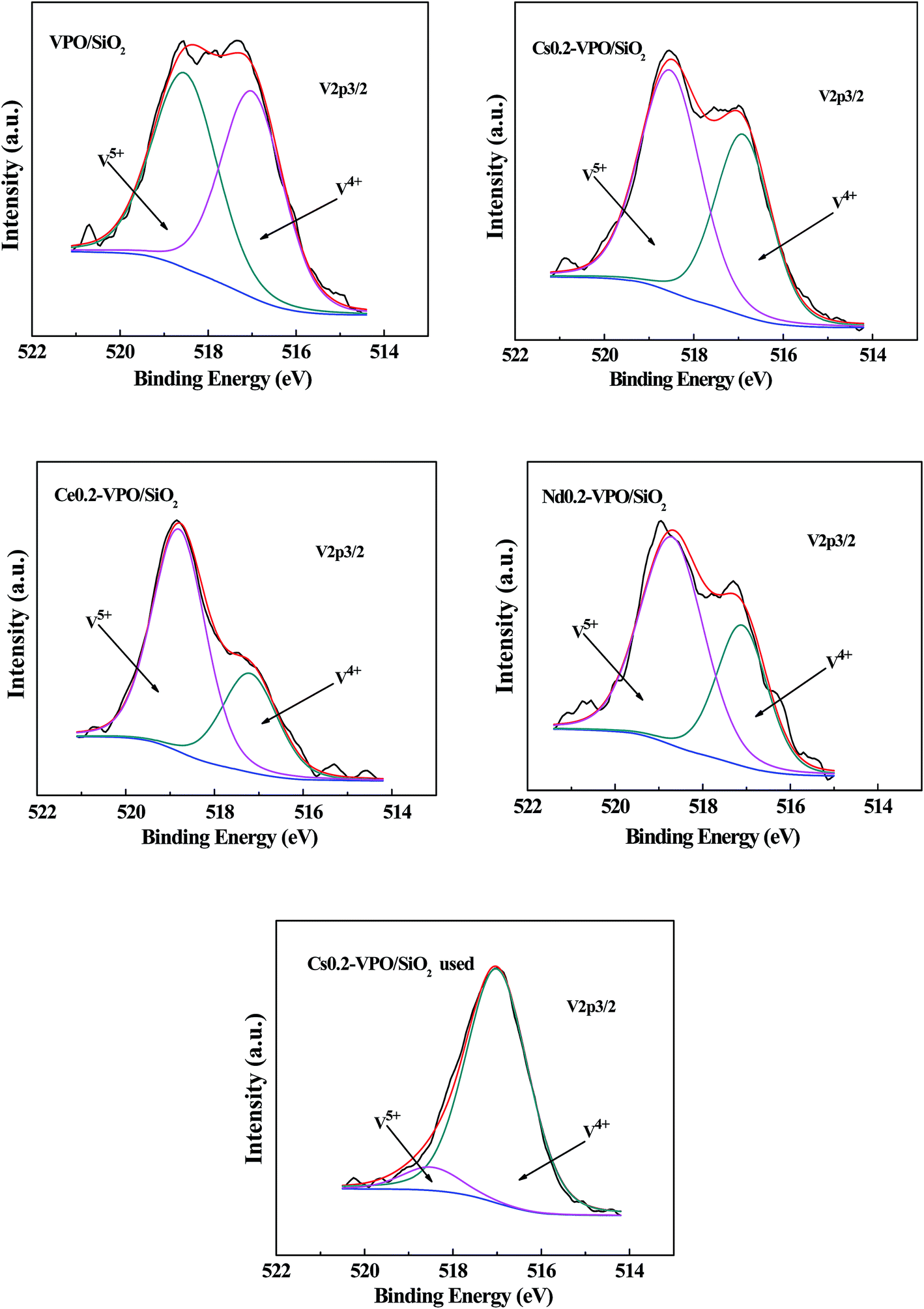 | ||
| Fig. 4 XPS spectra of V 2p3/2 of the VPO/SiO2, Cs0.2-VPO/SiO2, Nd0.2-VPO/SiO2, Ce0.2-VPO/SiO2, and used Cs0.2-VPO/SiO2 catalysts. | ||
3.3 N2 adsorption/desorption analysis
The specific surface areas and average pore diameters of the catalysts were determined by the N2 adsorption/desorption technique and the data are listed in Table 1. For the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts, their specific surface areas were 115.2, 70.9–104.1, 80.6–108.6, and 88.7–113.6 m2 g−1, respectively. Their average pore diameters were around 5 nm. The addition of metallic cations decreased the surface area and average pore size. It was reported that pure VPO catalyst has the specific surface area of ca. 30 m2 g−1.17 The silica aerogel support endowed the VPO catalysts with larger surface areas.3.4 CO2-TPD and NH3-TPD analyses
CO2-TPD and NH3-TPD analyses were conducted to compare the surface basicities and acidities of the VPO/SiO2 and metallic cation-modified VPO/SiO2 catalysts (Fig. 5 and 6). The basicities and acidities of the catalysts calculated according to their CO2 and NH3 desorption peak areas are listed in Table 1. The CO2-TPD profiles of the metallic cation-modified VPO/SiO2 catalysts show the CO2 desorption peaks appearing at ca. 690 °C, indicating that these catalysts had high base strength.17 The basicities of the metallic cation-modified VPO/SiO2 catalysts increased upon the increase in the metallic cation contents. Their basicities were in an order of Ce-VPO/SiO2 > Nd-VPO/SiO2 ≈ Cs-VPO/SiO2 > VPO/SiO2.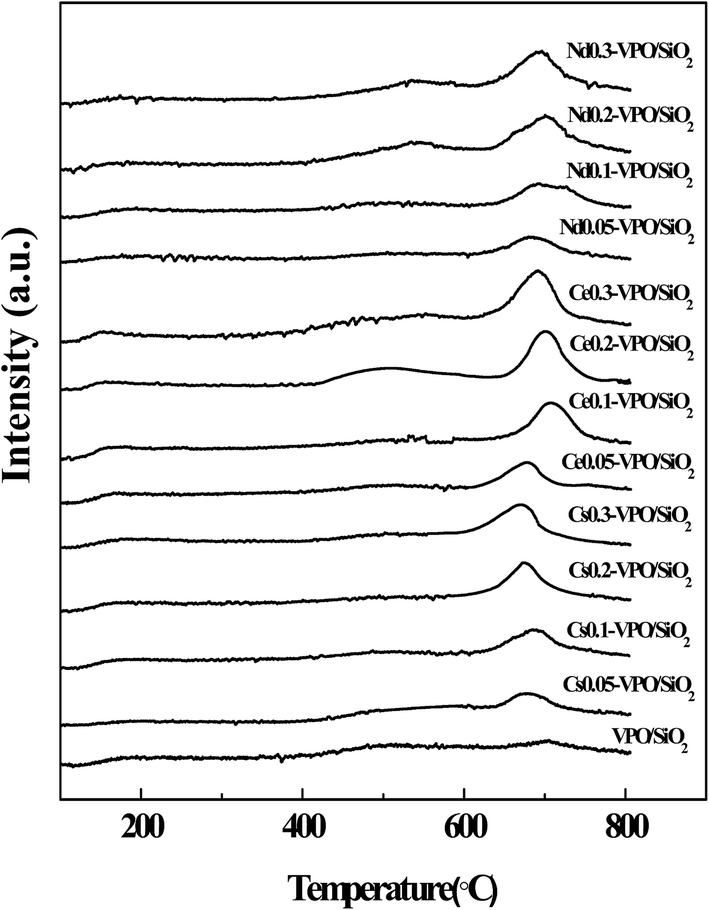 | ||
| Fig. 5 CO2-TPD profiles of the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts. | ||
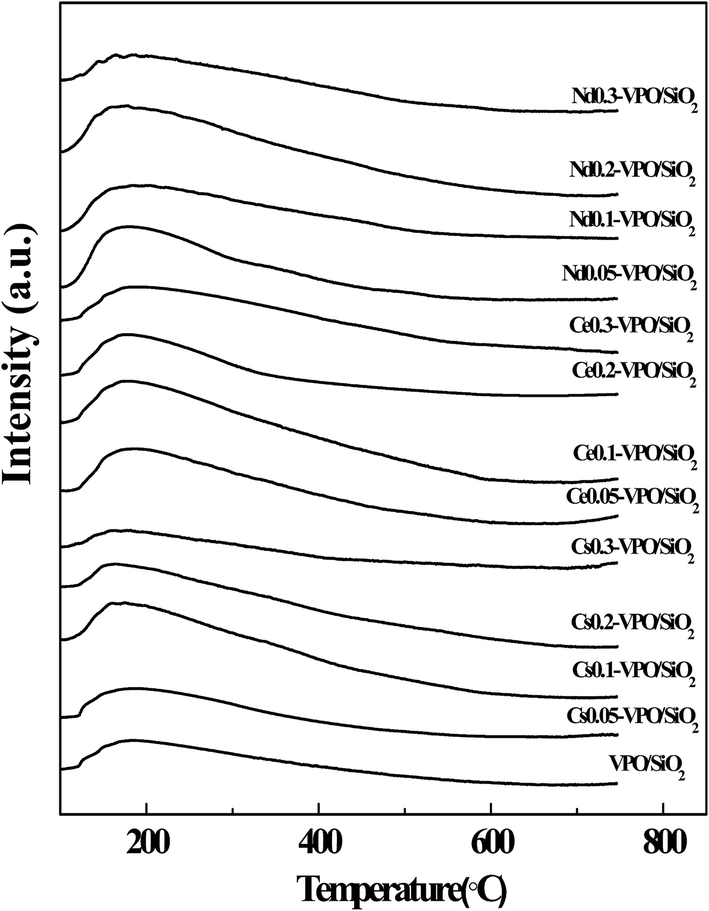 | ||
| Fig. 6 NH3-TPD profiles of the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts. | ||
The NH3-TPD profiles of the metallic cation-modified VPO/SiO2 catalysts show that the maximum NH3 desorption peaks appeared at ca. 180 °C. However, the temperatures of these NH3 desorption profiles were in a wide range of 100–600 °C, indicating that all the catalysts had weak-, mild- and strong acid sites.17,38 It was found that at lower ratios of metallic cation to vanadium ranging from 0.05 to 0.1 for Cs and Nd and from 0.05 to 0.2 for Ce, the metallic cation-modified VPO/SiO2 catalysts exhibited higher acidities than the VPO/SiO2 catalyst. Upon further increasing the metallic cation/vanadium ratio to 0.3, the acidities of the metallic cation-modified VPO/SiO2 catalysts decreased. The acidities of the metallic cation-modified VPO/SiO2 catalysts were comparable.
3.5 Catalytic performance
To evaluate the effect of the types of metal cations and their loadings on the aldol condensation of acetic acid with formaldehyde, a series of experiments were carried out at the reaction temperatures ranging from 320 to 400 °C. In our present work, acrylic acid was detected as the main product. Methyl acrylate, acetone, and CO2 were detected as the byproducts. The conversion of formaldehyde, the selectivity of acrylic acid, and the yield of acrylic acid based on formaldehyde were used as the parameters to evaluate the catalytic performances of the catalysts. The experimental results are shown in Fig. 7–9.When the reaction was carried out over the VPO/SiO2 and metallic cation-modified VPO/SiO2 catalysts, the formaldehyde conversion increased upon increasing the reaction temperature (Fig. 7a). Over the VPO/SiO2 catalyst, the conversion of formaldehyde increased to 58.7% with increasing the reaction temperature to 400 °C. All the metallic cation-modified VPO/SiO2 catalysts exhibited higher formaldehyde conversions than the VPO/SiO2 catalyst. It was found that the Cs0.2-VPO/SiO2, Ce0.1-VPO/SiO2, and Nd0.05-VPO/SiO2 catalysts exhibited high formaldehyde conversions in the Cs, Ce, and Nd cation-modified VPO/SiO2 catalysts, respectively. When the reaction temperature was above 320 °C, the formaldehyde conversions over the Cs0.2-VPO/SiO2, Ce0.05(0.1)-VPO/SiO2, and Nd0.05-VPO/SiO2 catalysts were more than 66%, 72%, and 66%, respectively. From among them, the Ce-VPO/SiO2 catalysts exhibited better catalytic activity for the conversion of formaldehyde in a wide range of Ce/V ratios. The types and contents of metallic cations present in the metallic cation-modified VPO/SiO2 catalysts affected their catalytic activities for the conversion of formaldehyde.
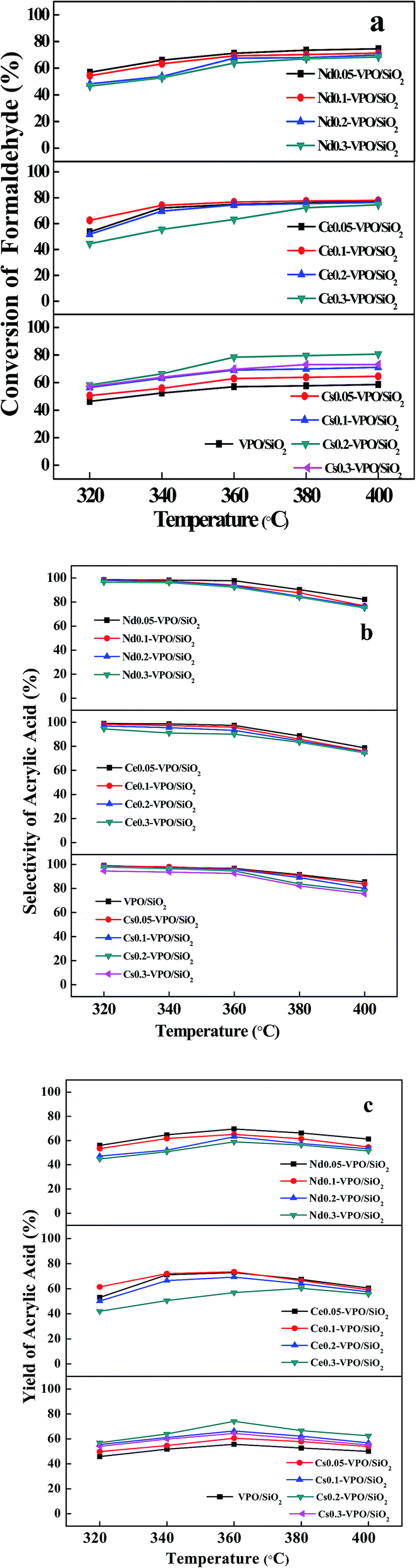 | ||
| Fig. 7 (a) Conversion of formaldehyde, (b) selectivity of acrylic acid, and (c) yield of acrylic acid over the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts. The acrylic acid selectivity and yield were based on formaldehyde. | ||
The surface acidity and basicity analyses showed that the Cs0.2-VPO/SiO2, Ce0.05(0.1)-VPO/SiO2, and Nd0.05-VPO/SiO2 catalysts had high acidities, revealing that high acidity favored the conversion of formaldehyde. Furthermore, it was found that the Ce-VPO/SiO2 catalysts had higher basicities than the Cs-VPO/SiO2 and Nd-VPO/SiO2 catalysts. Meanwhile, the former exhibited higher catalytic activity than the latter. The result revealed that the basicity of the catalyst also played an important role in the reaction.
The selectivities of acrylic acid over the VPO/SiO2 and metallic cation-modified VPO/SiO2 catalysts are shown in Fig. 7b. With increasing the reaction temperatures from 320 to 400 °C, the selectivities of acrylic acid over the VPO/SiO2 catalyst decreased from 98.9% to 85.4%. At 320 °C, the selectivities of acrylic acid over the Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts slightly decreased from 98.6% to 94.4%, 98.9% to 94.4%, and 98.5% to 96.4%, respectively, upon increasing the ratios of metallic cation to vanadium from 0.05 to 0.3. At 400 °C, the selectivities of acrylic acid decreased from 83.6% to 75.4%, 78.6% to 74.6%, and 82.1% to 75.0%. The addition of a larger amount of metallic cation in VPO/SiO2 catalyst obviously caused the decrease in acrylic acid selectivity. When the Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts had the same metallic cation content, they exhibited comparable acrylic acid selectivity.
The acrylic acid yield was also used as a parameter to evaluate the catalytic performance of the metallic cation-modified VPO/SiO2 catalyst (Fig. 7c). For the VPO/SiO2 catalyst, the yields of acrylic acid were less than 56% at the reaction temperatures ranging from 320 to 400 °C. When the Cs-VPO/SiO2 catalysts with the Cs/V ratios of 0.1–0.2 were used at the reaction temperatures of 340–380 °C, the yields of acrylic acid ranged from 61.1% to 74.2%. When the Ce-VPO/SiO2 catalysts with the Ce/V ratios of 0.05–0.2 were used at the reaction temperatures of 340–380 °C, the yields of acrylic acid ranged from 63.9% to 73.9%. When the Nd-VPO/SiO2 catalysts with the Nd/V ratios of 0.05–0.1 were used at the reaction temperatures of 340–380 °C, the yields of acrylic acid ranged from 61.5% to 69.5%. The results showed that the acrylic acid yields were dependent on both catalyst composition and reaction temperature. The Ce-VPO/SiO2 catalysts with a wider range of Ce/V ratios exhibited better catalytic activity for the aldol condensation of acetic acid with formaldehyde to acrylic acid at a wider range of reaction temperatures. The maximum formation rate of acrylic acid over the Ce-VPO/SiO2 catalyst was 10.1 mmol gcat−1 h−1, which was higher than that over the VPO/SiO2 catalyst.
When the VPO/SiO2 was used as the catalyst, with increasing the reaction temperature to 400 °C, the methyl acrylate selectivity increased to 2.4% (Fig. 8). All the metallic cation-modified VPO/SiO2 catalysts exhibited similar catalytic activity toward the formation of methyl acrylate. The maximum methyl acrylate selectivity over these catalysts was ca. 3.0%.
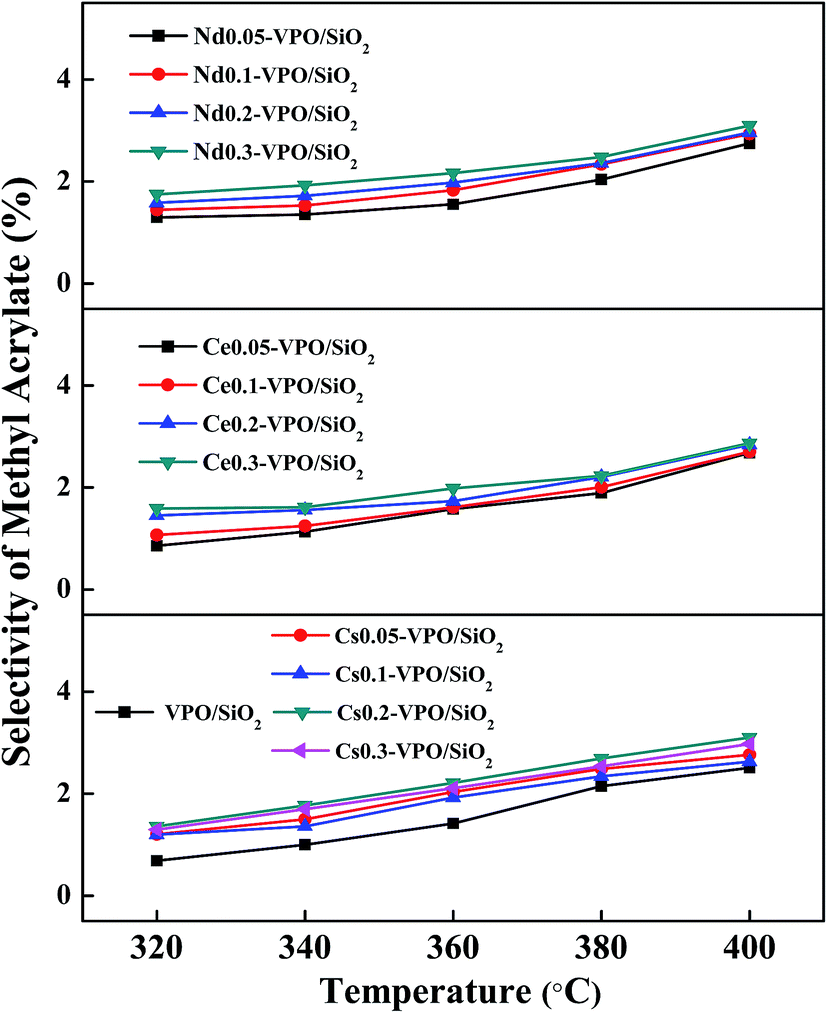 | ||
| Fig. 8 Selectivity of methyl acrylate over the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts. Methyl acrylate selectivity was based on formaldehyde. | ||
Acetone and CO2 were formed by the bimolecular dehydration of acetic acid.17,39
| 2CH3COOH = CH3COCH3 + CO2 + H2O | (6) |
The acetone yields based on acetic acid over different catalysts are shown in Fig. 9a. The acetone yields increased with the increase in the ratios of metallic cation to vanadium and the reaction temperatures. The maximum acetone yields of 2.5%, 4.2%, 3.6%, and 4.3% were obtained over the VPO/SiO2, Cs0.3-VPO/SiO2, Ce0.3-VPO/SiO2, and Nd0.3-VPO/SiO2 catalysts, respectively. The presence of the phosphate salts in the catalysts had a little effect on the dehydration of acetic acid to acetone.
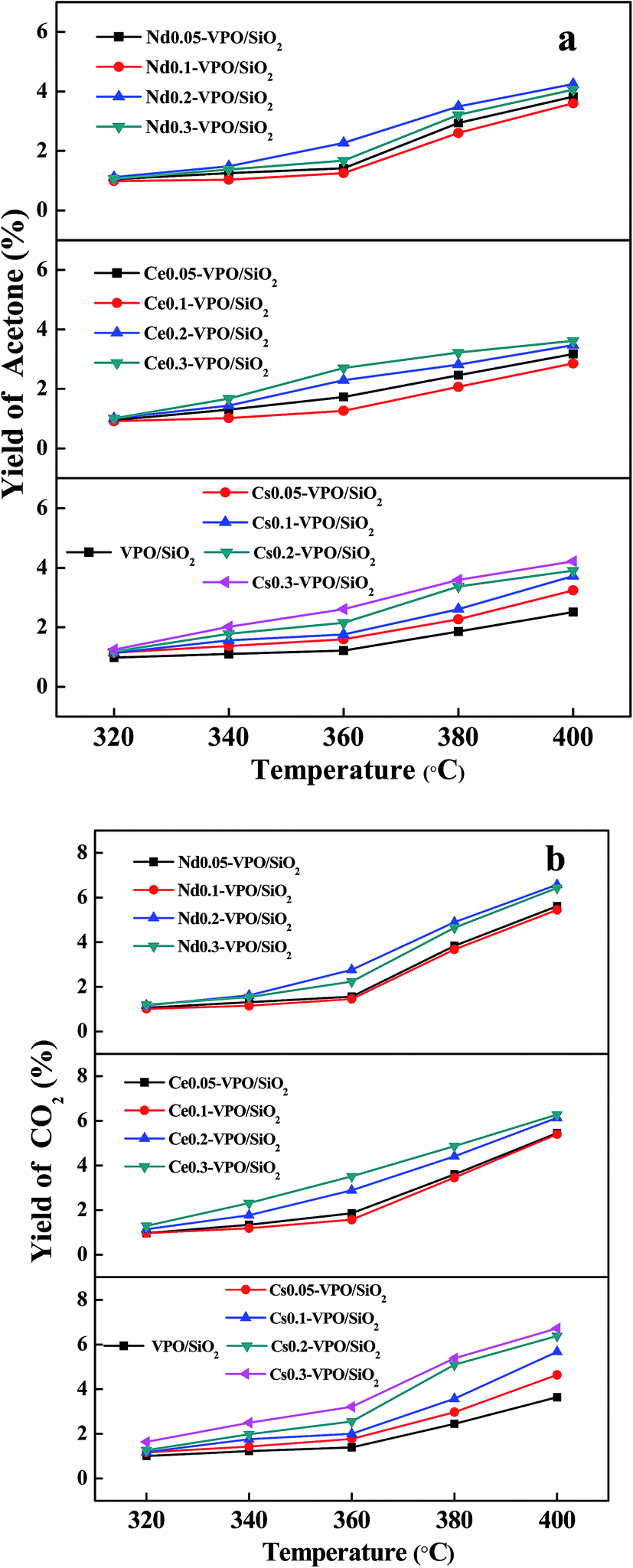 | ||
| Fig. 9 Yields of (a) acetone and (b) CO2 over the VPO/SiO2, Cs-VPO/SiO2, Ce-VPO/SiO2, and Nd-VPO/SiO2 catalysts. Yields of acetone and CO2 were based on acetic acid. | ||
The CO2 yields based on acetic acid over different catalysts are shown in Fig. 9b. The CO2 yields increased with the increase in the ratios of metallic cation to vanadium and the reaction temperatures. The maximum CO2 yields of 3.5%, 6.7%, 6.3%, and 6.6% were obtained over the VPO/SiO2, Cs0.3-VPO/SiO2, Ce0.3-VPO/SiO2, and Nd0.3-VPO/SiO2 catalysts, respectively.
It was found that the mole ratios of CO2 to acetone were larger than 1, indicating that CO2 was also formed through the decomposition of formaldehyde according to the following reactions.17,40
| 2CH2O + H2O = CHOOH + CH3OH | (7) |
| CHOOH = CO2 + H2 | (8) |
4. Conclusions
The VPO/SiO2 and Cs-, Ce-, and Nd-modified VPO/SiO2 catalysts were prepared by the deposition method. The VPO/SiO2 catalyst was composed of VOPO4 and (VO)2P2O7 phases. When Cs, Ce, and Nd cations were added in VPO/SiO2 catalyst, Cs4P2O7, CePO4, and NdPO4 were formed, respectively. When Nd cation was added in the catalyst, (VO)2P2O7 phase disappeared while a new αII-VOPO4 phase was formed. The addition of the metallic actions in the catalysts increased their basicities while their acidities increased at a lower ratio of metallic cation to vanadium. The presence of the metallic cations also increased the V5+/V4+ ratios.The Cs-, Ce-, and Nd-modified VPO/SiO2 catalysts exhibited higher catalytic activities for the conversion of formaldehyde, giving higher acrylic acid yield than the VPO/SiO2 catalyst. The Ce-VPO/SiO2 catalysts with a wider Ce/V ratio exhibited good catalytic activity for the aldol condensation of acetic acid with formaldehyde to acrylic acid at a wider range of reaction temperatures. The Ce-VPO/SiO2 catalyst may have commercial application considering that the acrylic acid yield was up to 73.9% and the maximum formation rate of acrylic acid was 10.1 mmol gcat−1 h−1, higher than those over the pure VPO, VPO/SiO2, and alkali hydroxide catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21506078 and 21506082), Research Foundation of Jiangsu University (15JDG026), China Postdoctoral Science Foundation (2016M591786 and 2016M601739), and Jiangsu Planned Projects for Postdoctoral Research Funds (1601084B).References
- X. Zhang, L. Lin, T. Zhang, H. Liu and X. Zhang, Chem. Eng. J., 2016, 284, 934–941 CrossRef CAS.
- G. Näfe, M. López-Martínez, M. Dyballa, M. Hunger, Y. Traa, T. Hirth and E. Klemm, J. Catal., 2015, 329, 413–424 CrossRef.
- C. Peterson, J. Chapman and J. Gallacher, US Pat., 8864950 B2, 21 Oct 2014.
- C. Peterson and J. Chapman, US Pat., 20130085303 A1, 4 Apr 2013.
- H. S. Chu, J. H. Ahn, J. Yun, I. S. Choi, T. W. Nam and K. M. Cho, Metab. Eng., 2015, 32, 23–29 CrossRef CAS PubMed.
- X. Suo, H. Zhang, Q. Ye, X. Dai, H. Yu and R. Li, Chem. Eng. Res. Des., 2015, 104, 346 CrossRef CAS.
- A. N. Parvulescu, A. Lange de Oliveira, S. A. Schunk, N. T. Woerz, M. Hartmann, K. Amakawa, M. Goebel, Y. Liu and M. Lejkowski, US Pat., 20150344394 A1, 3 Dec 2015.
- B. Yan, A. Mahmood, Y. Liang and B.-Q. Xu, Catal. Today, 2016, 269, 65–73 CrossRef CAS.
- F. Wang, J. Dubois and W. Ueda, Appl. Catal., A, 2010, 376, 25–32 CrossRef CAS.
- R. Nebesnyi, East.-Eur. J. Enterp. Technol., 2015, 1, 13–16 CrossRef.
- J. F. Vitcha and V. A. Sims, Ind. Eng. Chem. Prod. Res. Dev., 1966, 5, 50–53 CrossRef CAS.
- M. Ai, Appl. Catal., A, 2005, 288, 211–215 CrossRef CAS.
- J. Yan, C. Zhang, C. Ning, Y. Tang, Y. Zhang, L. Chen, S. Gao, Z. Wang and W. Zhang, J. Ind. Eng. Chem., 2015, 25, 344–351 CrossRef CAS.
- M. Ai, J. Catal., 1987, 107, 201–208 CrossRef CAS.
- X. Feng, B. Sun, Y. Yao, Q. Su, W. Ji and C. Au, J. Catal., 2014, 314, 132–141 CrossRef CAS.
- D. Yang, C. Sararuk, K. Suzuki, Z. Li and C. Li, Chem. Eng. J., 2016, 300, 160–168 CrossRef CAS.
- J. Hu, Z. Lu, H. Yin, W. Xue, A. Wang, L. Shen and S. Liu, J. Ind. Eng. Chem., 2016, 40, 145–151 CrossRef CAS.
- V. A. Zazhigalov, J. Haber, J. Stoch, I. V. Bacherikova, G. A. Komashko and A. I. Pyatnitskaya, Appl. Catal., A, 1996, 134, 225–237 CrossRef CAS.
- B. T. Pierini and E. Lombardo, Mater. Chem. Phys., 2005, 92, 197–204 CrossRef CAS.
- X. Wang, L. Xu, X. Chen, W. Ji, Q. Yan and Y. Chen, J. Mol. Catal. A: Chem., 2003, 206, 261–268 CrossRef CAS.
- B. T. Pierini and E. A. Lombardo, Catal. Today, 2005, 107–108, 323–329 CrossRef CAS.
- V. Zazhigalov, I. Bacherikova, E. Stokh, G. Komashko and A. Pyatnitskay, Theor. Exp. Chem., 1994, 30, 65–68 CrossRef.
- C. Carrara, S. Irusta, E. Lombardo and L. Cornaglia, Appl. Catal., A, 2001, 217, 275–286 CrossRef CAS.
- L. Cornaglia, S. Irusta, E. Lombardo, M. Durupty and J. Volta, Catal. Today, 2003, 78, 291–301 CrossRef CAS.
- T. Shimoda, T. Okuhara and M. Misono, Bull. Chem. Soc. Jpn., 1985, 58, 2163–2171 CrossRef CAS.
- F. Trifirò and R. Grasselli, Top. Catal., 2014, 57, 1188–1195 CrossRef.
- J. Frey, C. Lieder, T. Schölkopf, T. Schleid, U. Nieken, E. Klemm and M. Hunger, J. Catal., 2010, 272, 131–139 CrossRef CAS.
- W. E. Morgan, J. R. Van Wazer and W. J. Stec, J. Am. Chem. Soc., 1973, 95, 751–755 CrossRef CAS.
- J. M. Pemba-Mabiala, M. Lenzi, J. Lenzi and A. Lebugle, Surf. Interface Anal., 1990, 15, 663–667 CrossRef CAS.
- Z. Z. Wang, X. W. Tao, X. B. Zhang, Z. X. Ba and Q. Wang, Mater. Technol., 2015, 30, 321–326 CrossRef CAS.
- F. Rivera-López and M. Pérez, Surf. Interface Anal., 2012, 44, 927–930 CrossRef.
- H. Guan and Y. Zhang, J. Solid State Chem., 2004, 177, 781–785 CrossRef CAS.
- L. Cornaglia and E. Lombardo, Appl. Catal., A, 1995, 127, 125–138 CrossRef CAS.
- L. Shen, H. Yin, A. Wang, X. Lu and C. Zhang, Chem. Eng. J., 2014, 244, 168–177 CrossRef CAS.
- L. M. Cornaglia and E. A. Lombardo, J. Phys.: Condens. Matter, 1993, 5, A225–A226 CrossRef.
- X.-K. Li, W.-J. Ji, J. Zhao, Z.-B. Zhang and C.-T. Au, J. Catal., 2006, 238, 232–241 CrossRef CAS.
- H. Igarashi, K. Tsuji, T. Okuhara and M. Misono, J. Phys. Chem., 1993, 97, 7065–7071 CrossRef CAS.
- F. Jing, Y. Zhang, S. Luo, W. Chu, H. Zhang and X. Shi, J. Chem. Sci., 2010, 122, 621–630 CrossRef CAS.
- F. C. Calaza, T.-L. Chen, D. R. Mullins, Y. Xu and S. H. Overbury, Catal. Today, 2015, 253, 65–76 CrossRef CAS.
- X. D. Peng and M. A. Barteau, Langmuir, 1989, 5, 1051–1056 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |