
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the excited-state double proton transfer mechanisms of doxorubicin in acetonitrile solvent†
Xiaoyan Liu,
Jinfeng Zhao and
Yujun Zheng *
*
School of Physics, Shandong University, Jinan 250100, China. E-mail: yzheng@sdu.edu.cn
First published on 3rd November 2017
Abstract
Doxorubicin (DXR), having double intramolecular hydrogen bonds, is theoretically investigated with an aim to explore the excited-state intramolecular double proton transfer (ESIDPT) mechanism regarding stepwise versus synchronous double proton transfer. The changes in the calculated primary bond lengths and bond angles testify that the intramolecular hydrogen bonds (O1–H2⋯O3 and O4–H5⋯O6) are both strengthened in the S1 state (ca. the S0 state), which provides the possibility for the ESIDPT reaction. We adopt the TDDFT and CIS methods coupled with transition state (TS) theory to further elucidate the ESIDPT mechanism. The potential energy surfaces (PESs) are successfully constructed using different basis sets. Three kinds of stable structure are found on the S1 PES. Herein, we establish a new elaborated ESIDPT mechanism for DXR. This new mechanism unambiguously excludes synchronous double proton transfer, while the stepwise double proton transfer mechanism is theoretically confirmed. This systematic investigation into the photophysical properties of the antineoplastic drug DXR may be significant in obtaining fundamental insight into the transport mechanism of DXR inside cultured cells.
1 Introduction
Ever since Weller’s first report on excited-state intramolecular proton transfer (ESIPT) reactions based on salicylic acid and its derivatives,1 excited-state intramolecular reaction dynamics are overwhelmingly studied both experimentally and theoretically.2–5 In particular, the proton transfer reaction triggers enormous interest due to the property of charge redistribution, which can be applied to molecular switches,6,7 laser dyes,8,9 chemosensors,10,11 UV-photostabilizers,12,13 molecular recognition,14,15 and so on. Moreover, the tautomer generally exhibits a large Stokes shifted emission, which is essential for the fabrication of organic light-emitting devices.16–18 From a molecular point of view, proton transfer can, in general, be divided into intermolecular and intramolecular cases. ESIPT, compared with the former, takes place on a unimolecular basis which avoids bimolecular complicacy, most intermolecular interactions, and most entropic effects. The prerequisite for ESIPT lies in the formation of intramolecular hydrogen bonds. Recent advances reported by Han and co-workers show that excited-state hydrogen bond behaviour plays a vital role in photochemical reactions (e.g. fluorescence quenching, spectral shifts, excited-state proton transfer, etc.).19–21 For example, Zhao et al. systematically demonstrate that the intermolecular hydrogen bond is observably strengthened in the excited-state using coumarin 102 and phenol as a hydrogen bonding (H-bonding) prototype.22 This strong intramolecular hydrogen bond provides the possibility for an ESIPT reaction.Most ESIPT reactions involve a single-proton transfer.23–25 However, in biological systems, there exist many double- or multiple-proton transfer cases. In these cases, the single-proton transfer case is not sufficient to mimic the aforementioned proton transfer reactions. Unfortunately, to the best of our knowledge, reports referring to the ESIDPT mechanism of biological systems containing two protons are sparse.3–5 Anthracycline antitumor antibiotics (ACAAs), which are glycosides produced by actinomycetes, are clinically one of the most widely used anticancer drugs. An ACAA is an anthracene ring attached to a six-membered ring-based side chain and an aminosaccharide. It can intercalate into DNA double-stranded polynucleotides, increasing the distances between adjacent DNA base pairs and causing DNA fragmentation.26 Doxorubicin (DXR), a member of the ACAA family, is a topoisomerase II (Top 2)-targeting drug, which is one of the most effective anticancer drugs used in clinical applications.27 However, DXR-based chemotherapy can increase the risk of developing cardiac toxicities, including cardiomyopathy and congestive heart failure.27–29 Over the years, considerable efforts have focused on devising a drug delivery system that can eliminate these adverse affects, including liposome, hydrogel and nanoparticle (NP) systems.30–32 For instance, Nowicka et al. report magnetic ferrite nanoparticles (NPs) which are of a relatively low toxicity.33 The experimental results indicate that after binding to the magnetic NPs, the DXR molecules are still capable of intercalating the dsDNA helices. That is to say, the DXR-NP system can improve survival and efficacy in vivo. Herein, the fluorescence characteristic of DXR-delivery systems acquires extensive attention.34,35 Unfortunately, studies into the photophysical properties of DXR are sparse. Sturgeon and Schulman observe the protolytic equilibria of DXR at different pH values through spectrophotometric determination.36 Subsequently, the phenomenon of intramolecular double proton transfer in DXR was systematically investigated for the first time by Kumar and co-workers.37 Kumar et al. experimentally observe dual fluorescence bands, and prove theoretically a hypothesis about the ESIDPT mechanism of DXR.
Considering this ambiguous and deficient mechanism, we report the comprehensive photophysical properties and ESIDPT mechanisms for DXR in acetonitrile in this paper. On the one hand, DXR has 4 six-membered rings. One of them is a nonplanar, nonaromatic ring attached to an aminoglycosidic side chain. Also, the light-absorbing moiety (i.e. the chromophore) is three planar and aromatic hydroxy anthraquinonic rings. The presence of multiple functional groups makes DXR show excellent photophysical properties. On the other hand, DXR possesses a hydroxyl group and a carbonyl oxygen in close proximity, which promotes the formation of intramolecular double hydrogen bonds. Interestingly, the results of this study reveal that the ESIDPT reaction can only occur in a stepwise manner. Details of the results and discussions are elaborated as follows.
2 Computational details
The structures of doxorubicin (DXR) and its tautomers in acetonitrile (ACN) are optimized by the long-range-corrected (LRC) functional CAM-B3LYP38 with the 6-311+G(d,p) basis set for the ground-state (S0) and by the time-dependent (TD) functional CAM-B3LYP with the 6-311+G(d,p) basis set for the first singlet excited-state (S1). The vertical S0 → S1 excitation and S1 → S0 emissions are calculated by time-dependent density functional theory (TD-DFT) at the CAM-B3LYP/6-311+G(d,p) level. Vibrational frequency calculations of the optimized structures are carried out specifically to see if any imaginary frequency is obtained. The solvation effects in ACN are considered using the integral equation formalism variant of the polarizable continuum model (IEFPCM).39 All of the single-point energies, NPA charges and ESP are calculated based on the CAM-B3LYP/6-311+G(d,p) method. Furthermore, the above CAM-B3LYP/6-311+G(d,p) calculations are checked by full calculations using the ωB97XD40 functional with a 6-311+G(d,p) basis set.The potential energy curves (PECs) of the S1 state are calculated by setting the bond distances of the corresponding O–H to a series of various values while the other atoms are relaxed. The PECs thus-obtained give a clearer picture of the stepwise nature of this ESIDPT reaction. The excited-state stepwise mechanism is confirmed by the S1 PEC calculations at the ωB97XD/6-311+G(d,p) level. In addition, transition state (TS) theory is also applied to verify the stepwise ESIDPT process. Firstly, we search the TS structures in the S1 state using the CIS/6-311+G(d,p) method, starting from an educated guess based on placing the hydrogen atom being transferred between the hydrogen donor and acceptor, and on adjusting the bond lengths affected by the proton transfer. Then, Hessian calculations are carried out at the guessed TS structure to obtain the force constants.41 By the Berny optimization method, our obtained TS structures are confirmed to be only one imaginary frequency, and its vibrational eigenvector points in the correct direction. All the theoretical calculations are carried out using the Gaussian 09 program.42 The above methods are successfully accomplished to elaborate the ground-state and excited-state proton transfer processes.43–46
3 Results and discussion
3.1 Structural characterizations
Based on the DFT and TD-DFT methods for the S0 and S1 states, four stable structures are found, i.e., the normal form (DXR-N), the single proton transfer forms (DXR-AS and DXR-BS), and the double proton transfer form (DXR-D) for the DXR molecule. Fig. 1 shows the optimized structures of DXR and its proton-transfer tautomers, while the corresponding structural parameters are summarized in Table 1. It should be noted that the calculated bond lengths of O1–H2, H2⋯O3, O4–H5 and H5⋯O6 of the DXR-N form are 0.999, 1.649, 1.002 and 1.618 Å in the S0 state, respectively. Upon excitation to the S1 state, these bond lengths change to be 1.024, 1.559, 1.035 and 1.503, respectively. Obviously, the hydrogen bonds (H2⋯O3 and H5⋯O6) are shortened, which indicates that these two intramolecular bonds (i.e., O1–H2⋯O3 and O4–H5⋯O6) are strengthened in the S1 state. And δ(O1–H2⋯O3) changes from 147.8° in the S0 state to 152.3° in the S1 state. Herein, the bond angle in the S1 state is closer to 180° than it is in the S0 state. In addition, in view of the DXR-D form, the shortening of the lengths of the O1⋯H2 and O4⋯H5 bonds with the concomitant enlargement of the O1–H2–O3 and O4–H5–O6 angles indicates that the two intramolecular hydrogen bonds (i.e., O1⋯H2–O3 and O4⋯H5–O6) are more stable in the S1 state than the S0 state. It is not strange that some of the proton-transfer tautomers exist only in the S1 state but not in the S0 state if the normal form or other tautomers are much more stable in S0 state. For the single proton transfer forms (DXR-AS and DXR-BS), only the stable structures exist in the S1 state (details vide infra).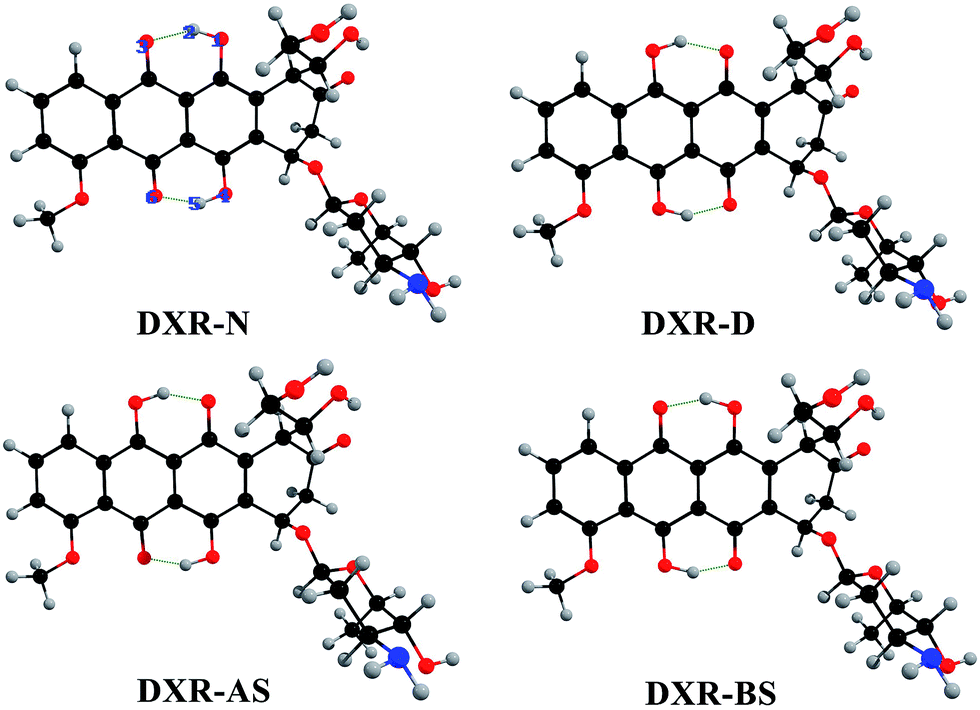 | ||
| Fig. 1 Optimized structures of the DXR, DXR-AS, DXR-BS and DXR-D forms at the CAM-B3LYP/6-311+G(d,p)/IEF-PCM(acetonitrile) theoretical level. Red: O; gray: H; blue: N; and black: C. | ||
| DXR-N | DXR-D | DXR-AS | DXR-BS | |||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S1 | S1 | |
| O1–H2 | 0.999 | 1.024 | 1.623 | 1.603 | 1.572 | 1.019 |
| H2⋯O3 | 1.649 | 1.559 | 1.006 | 1.011 | 1.021 | 1.572 |
| O4–H5 | 1.002 | 1.035 | 1.584 | 1.573 | 1.029 | 1.535 |
| H5⋯O6 | 1.618 | 1.503 | 1.010 | 1.013 | 1.524 | 1.024 |
| δ(O1–H2···O3) | 147.8° | 152.3° | 149.0° | 150.6° | 151.9° | 151.8° |
| δ(O4–H5···O6) | 148.7° | 153.7° | 151.7° | 152.7° | 152.9° | 154.0° |
3.2 Electronic spectra and charge distributions
It is known that B3LYP can be quite accurate for aromatic molecules, but it can sometimes lose efficacy when a charge-transfer (CT) state is involved.47 Recently, a series of so-called LRC functionals were widely used to investigate the excited-state properties of large molecules.48,49 Hence, absorption and emission spectra of DXR simulated at both the B3LYP and CAM-B3LYP functionals are shown in Fig. S1 (ESI†) and are compared with the experimental results.37For the absorption spectra of DXR, the simulated result with the B3LYP functional is in good agreement with the experimental absorption band localized at 475 nm, while the peaks calculated with the CAM-B3LYP functional are slightly blue-shifted (see Fig. S1†). This could be caused by the LRC, which can avoid underestimating the energy of the excited CT state but also raises the energies of the non-CT state. When the ESIPT reaction is associated with CT, the B3LYP functional predictably loses efficacy.49 The fluorescence peak of DXR-D is calculated as 680 nm at the B3LYP level, which is great different to the 575 nm in the experimental spectrum. However, the fluorescence peak at 577 nm simulated by CAM-B3LYP agrees well with the experimental result (Fig. S1†). Although the peaks in the absorption and emission spectra of DXR are slightly different from the experimental spectra, CAM-B3LYP would clearly be the better choice in this work.
In addition, we calculate the fluorescence peaks of the single and double proton-transfer tautomers based on the TD-CAM-B3LYP method, which are at 578, 572 and 577 nm, respectively (Fig. 2). Note that the peak values (i.e., 578, 572 and 577 nm) may all correspond to the experimental value 575 nm. In the experimental study, Kumar et al. report that the 575 nm peak is attributed to the double proton-transfer form DXR-D.37 Furthermore, the electronic spectra simulated at the ωB97XD/6-311+G(d,p) level are consistent with the above results (Fig. S2, ESI†). According to our simulation, because of the tiny difference between the three fluorescence peaks, it is hard to distinguish and attribute to the DXR-AS, DXR-BS or DXR-D forms. Herein, we expect that additional experimental study will corroborate or refute our computed result.
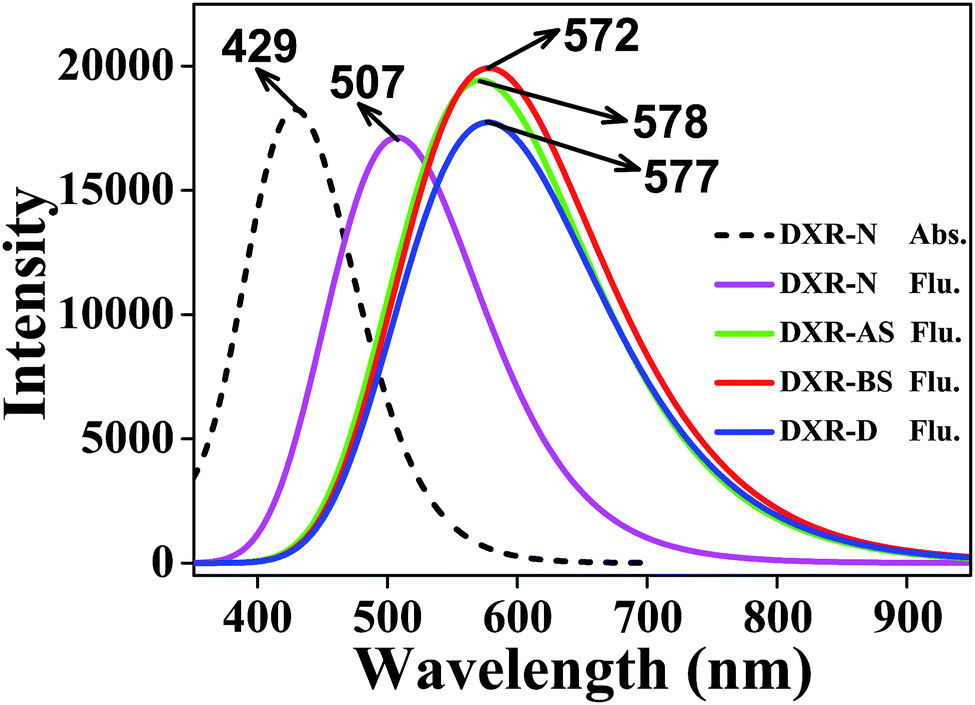 | ||
| Fig. 2 The absorption and fluorescence spectra of DXR in acetonitrile solvent. | ||
The above spectroscopic results lead to two cases. One is that the potential barrier between DXR and DXR-D is too high to cross, so that only the stepwise ESIDPT occurs, generating two intermediates (i.e. DXR-AS and DXR-BS). The other case is that the potential barrier between these two forms is not too high to cross, so that the ESIDPT reaction can take place in a synergic manner. Which pathway is more dominant? We will elaborate in the following discussion about the potential energy curves.
Furthermore, the electron density difference (EDD) maps between the S0 and S1 states as well as the HOMO and LUMO maps involved in the S0 → S1 transition of DXR-N and DXR-D systems in acetonitrile solvent are plotted in Fig. 3. Table 2 indicates that the S0 → S1 transition is of the HOMO → LUMO type. The EDD map of DXR-N shows that there is a net electron density shift from the hydroxyl group to the carbonyl oxygen, which facilitates the ESIDPT reactions. The results obtained from the HOMO–LUMO maps clearly indicate that upon excitation of DXR, the hydroxyl proton is expected to be more acidic, whereas the carbonyl oxygen is more basic (ca. the S0 state), driving the ESIDPT reactions.
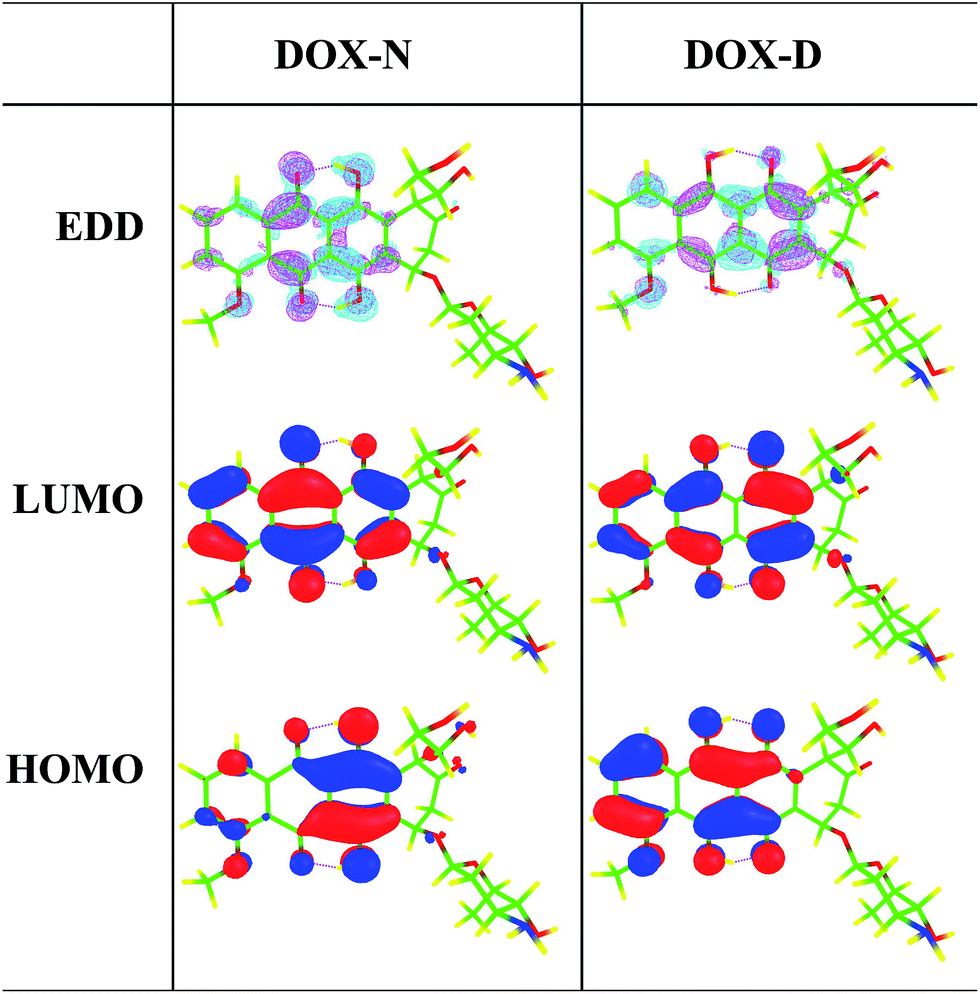 | ||
| Fig. 3 Calculated electron-density difference (EDD) map between the S0 and S1 states and the HOMO–LUMO maps for DXR-N and DXR-D systems in acetonitrile solvent. In the EDD maps, from S0 to S1, the regions with increasing electron density are shown in magenta, whereas the regions with decreasing density are shown in cyan. | ||
| Transition | λ (nm) | f | Composition | CI (%) | |
|---|---|---|---|---|---|
| DXR | S0 → S1 | 429.9 | 0.4472 | H → L | 97.45 |
| S0 → S2 | 358.1 | 0.0002 | H-10 → L+1 | 3.99 | |
| H-6 → L | 60.77 | ||||
| H-6 → L+1 | 2.15 | ||||
| H-5 → L | 22.60 |
In addition, the electrostatic potential (ESP) and natural bond orbital (NBO) analyses for DXR-N and DXR-D are obtained for a preliminary prediction of the potential H-bonding sites. As plotted in Fig. 4, for DXR-N and DXR-D, the positive charge zones mostly focus on the H atoms of the hydroxyl group, which indicates that they are clearly a series of H-bonding donating sites. The negative charge zones are mainly located near the O atoms of the hydroxyl groups and carbonyl groups, which demonstrates that these regions will be the potential H-bonding accepting sites (i.e. binding sites). For DXR-N, NPA charges depict that the H atoms on the hydroxyl groups (H2 and H5) possess much larger positive charges than the other hydrogen atoms, which further demonstrates that H2⋯O3 and H5⋯O6 may form strong intramolecular hydrogen bonds. Similar results are also reflected at the ωB97XD/6-311+G(d,p) level (Fig. S3 and S4, ESI†).
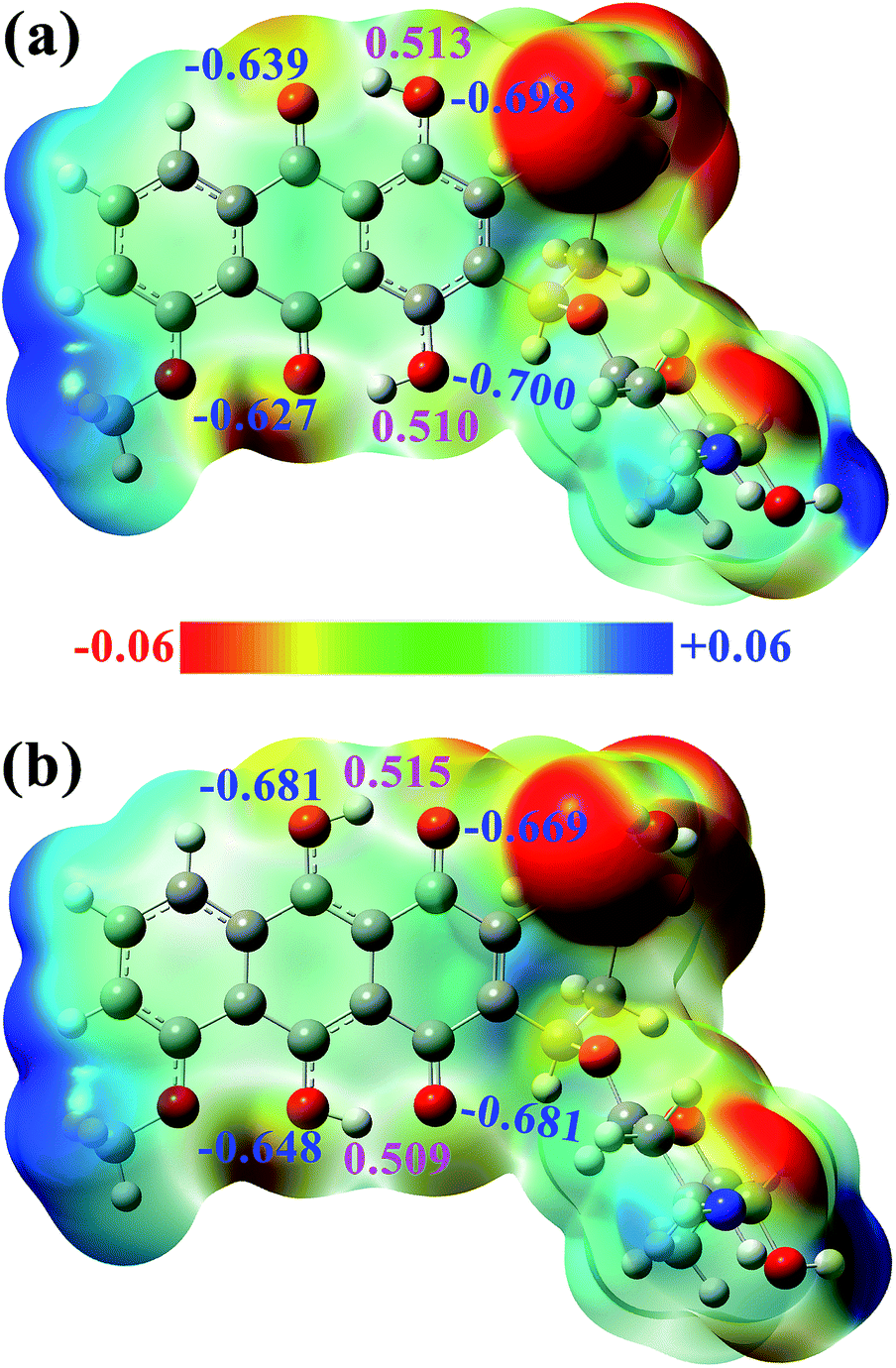 | ||
| Fig. 4 ESP of the (a) DXR-N and (b) DXR-D forms, NPA charges of the selected atoms are given in blue and magenta fonts. | ||
3.3 Mechanism analyses
To explore whether single proton transfer (SPT) or double proton transfer (DPT) happens, and to further reveal the detailed mechanism of the ESIDPT process of DXR in acetonitrile, the S0 and S1 state geometrical structures are optimized with fixed O1–H2 and O4–H5 bond lengths at the CAM-B3LYP/TD-CAM-B3LYP levels. Herein, we construct the PECs of the S1 state, as shown in Fig. 5. The potential barriers among the minima in the S1 state are calculated as follows: 0.00 kcal mol−1 is needed to cross from point A to point C and from point C to point B 2.02 kcal mol−1 is needed; 0.46 kcal mol−1 is needed to cross from point A to point D and from point D to point B 1.07 kcal mol−1 is needed; and an 11.03 kcal mol−1 potential barrier separates point A from point B. It can be seen clearly that compared with the SPT processes, the DPT process needs to cross a higher barrier (>11 kcal mol−1). That is to say, the synchronous ESIDPT is not possible. Subsequently, we simulate the PESs using the different functionals B3LYP and ωB97XD, as shown in Fig. S5 and S6 (ESI†), while the corresponding data are summarized in Table 3. Obviously, whether it is the SPT or the DPT processes, there exist high barriers (>5 kcal mol−1) in the S0 state (Fig. S5(c and d)†). Upon electronic excitation, the SPT and DPT potential energy barriers dramatically decrease (Fig. S5(a and b)†). However, compared with the SPT process, the energy barrier of the DPT process is slightly higher (<2 kcal mol−1), which is not convincing enough to rule out the DPT mechanism. Hence, the simulated results are fitted with the DXR system based on the long-range-hybrid functionals. This phenomenon is consistent with the discussion in section 3.2. Moreover, the calculated PECs (Fig. S6†) verify the above ESIDPT mechanism at the ωB97XD/6-311+G(d,p) level.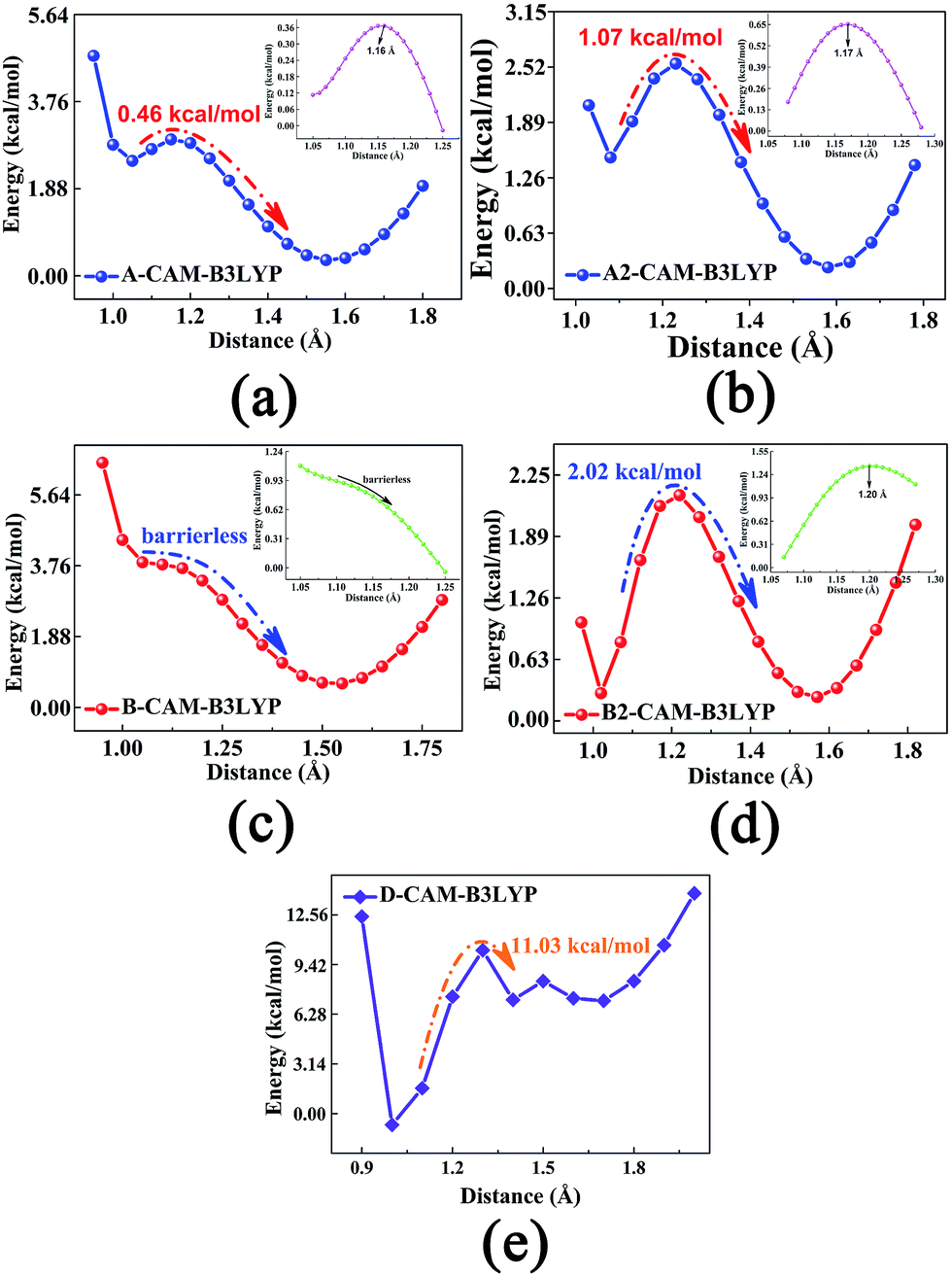 | ||
| Fig. 5 The excited-state PECs of the DXR system in ACN solvent at the CAM-B3LYP/6-311+G(d,p) level. (a) and (b) The stepwise single proton (H2) transfer processes; (c) and (d) the stepwise single proton (H5) transfer processes; (e) the synchronous double proton (H2 and H5) transfer process. Inset: the PECs around inflection points along a smaller step size (i.e., 0.01 Å). | ||
| A → C | C → B | A → D | D → B | A → B | |
|---|---|---|---|---|---|
| CAM-B3LYP | 0.00 | 2.02 | 0.46 | 1.07 | 11.03 |
| B3LYP | 1.53 | 0.54 | 1.55 | 0.58 | 2.86 |
| ωB97XD | 0.00 | 2.62 | 0.75 | 1.50 | 12.63 |
| B → C | C → A | B → D | D → A | B → A | |
|---|---|---|---|---|---|
| CAM-B3LYP | 2.06 | ∞ | 2.31 | 2.29 | 3.19 |
| B3LYP | 1.59 | 1.22 | 2.59 | 0.22 | 2.81 |
| ωB97XD | 2.40 | ∞ | 2.50 | 3.06 | 3.19 |
Therefore, we delineate the stepwise double proton transfer process as follows: firstly, the DXR molecule is excited from S0 to S1 with the structure identified as point A; the O1–H2⋯O3 and O4–H5⋯O6 hydrogen bonds are strengthened after excitation. Then, due to negligible barriers, the point A structure can transfer one proton from the hydroxyl O to the carbonyl O atom, thereby forming the C or D isomer. Subsequently, the other proton is transferred due to the 1–2 kcal mol−1 potential barriers, forming the B structure. It should be noted that the two successive single proton transfer processes (i.e. A → C → B and A → D → B) are competitive in the S1 state. Compared with each other, the reverse proton transfer processes among these minimum points can also occur through a synchronous double proton transfer process (i.e. B → A), summarized in Table 3. In turn, through the S1 → S0 internal conversion (IC) process, B, C and D structures can emit fluorescences of 578, 572 and 577 nm, respectively. Due to the low barriers, after the radiative transition, the unavailable B structure (shown in Fig. 5(b) and (d)) can reverse either single proton forming the C or D structure, or double protons forming the A structure. Similarly, the C and D structures can also reverse the other proton, forming a structure because of the low barriers.
To obtain more accurate potential energy barriers, the CIS/6-311+G(d,p) method is used to search the excited-state TS structures, and all of the TS structures are shown in Fig. S7 (ESI†). By the Berny optimization method, our calculated TS structures are confirmed to be only one imaginary frequency, and its vibrational eigenvector points in the correct direction. Zero-point energy corrections are also performed according to the harmonic vibrational frequencies. All the potential energy barriers are shown in Table 3. The corresponding imaginary frequencies for the TS structures are listed in Table S1 (ESI†). In fact, only the TS structures can be found along with the stepwise ESIDPT path in our calculations, which is consistent with the above conclusion that the ESIDPT mechanism of DXR is sequential rather than concerted in the S1 state.
4 Conclusion
Based on multiple functional simulations, we show clearly that DXR, containing double intramolecular hydrogen bonds, undergoes a stepwise double proton transfer reaction. Unfortunately, as shown in the S1 PECs, the sequence of the two proton (H2 and H4) transfers is uncertain because of the tiny difference between the potential barriers. That is to say, two successive single proton transfer pathways (i.e. A → C → B and A → D → B) are competitive in the S1 state. Nevertheless, the synchronous double proton transfer mechanism can be really ruled out due to non-negligible barriers. What’s more, the systematic investigation into the ESIDPT mechanism of DXR provides fundamental insight into the binding of DXR to DNA and the photophysical properties of DXR inside cultured cells.In addition, this theoretical research complements earlier related work,37 and predicts two experimentally unobserved fluorescence peaks at 578 and 572 nm (calculated values) of the single proton transfer tautomers. The reason this is neglected experimentally is that it is difficult to distinguish between the tiny difference of the fluorescence peaks in tautomers. We hope that this study will encourage additional experimental work to corroborate or refute our prediction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation (No. 11374191 and 11604333) and the National Basic Research Program of China (No. 2015CB921004).References
- A. Weller, Z. Electrochem, 1956, 60, 1144–1147 CAS.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531–534 CrossRef CAS PubMed.
- C. Y. Peng, J. Y. Shen, Y. T. Chen, P. J. Wu, W. Y. Hung, W. P. Hu and P. T. Chou, J. Am. Chem. Soc., 2015, 137, 14349–14357 CrossRef CAS PubMed.
- J. F. Zhao, J. S. Chen, J. Y. Liu and M. R. Hoffmann, Phys. Chem. Chem. Phys., 2015, 17, 11990–11999 RSC.
- J. F. Zhao and Y. J. Zheng, Sci. Rep., 2017, 7, 44897 CrossRef CAS PubMed.
- S. J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS PubMed.
- M. F. Rode and A. L. Sobolewski, J. Phys. Chem. A, 2010, 114, 11879–11889 CrossRef CAS PubMed.
- P. T. Chou, M. L. Martinez and J. H. Clements, Chem. Phys. Lett., 1993, 204, 395–399 CrossRef CAS.
- D. Ma, F. Liang, L. Wang, S. Lee and L. Hung, Chem. Phys. Lett., 2002, 358, 24–28 CrossRef CAS.
- B. Liu, H. Wang, T. S. Wang, Y. Y. Bao, F. F. Du, J. Tian, Q. B. Li and R. K. Bai, Chem. Commun., 2012, 48, 2867–2869 RSC.
- J. Z. Zhao, S. M. Ji, Y. H. Chen, H. M. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- J. Catalan and J. C. del Valle, J. Am. Chem. Soc., 1993, 115, 4321–4325 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2006, 8, 3410–3417 RSC.
- Y. Kubo, S. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–523 CrossRef CAS.
- H. C. Chou, C. H. Hsu, Y. M. Cheng, C. C. Cheng, H. W. Liu, S. C. Pu and P. T. Chou, J. Am. Chem. Soc., 2004, 126, 1650–1651 CrossRef CAS PubMed.
- K. C. Tang, M. J. Chang, T. Y. Lin, H. A. Pan, T. C. Fang, K. Y. Chen, W. Y. Hung, Y. H. Hsu and P. T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. A. Chen, W. Y. Hung, W. F. Tang, Y. H. Hsu, C. L. Chen, F. Y. Meng and P. T. Chou, Chem. Mater., 2016, 28, 8815–8824 CrossRef CAS.
- Q. Fei, X. F. Gu, Y. J. Liu, B. Shi, H. Y. Liu, G. Xu, C. B. Li, P. Shi and C. C. Zhao, Org. Biomol. Chem., 2017, 15, 4072–4076 CAS.
- F. B. Yu, P. Li, G. J. Zhao, T. S. Chu and K. L. Han, J. Am. Chem. Soc., 2011, 133, 11030–11033 CrossRef CAS PubMed.
- G. J. Zhao, J. Y. Liu, L. C. Zhou and K. L. Han, J. Phys. Chem. B, 2007, 111, 8940–8945 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, J. Phys. Chem. A, 2007, 111, 2469–2474 CrossRef CAS PubMed.
- Y. M. Dai, J. F. Zhao, Y. L. Cui, Q. Y. Wang, P. Song, F. C. Ma and Y. Y. Zhao, Spectrochim. Acta, Part A, 2015, 144, 76–80 CrossRef CAS PubMed.
- C. L. Chen, H. W. Tseng and P. T. Chou, et al., J. Phys. Chem. A, 2016, 120, 1020–1028 CrossRef CAS PubMed.
- Y. A. Chen, F. Y. Meng, Y. H. Hsu, C. H. Hung, C. L. Chen, K. Y. Chung, W. F. Tang, W. Y. Hung and P. T. Chou, Chem.–Eur. J., 2016, 22, 14688–14695 CrossRef CAS PubMed.
- G. E. Kellogg, J. N. Scarsdale and F. A. Fornari, Nucleic Acids Res., 1998, 26, 4721 CrossRef CAS PubMed.
- Y. L. Lyu, J. E. Kerrigan, C. P. Lin, A. M. Azarova, Y. C. Tsai, Y. Ban and L. F. Liu, Cancer Res., 2007, 67, 8839–8846 CrossRef CAS PubMed.
- K. B. Wallace, Basic Clin. Pharmacol. Toxicol., 2003, 93, 105–115 CrossRef CAS.
- S. Mouli, G. Nanayakkara, A. AlAlasmari, H. Eldoumani, X. Fu, A. Berlin, M. Lohani, B. Nie, R. D. Arnold, A. Kavazis, F. Smith, R. Beyers, T. Denney, M. Dhanasekaran, J. Zhong, J. Quindry and R. Amin, Am. J. Physiol.: Heart Circ. Physiol., 2015, 309, H844–H859 CAS.
- M. D. Smet, S. Langereis, S. van den Bosch and H. Grull, J. Controlled Release, 2010, 143, 120–127 CrossRef PubMed.
- H. T. Ta, C. R. Dass, I. Larson, P. F. Choong and D. E. Dunstan, Biomaterials, 2009, 30, 3605–3613 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- A. M. Nowicka, A. Kowalczyk, M. Donten, P. Krysinski and Z. Stojek, Anal. Chem., 2009, 81, 7474–7483 CrossRef CAS PubMed.
- H. Ali-Boucetta, K. T. Al-Jamal, D. McCarthy, M. Prato, A. Bianco and K. Kostarelos, Chem. Commun., 2008, 4, 459–461 RSC.
- N. S. H. Motlagh, P. Parvin, M. Refahizadeh and A. Bavali, Appl. Opt., 2017, 56, 7498–7503 CrossRef PubMed.
- R. J. Sturgeon and S. G. Schulman, J. Pharm. Sci., 1977, 66, 958–961 CrossRef CAS PubMed.
- D. R. Kumar, S. Dhar, A. Sarkar and S. B. Chandra, J. Phys. Chem. A, 2011, 115, 9169–9179 CrossRef PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- S. Chai, G. J. Zhao, P. Song, S. Q. Yang, J. Y. Liu and K. L. Han, Phys. Chem. Chem. Phys., 2009, 11, 4385–4390 RSC.
- P. Song and F. C. Ma, Int. Rev. Phys. Chem., 2013, 32, 589–609 CrossRef CAS.
- J. F. Zhao and P. Li, RSC Adv., 2015, 5, 73619–73625 RSC.
- J. F. Zhao, X. Y. Liu and Y. J. Zheng, J. Phys. Chem. A, 2017, 121, 4002–4008 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- Y. H. Liu, S. C. Lan, C. Zhu and S. H. Lin, J. Phys. Chem. A, 2015, 119, 6269–6274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08945g |
| This journal is © The Royal Society of Chemistry 2017 |