
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemporal dynamics of bacterial communities and predicted nitrogen metabolism genes in a full-scale wastewater treatment plant†
Xiao-Yan Fan,
Jing-Feng Gao *,
Kai-Ling Pan,
Ding-Chang Li and
Hui-Hui Dai
*,
Kai-Ling Pan,
Ding-Chang Li and
Hui-Hui Dai
National Engineering Laboratory for Advanced Municipal Wastewater Treatment and Reuse Technology, Beijing University of Technology, Beijing 100124, China. E-mail: gao.jingfeng@bjut.edu.cn; gao158@gmail.com; Fax: +86-10-67391983; Tel: +86-10-67391918
First published on 15th December 2017
Abstract
The dynamics of bacterial communities in a full-scale wastewater treatment plant (WWTP) were investigated using Illumina MiSeq sequencing and PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) over a one-year period. Results showed that the temporal dynamics of structure, diversity and abundance of bacterial community were not obvious. The persistent species had low diversity but high relative abundance. Dechloromonas, Thauera, Nitrosomonas and Nitrospira were the four dominant persistent genera. The intermittent and transient genera showed different dynamics with persistent genera. Variation partitioning analysis suggested that water quality indexes (13.40%) had the greatest contribution to bacterial community variation, followed by temperature (9.20%). Quantitative polymerase chain reaction analysis revealed the stable abundances of nitrifying and denitrifying bacteria, and the occurrence of complete ammonia oxidizers (Comammox) in the full-scale WWTP. Functional prediction showed that the bacterial functional compositions were also relatively stable, and metabolism was the most dominant functional category. Many genes involved in the nitrogen metabolism pathway were predicted to be present with high gene counts, e.g., nirB, nirD, nasA, norB, nirK, nosZ and nifH. Most of these genes were predicted to be mainly contributed by Rhizobiales, Burkholderiales, Rhodocyclales and Actinomycetales. Overall, the composition and function of bacterial community in a long-term monitoring were relatively stable, which is important for maintaining the high wastewater treatment capability in full-scale WWTPs.
1. Introduction
Biologically activated sludge processes have been extensively used for treating municipal and industrial wastewater due to their high removal efficiency of nutrients and low operational cost. The performance and stability of these processes rely on the structure and function of microorganisms within the activated sludge. Bacteria, protozoans, fungi and filamentous organisms, the main groups of activated sludge, have been shown to actively participate in the biological treatment of wastewater.1 Especially, bacteria play important roles in the removal of pollutants.2 The bacterial community structure and composition within or between activated sludge systems have been widely investigated.3,4 Although some significant bacterial community members have been identified, such studies do not capture the dynamics of bacterial community structure. Moreover, the bacterial community dynamics are thought to be influenced by changing environmental or operational variables.5 Furthermore, variations in microbial community structure are often closely associated with changes in the functions of microbes.6 Study on temporal dynamics is an alternative way for correlating environmental or operational variables with bacterial community and function, which is critical for improving treatment performance and predicting responses to unexpected environmental shifts.Previously, regarding the temporal bacterial community dynamics in activated sludge systems, researchers mainly focus on specific microbes, e.g., nitrifiers7 or denitrifiers8 in lab-scale bioreactors or pilot-scale wastewater treatment plants (WWTPs), lacking an overview of the bacterial community dynamics in full-scale WWTPs. In recent years, the dynamics of bacterial community in full-scale WWTPs were investigated using conventional molecular technologies and next-generation sequencing (NGS). The one-year dynamics of bacterial communities in full-scale WWTPs have been investigated using terminal restriction fragment length polymorphism9 and pyrosequencing.6 Moreover, a few researchers focus on the long-term studies of the bacterial community using NGS. Pyrosequencing has been applied to investigate the 5-year temporal dynamics of bacterial community in a full-scale WWTP treating saline domestic sewage containing ∼30% seawater, suggesting there is no significant seasonal succession.10 Recently, the metagenomic technique is employed to investigate both the bacterial community and function in a municipal WWTP over 4 years, but this study only focused on the dynamics of two seasons (winter and summer).11 Compared with metagenomic technique, a relatively uncomplicated and effective way to investigate the bacterial function is using PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) based on 16S rRNA gene. PICRUSt, as a bioinformatics tool, has been applied to predict the bacterial functional composition in some simple habitats, such as human and animal gut.12 Recently, PICRUSt is also used to study bacterial functional composition in some complex environments, including soil,13 sediment,14 wastewater15 and activated sludge.16,17 Overall, the combination of NGS and PICRUSt will be an effective method to investigate the dynamics of bacterial community and function in full-scale WWTPs, and could provide useful information for improving treatment performance.
The biological technology for nitrogen removal aimed to protect water bodies from eutrophication is of great interest for many researchers. Biological nitrification and denitrification are the widely used methods for nitrogen removal in WWTPs. Nitrification is catalyzed by ammonia oxidizing bacteria (AOB), ammonia oxidizing archaea (AOA), complete ammonia oxidizers (Comammox) and nitrite oxidizing bacteria (NOB). Denitrification is performed by denitrifying bacteria (DNB). Nitrification failure occurs frequently in many WWTPs because the nitrifiers (especially AOB) are highly sensitive to several environmental factors.18 Denitrification is often limited by lack of available organic carbon source for the wastewater with low C/N ratio.19 Therefore, a better understanding of the microbial ecology of nitrifiers and DNB in WWTPs is important for the complete nitrogen removal from wastewater. In previous studies, the diversity and abundance of AOB and AOA in full-scale WWTPs have been well investigated,20,21 however, little is known about their temporal dynamics. The newly discovered Comammox are widespread in both natural and engineered ecosystems, including soils, freshwater ecosystems, drinking water systems and WWTPs.22,23 The occurrence of Comammox raises many fundamental questions to be further solved, e.g., the diversity and abundance of Comammox, the contributions of Comammox to nitrification and the re-evaluation of earlier studies regarding nitrification.24 Although Comammox have been successfully detected in nine full-scale WWTPs,25 the dynamics of them are still unknown. Compared with DNB in natural habitats, only a few studies investigate the diversity and abundance of DNB in WWTPs.26 Overall, a better insight into the dynamics of nitrifiers and DNB in full-scale WWTPs allow researchers to look beyond a mere engineering problem in order to develop promising strategies for improved process performance.
Bulking and foaming are two of the most frequently occurring operational problems in full-scale WWTPs, and these problems are mainly associated with the excessive growth of bulking and foaming bacteria (BFB).27,28 In early studies, BFB were identified by microscopy27 or conventional molecular techniques.28,29 Recently, some NGS techniques, e.g., Ion Torrent platform and pyrosequencing, have been applied to investigate the BFB community in Macau Peninsula WWTP30 and 14 global WWTPs.31 Other than their notoriety, BFB also play important roles in normal activated sludge because they are significant builder for bioflocs.31 Therefore, BFB play double-edged roles in activated sludge. Previous studies mainly focused on the BFB community in bulking or foaming activated sludge samples, but their compositions and dynamics have not been comprehensively characterized in normal activated sludge samples.31
In this study, Illumina MiSeq sequencing and PICRUSt were employed to characterize temporal dynamics of composition and predicted functional ecology of bacterial community in a full-scale WWTP. The detailed objectives were: (1) to investigate the dynamics of bacterial community structure and diversity; (2) to investigate the dynamics of genera related to nitrogen removal and BFB; (3) to assess the relationships between bacterial community and environmental or operational variables; (4) to explore the dynamics of the absolute abundance of bacterial 16S rRNA gene and functional genes related to nitrogen removal; (5) to elucidate the dynamics of bacterial functional composition and the variations in genes involved in nitrogen metabolism pathway.
2. Materials and methods
2.1 Sample collection
The sampling site is a full-scale WWTP (YF-WWTP) treating municipal wastewater in Beijing, China (40.05° N, 116.26° E). The plant is designed as a Carrousel 3000 oxidation ditch process for carbon and nitrogen removal. The treatment capacity was 2 × 104 m3 d−1, and the dissolved oxygen (DO) in outer, middle and inner channel was 0.2–0.3, 0.5–1.5 and 1.5–2.5 mg l−1, respectively. Activated sludge sample (∼200 ml) was collected monthly from the outer channel of the oxidation ditch process with water sampler from March 2015 to February 2016, and twelve samples were obtained. At sampling, the samples were fixed in 50% (v/v) ethanol, kept in 50 ml sterilized centrifuge tubes, stored on ice and delivered to the laboratory within 2 h. Subsequently, the samples were freeze-dried by using Labconco Freezone 1 L (Labconco, USA) and stored at −20 °C until DNA extraction. The 12 activated sludge samples were named as YF.1, YF.2, YF.3, YF.4, YF.5, YF.6, YF.7, YF.8, YF.9, YF.10, YF.11 and YF.12, and the numbers presented the month of sampling. Moreover, some important environmental and operational variables were monitored, including influent and effluent BOD, COD, NH4+–N, mixed liquid suspended solids (MLSS), sludge retention time (SRT), hydraulic retention time (HRT), dissolved oxygen (DO) and temperature (T) (Table S1, in ESI†).2.2 DNA extraction, Illumina MiSeq sequencing and sequence processing
For each sample, microbial genomic DNA was extracted from 0.05–0.10 g of dried activated sludge using a Fast DNA® SPIN Kit for soil (Qiagen, CA, USA) according to the manufacturer's instructions. The concentration and purity of DNA extracted were determined by Nanodrop-1000 (Thermo Fisher Scientific, USA).Illumina MiSeq sequencing was applied to investigate the dynamics of bacterial community structure and composition. The hypervariable regions V3–V4 of bacterial 16S rRNA gene were PCR-amplified with primer set 338F and 806R in an Applied Biosystems 9700 PCR system (ABI GeneAmp® 9700). Each sample was amplified in triplicates. The amplicons were pooled, purified with AxyPrep DNA Gel Extraction Kit (Axgen, USA) and quantified using a QuantiFluor™-ST (Promega, USA). After purification, the amplicons from individual products were pooled and normalized in equimolar amounts. Finally, the library was constructed and sequenced on Illumina MiSeq PE300 sequencer (Illumina, USA) in Majorbio Co., Ltd (Shanghai, China). All sequences obtained have been deposited in the Sequence Read Archive (SRA) database under accession number of SRP093567.
After sequencing, the barcodes, primers and adapters were removed. The raw paired-ended (PE) reads were merged using Fast Length Adjustment of SHort reads (FLASH).32 Quantitative Insights Into Microbial Ecology (QIIME) pipeline package was applied for post-sequencing analysis.33 Sample was rarified to 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 230 sequences per sample. The normalized sequences of all samples were clustered into operational taxonomic units (OTUs) using the UCLUST algorithm by setting 97% similarity. The taxonomic classification of OTUs were performed using the GreenGenes database. Subsequently, the alpha diversity indices, including OTU number, Chao 1, ACE, Shannon index and Simpson index and the rarefaction curves were generated to evaluate the richness and diversity of bacterial communities. Beta diversity, including principal coordinate analysis (PCoA) and cluster analysis (CA) based on the weighted UniFrac distance metrics were used to compare bacterial composition and structure across different samples. The dynamics of bacterial populations were investigated based on the occurrence frequency of OTUs. In this study, the occurrence frequency was calculated by dividing the number of months in which an OTU was detected by the number of total months. The AOB, NOB, DNB and BFB that have been reported previously were selected to further investigate their dynamics in the full-scale WWTP.4,10,11,28,29
230 sequences per sample. The normalized sequences of all samples were clustered into operational taxonomic units (OTUs) using the UCLUST algorithm by setting 97% similarity. The taxonomic classification of OTUs were performed using the GreenGenes database. Subsequently, the alpha diversity indices, including OTU number, Chao 1, ACE, Shannon index and Simpson index and the rarefaction curves were generated to evaluate the richness and diversity of bacterial communities. Beta diversity, including principal coordinate analysis (PCoA) and cluster analysis (CA) based on the weighted UniFrac distance metrics were used to compare bacterial composition and structure across different samples. The dynamics of bacterial populations were investigated based on the occurrence frequency of OTUs. In this study, the occurrence frequency was calculated by dividing the number of months in which an OTU was detected by the number of total months. The AOB, NOB, DNB and BFB that have been reported previously were selected to further investigate their dynamics in the full-scale WWTP.4,10,11,28,29
2.3 Quantitative analysis of microbial communities
The abundances of bacteria, AOA, AOB, Candidatus Nitrospria inopinata, NOB and DNB were quantified by quantitative polymerase chain reaction (qPCR) in a MX3005P Thermocycler (Agilent Technologies, USA) with GoTaq® qPCR Master Mix (Promega, USA). Ca. N. inopinata is a Comammox enrichment culture.22,23 The qPCR was performed in a 20 μl reaction mixture consisting of 7.2 μl of aseptic water, 0.4 μl of each primer (10 μM), 10 μl of GoTaq® qPCR Master Mix (Promega, USA) and 2 μl of template DNA (∼20 ng). Each reaction was performed in triplicate. The primer sets and protocols for qPCR are summarized in Table S2 (in ESI†). Plasmids containing target genes were used as standards. The standard curves were generated from 10-fold serial dilutions of plasmids, and the copy number ranges of them are listed in Table S2 (in ESI†). The amplification efficiencies of qPCR assays ranged from 90.0 to 112.0%, and R2 values were in the range of 0.994 to 0.999.2.4 Statistical analysis and network analysis
The statistical significance among datasets, e.g., structure, diversity and abundance of bacterial community, was assessed by PerMANOVA in PAST (http://folk.uio.no/ohammer/past/). Correlations between bacterial community and some important variables were investigated by calculating their Spearman's rank correlation coefficients (SRCCs). A correlation between two items was considered statistically robust if the SRCC ≥0.6 or ≤−0.6 and the P-value ≤0.05. Cytoscape (version 3.0.2) software was used to visualize the network graphs.34 In the correlation network, each node stands for one genus or one variable, and each node-connecting edge represents a positive or negative correlation between nodes. For nodes representing genera, the positive correlations suggest their co-occurrences. For genera and variables, the positive correlations indicate that the variables may be beneficial for the accumulation of these genera. Furthermore, the contributions of the environmental variables to the core genera structure were quantified by variation partitioning analysis (VPA) using R (version 2.15).2.5 Functional prediction analysis
The PICRUSt v1.0.0 pipeline was applied to predict the putative metabolic functions of bacterial communities in activated sludge samples based on the 16S rRNA-derived OTUs.12 Firstly, the OTU table was normalized by dividing each OTU abundance by the known or predicted 16S copy number abundance with the script normalize_by_copy_number.py. Secondly, the final functions for metagenome were predicted with the script predict_metagenomes.py, generating a table of Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologs (KOs). Thirdly, the predicted metagenomes were collapsed into higher categories (e.g. KOs into KEGG pathways) with the script categorize_by_function.py. PCoA based on the Bray–Curtis distances was created to compare the bacterial functions among samples with QIIME.33 Finally, the predictive genes involved nitrogen metabolism pathway and the bacterial orders contributed to these genes were further investigated using the script metagenome_contributions.py script with the – l option. The KOs of nitrogen cycle used in the script are summarized in Table S3 (in ESI†). Moreover, the weighted Nearest Sequenced Taxon Index (NSTI) score for each sample was further calculated to evaluate the accuracy of the metagenome predictions using the – a option of predict_metagenomes.py.3. Results and discussion
3.1 Dynamics of bacterial community structure and diversity
Dynamics in bacterial community structure were evaluated by PCoA (Fig. 1). The bacterial communities in 12 activated sludge samples were separated into five Groups. Especially, Group IV contained eight samples collected from different seasons, suggesting there was similar bacterial community structure. For the other four Groups, only one sample was included in each Group, and these samples were discrete, suggesting their dissimilar bacterial community structure. The results were further supported by CA analysis (Fig. S1, in ESI†). However, the dissimilarity of the five Groups was not significant (PERMANOVA p > 0.05). These results suggest that the dynamics of bacterial community structure in the full-scale WWTP was not obvious. The 5-year temporal dynamics of bacterial community also show no significant seasonal succession.10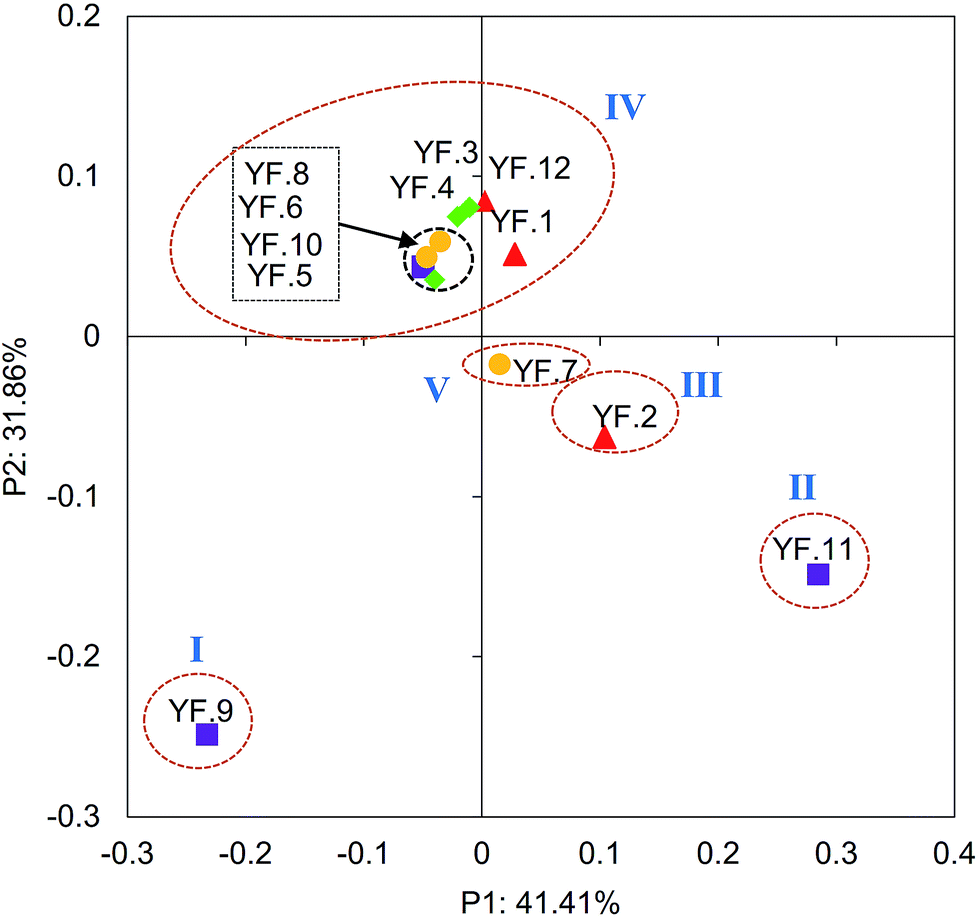 | ||
| Fig. 1 Dynamics of bacterial community structure of 12 activated sludge samples collected monthly in a full-scale WWTP as revealed by principal coordinate analysis. | ||
The bacterial diversity indexes of the five Groups based on OTUs, Chao 1, ACE, Shannon index and Simpson index revealed that there were no obvious temporal variations of the richness and diversity of bacterial communities (PERMANOVA p > 0.05) (Table S3, in ESI†). Moreover, all the rarefaction curves arrived to plateau phases and the Good's coverage values were above 95%, suggesting that most of the microorganisms had been detected by Miseq sequencing.
3.2 Dynamics of bacterial populations based on the occurrence frequency of OTUs
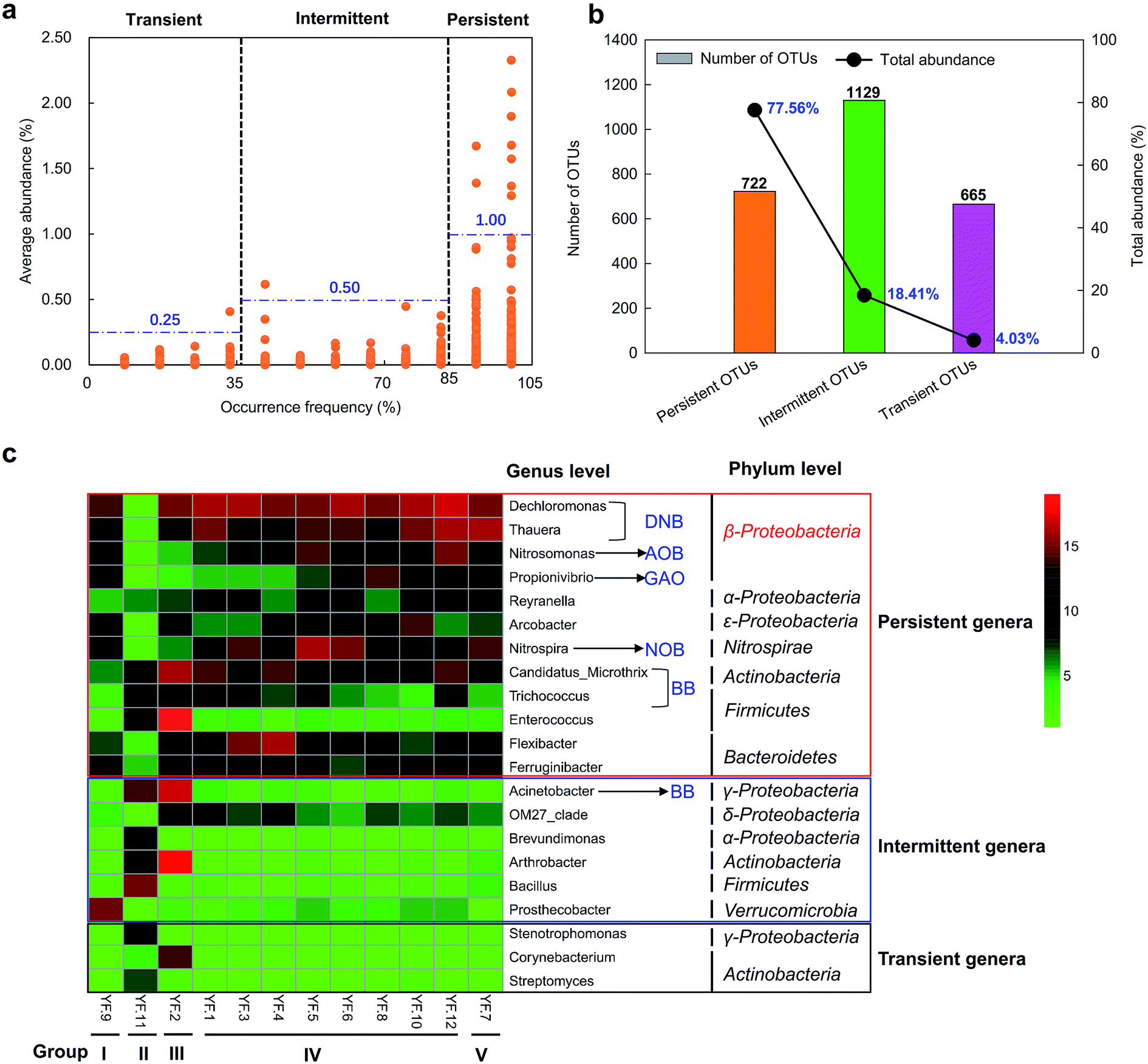 | ||
| Fig. 2 Defining the one-year activated sludge microbiome in a full-scale WWTP by their occurring frequency. The mean relative abundance of OTUs (a) is shown relative to the 0.97-OTU's percentage occurrence (X axis). The number and relative abundance of OTUs of different occurrence frequencies (persistent, intermittent and transient) are shown in (b). The genera with relative abundance higher than 1% of total sequences at least one sample are shown in (c). The occurrence frequency was calculated by dividing the number of months in which an OTU was detected by the number of total months. | ||
In this study, 12 dominant persistent genera were detected. Among them, Dechloromonas and Thauera showed high relatively abundance in each Group, except Group II. Nitrosomonas and Nitrospira were more abundant in Group I, IV and V. Dechloromonas and Thauera have been reported as DNB to remove nitrate.36 Members of Thauera exhibit high denitrification rate.37 Nitrosomonas is known to play an important role in ammonia oxidation in WWTPs.20,21 Nitrospira is widely considered to be an important NOB to perform nitrite oxidation. These four dominant persistent genera are important for the nitrogen removal in the full-scale WWTP. Candidatus_Microthrix, one of the bulking bacteria (BB), was abundant in each Group. This genus is often responsible for bulking in activated sludge, but is still the natural member of the bacterial community in activated sludge.28 Flexibacter showed higher relative abundance in Group III–IV, especially, it was abundant in sample YF. 3 and YF.4. Except these six genera, the other six persistent genera only abundant in some of the five Groups, including Propionivibrio, Reyranella, Arcobacter, Trichococcus, Enterococcus and Ferruginibacter. Propionivibrio is a novel glycogen accumulating organism (GAO), which is closely related to Candidatus Accumulibacter, but do not store excessive amounts of polyphosphate.38 Arcobacter was abundant in Group III and some of samples in Group IV. Enterococcus showed highest relative abundance in Group III. Some species of Arcobacter and Enterococcus are a cause of some serious diseases not only for human but also for animals.39 Trichococcus is also the main BB, which was abundant in Group II–III and two samples in Group IV. Overall, these dominant persistent genera may play critical roles in the removal of pollutants, and showed different dynamics in the full-scale WWTP.
The dominant intermittent and transient genera were more abundant in Group I–III. There were six dominant intermittent genera, including Acinetobacter, OM27_clade, Brevundimonas, Arthrobacter, Bacillus and Prosthecobacter. Prosthecobacter showed the highest relative abundance in Group I. Acinetobacter, Brevundimonas, Arthrobacter and Bacillus were abundant in Group II. Acinetobacter, OM27_clade and Arthrobacter were the main intermittent genera in Group III. Only three dominant transient genera were detected. Stenotrophomonas and Streptomyces showed highest relative abundance in Group II. Corynebacterium was most abundant in Group III. In summary, the persistent, intermittent and transient genera showed different temporal dynamics.
3.3 Dynamics of denitrifying bacteria and bulking and foaming bacteria
Nitrogen removal is one of the main tasks of WWTPs. The dynamics of genera related to nitrification and denitrification need to be further investigated. In this study, Nitrosomonas and Nitrospira were the genera related to nitrification, and their dynamics have been given in section 3.2. Thus, here, we only focused on the dynamics of DNB, except the DNB mentioned above. As shown in Fig. 3, eight DNB were detected, including Hyphomicrobium, Comamonas, Zoogloea, Paracoccus, Thermomonas, Acidovorax, Pseudomonas and Azoarcus. Hyphomicrobium was widely distributed in each group with higher relative abundance than the other DNB. Comamonas and Zoogloea were most abundant in Group II and Group I, respectively. Compared with the other four Groups, Paracoccus was also more abundant in Group II. Whereas, the relative abundance of the other four DNB was lower than 0.04%. | ||
| Fig. 3 Distributions of denitrifying bacteria (DNB), bulking bacteria (BB) and foaming bacteria (FB) across different Groups in the full-scale WWTP. The mean relative abundance of each genera is shown in the figure. | ||
Moreover, in addition to causing bulking or foaming problems, many of BFB are natural members of the communities in WWTPs and have versatile roles, including lipid, polysaccharides and protein degradation and polyhydroxyalkanoates storage.40 Therefore, the BB and foaming bacteria (FB) were further investigated, except the BFB mentioned in section 3.2. Ten BB were detected in the full-scale WWTP with the relative abundance lower than 1.06%. Mycobacterium was more abundant in Group II–V than that Group I. The relative abundance of Roseiflexus and Chryseobacterium was highest in Group I and Group II, respectively. Defluviicoccus was more abundant in Group II and V than the other three Groups. Haliscomenobacter showed higher relative abundance in Group IV, V and I than Group II and III. The other five BB with relative abundance lower than 0.13% were Beggiatoa, Streptococcus, Thiothrix, Sphingobacterium and Acidovorax. Only three FB were found, including Bifidobacterium, Gordonia and Rhodococcus, and their relative abundances were lower than 0.40%. Bifidobacterium was detected in all the five Groups, and showed higher relative abundance in Group II–V. Gordonia and Rhodococcus were more abundant in Group II and III. The BFB observed in the full-scale WWTP may be opportunistic BFB, which could cause bulking or foaming problems only under certain operational conditions.
3.4 Network analysis and variation partitioning analysis of genera and environmental variables
Network interface was applied to investigate the relationships between environmental and operational variables and some genera. The results of network analysis can provide an awareness of the variables that favor or disfavor particular microbes,4,10 which is helpful for guiding the operation of WWTPs. As shown in Fig. 4a, the network had 34 nodes and 33 edges. MLSS showed positive correlations with Dechloromonas, Ferruginibacter, Anaerolinea, OM27_clade and Nitrosomonas, suggesting that the increase in MLSS may promote the accumulation of these genera. HRT was positively correlated with Nitrosomonas and Nitrospria, indicating the long HRT is in favor of the accumulation of nitrifiers. In addition to MLSS and HRT, the positive correlation was observed between T and Nitrospria, suggesting increases in MLSS, HRT and T play important roles in keeping good nitrification performance in WWTPs. Negative correlations were observed between T and most of the BFB, e.g., Acidovorax, Streptococcus, Beggiatoa, Bifidobacterium and Trichococcus. Also, DO was negatively correlated with Candidatus_Microthrix. The results suggest that low T or low DO may be in favor of the accumulation of BFB. Therefore, T and DO are important factors for the occurrence of bulking or foaming problems in full-scale WWTPs. These findings were consistent with previous studies.41–44 Most of the sludge bulking events have often been observed at times when the T is low (e.g., winter and spring) because low T is advantageous for the growth of BFB.41,42 Also, low DO has been confirmed to have a strong negative effect on sludge settleability, leading to the proliferation of BFB.43,44 However, a few studies suggested that high T also could cause filamentous bulking and biological foaming,30,45 which was different from this study. Moreover, the influent BOD and COD concentrations showed positive correlations with Dechloromonas and Thauera, two abundant DNB in the WWTP, suggesting that carbon source is an important factor for denitrification. Effluent BOD concentrations were positively correlated with some genera, e.g., Kurthia, Carnobacterium and Brevundimonas. Overall, MLSS, HRT, DO, T and some important water quality indexes (WQI) of wastewater are important for the stable performance of WWTPs. | ||
| Fig. 4 Relationships between environmental factors and dominant genera. (a) Network analysis. (b) Variation partitioning analysis. Each diagram represents the biological variation partitioned into the relative effects of each factor or a combination of factors. WQI: water quality indexes; OP: operational parameters; T: temperature. | ||
VPA was further applied to quantitatively assess the contributions of WQI (influent and effluent BOD, COD, NH4+–N), operational parameters (OP: MLSS, SRT, HRT and DO) and T to bacterial community dynamics. Fig. 4b shows that 35.20% of the variations could be explained by these three components. WQI had the greatest contribution (13.4%) to the variation of bacterial communities, followed by T (9.20%) and OP (8.46%). In a previous study,6 wastewater characteristics (20.30%) have been confirmed to be the greatest contributors to bacterial community variance, followed by OP (19.9%) and bioreactor scale (3.6%). Moreover, the contribution of WQI and OP to bacterial community variations was only 3.62%, and it was lower than 0.35% for the other two or three major components, suggesting that interactions among the three major components had less influence than the individual components. This agreed with the finding of Wang et al.46 and Wen et al.,47 who suggest that the contributions of interactions among WQI, OP and geographic locations to the bacterial community variations in different full-scale WWTPs are much lower than individual components. However, 64.80% of the core genera communities could not be explained by these three components. In previous studies, about 30–50% of the bacterial community variance could not be explained by the similar components.46,47 It is reasonable to expect that some additional factors, such as unmonitored WQI and OP, stochastic dispersal and immigration,48 phage predation49 and inhibitory chemicals46 may play an influential role in shaping bacterial communities in full-scale WWTPs. Overall, the relative influence of deterministic and stochastic factors in structuring microbial communities needs future investigation.
3.5 Quantification of bacterial 16S rRNA gene and functional genes of nitrogen removal microorganisms
qPCR was further applied to investigate the dynamics of the absolute abundance of bacterial 16S rRNA gene and functional genes related to nitrification and denitrification. As shown in Fig. 5, the abundance of bacterial 16S rRNA gene in the five Groups was close, ranging from 1.31 × 1011 (Group III) to 8.83 × 1011 copies per g sludge (Group II). The abundance of AOB amoA gene was varied between 7.76 × 106 (Group III) and 8.83 × 107 copies per g sludge (Group III), and it was nearly one order of magnitude higher than AOA amoA gene. The abundance of AOA amoA gene in Group II was lower than that in the other Groups. The average abundance of AOA amoA gene in the full-scale WWTP was 1.39 × 106 copies per g sludge. In addition to AOB and AOA, nitrification also could be performed by Comammox, which can completely oxidize ammonia to nitrate. In this study, Ca. N. inopinata (a Comammox enrichment culture) was successfully quantified in the five Groups, suggesting the occurrence of Comammox in the full-scale WWTP. The abundance of Ca. N. inopinata amoA gene was higher in Group III, IV and V than Group I and II, ranging from 9.56 × 104 to 7.63 × 105 copies per g sludge. Moreover, only NOB belonged to Nitrospira was successfully quantified, which was consistent with the results of MiSeq sequencing. The abundance of Nitrospira was high, ranging from 5.36 × 108 (Group II) to 1.49 × 1010 copies per g sludge (Group I). | ||
| Fig. 5 Quantification of total bacteria and genera related to nitrification and denitrification. Their abundances are quantified by qPCR based on 16S rRNA gene and functional genes. Ca. N. inopinata is a Comammox enrichment culture. For denitrification, only the abundance of two different nitrite reductase genes (nirK and nirS genes) was determined because nitrite reductase is the key enzyme in this process. | ||
For denitrification, nitrite reductase is the key enzyme.50 Therefore, the abundance of two different nitrite reductase genes (nirK and nirS genes) was determined. Except the Group II, the abundance of nirS gene was higher than that of nirK gene. Compared with Group I and V, the abundance of nirK gene was higher in the other three Groups. While, the abundance of nirS gene was close in different Groups. In this study, the average abundance of nirK and nirS genes was 1.40 × 109 and 4.67 × 109 copies per g sludge, respectively. Statistical analysis further suggests that there were no significant variations among the abundances of these genes in different Groups (PERMANOVA p > 0.05). Overall, the abundances of total bacteria and key genera related to nitrogen removal were relatively stable in one-year monitoring in the full-scale WWTP.
3.6 Predictive analysis of bacterial functions
PICRUSt was applied to infer the potential functions of bacterial communities. The predictive accuracy of PICRUSt can be evaluated by weighted NSTI score, which is a measure of the phylogenetic distance between the OTUs in a given sample and the reference OTUs.12 Here, the NSTI score of each activated sludge sample ranged from 0.11 (YF.11) to 0.25 (YF.9), with a mean of 0.18 ± 0.03. The prediction accuracy of PICRUSt decreases with increasing NSTI scores. However, PICRUSt still produce accurate metagenome predictions with a mean NSTI of 0.17.12 Thus, in this study, the PICRUSt predictions of activated sludge samples were sketchy and should be treated with caution. Whereas, higher mean NSTI values were also reported in complex environmental communities, e.g., Guerrero Negro hypersaline microbial mats (0.23)12 and coastal sediments (0.186).51 The mean NSTI value in activated sludge was comparable with that in coastal sediments. The lower degree of accuracy is mainly related to the unexplored diversity in complex environmental (lost in the closed reference OTU picking procedure).12,51 Overall, the predictive results still could provide some important insights into bacterial functions in the full-scale WWTP.Similar as the bacterial communities, the bacterial functions across samples were also grouped into five Groups as revealed by PCoA analysis (Fig. S2, in ESI†) and were relatively stable (PERMANOVA p > 0.05). This might be partly related to the functional redundancy. Complex microbial communities are believed to have functional redundancy and lead to stable function.52 In this study, metabolism (49.5–51.0%) was the most dominant functional category in different Groups, followed by genetic information processing (16.2–17.0%), environmental information processing (12.4–14.2%) and cellular processes (3.3–4.4%) (Fig. S3, in ESI†). Whereas, human diseases and organismal systems accounted for extremely low percentages. The top 35 bacterial potential functions in different Groups were further selected, and their relative abundances in each Group were close (Fig. S4, in ESI†). In general, transporters, ABC transporters, two-component system and secretion system were the main pathways under environmental information processing. DNA repair and recombination proteins and bacterial motility proteins were the main pathways under genetic information processing and cellular processes, respectively. 21 important metabolism pathways were found, e.g., oxidative phosphorylation, carbon fixation pathways in prokaryotes, pyruvate metabolism, methane metabolism and nitrogen metabolism. Overall, the dynamics of bacterial functions in the full-scale WWTP were not obvious.
Moreover, WWTPs are of great importance in nitrogen cycle, thus, the genes involved in nitrogen metabolism pathway (PATH: ko00910) were further investigated. In total, 24 functional genes were found. Fig. 6a shows the dynamics of counts for genes involved in nitrogen cycle. The biological processes were arranged according to the oxidation states of nitrogen (from +5 in nitrate to −3 in ammonia). For dissimilatory nitrate reduction, nirB gene was most abundant, showing higher gene count in Group II–V than Group I, followed by nirD, nrfA and narI/V genes. The nirD and nar I/narV genes were more abundant in Group III–V, while nrfA gene showed highest gene count in Group I. The nasA and nirA were the dominant genes responsible for assimilatory nitrate reduction in the full-scale WWTP, and their dynamics in different Groups was not obvious. For denitrification, eight genes were identified. The narH/narY/nxrB, nosZ, narG/narZ/nxrA and norB genes were predicted to be more abundant than nirK, norC, napA and napB genes. The narH/narY/nxrB and nosZ genes were more abundant in Group I. The other genes showed similar dynamics, and their gene counts were higher in Group IV and V than Groups I–III. The nifH, nifK and nifD genes were important for nitrogen fixation, showing more abundant in Group IV and V. The counts of genes involved in ammonia oxidation were much lower than the other genes mentioned above. Based on the results of qPCR, the abundances of AOB and AOA amoA genes were also lower than the genes involved in denitrification and nitrite oxidation, which were consistent with the predict results.
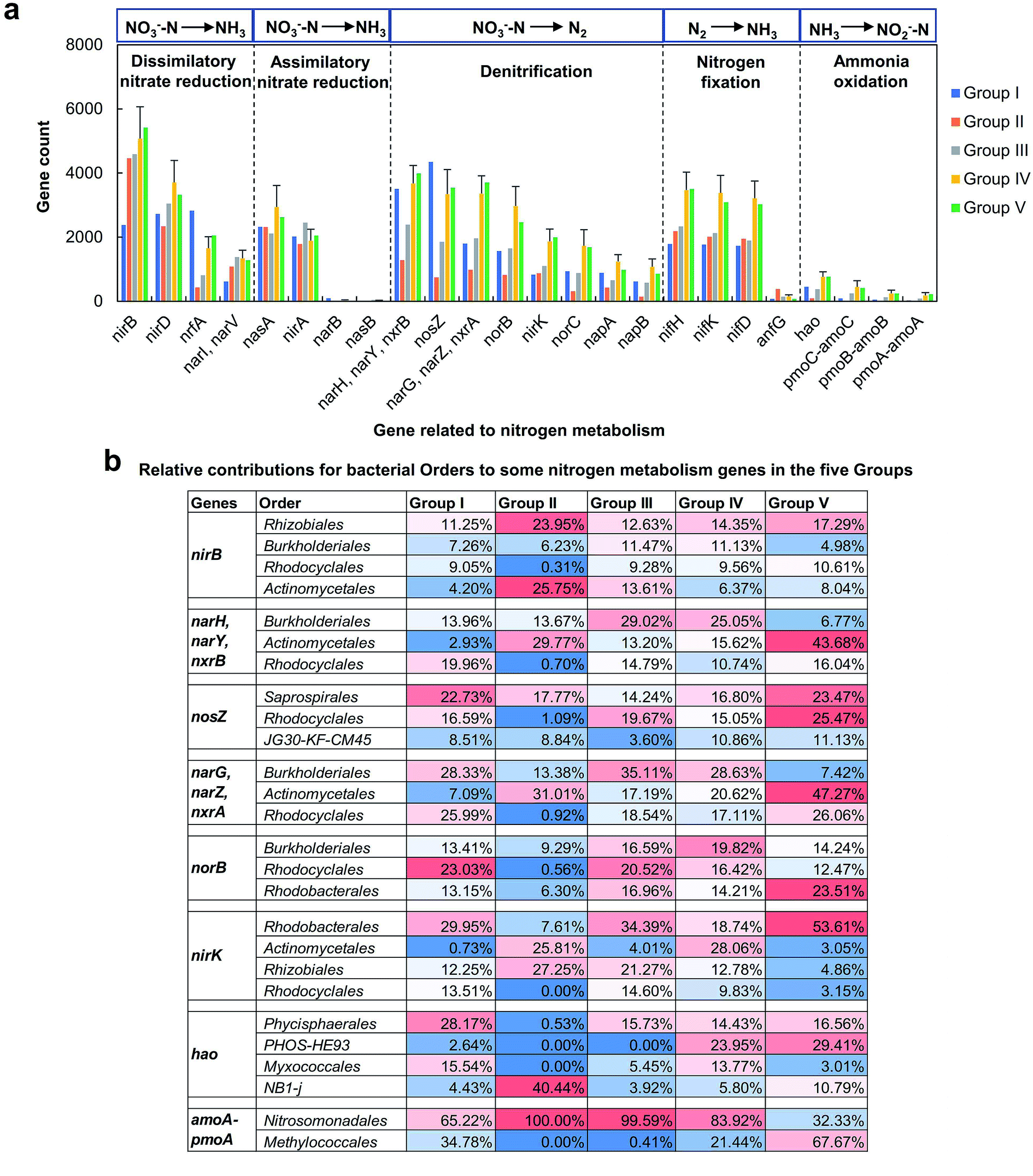 | ||
| Fig. 6 Predictive analysis of genes involved in nitrogen metabolism pathway. (a) The dynamics of counts for genes catalyzed different reactions in nitrogen cycle in different Groups. The biological processes are arranged according to the oxidation states of nitrogen (from +5 in nitrate to −3 in ammonia). (b) The relative contributions for bacterial orders to some nitrogen metabolism genes in different Groups. Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) was applied for the predictions of bacterial functional compositions. | ||
The relative contributions for bacterial orders to some genes of interest were further investigated (Fig. 6b). These genes appeared primarily among 13 taxonomically diverse orders, and many of them were predicted to be contributed by Rhizobiales, Burkholderiales, Rhodocyclales and Actinomycetales. Rhizobiales was important for the presence of nirB gene in each Group. The nirB gene also predicted to mainly occur in Burkholderiales and Rhodocyclales in Group III and IV, and this gene was largely contributed by Actinomycetales in Group II. Burkholderiales, Actinomycetales and Rhodocyclales contributed the most percentages to narH/narY/nxrB gene in Group III and V, Group II and V, and Group I, respectively. The orders contributed to narG/narZ/nxrA gene in each Group were similar as narH/narY/nxrB gene. Saprospirales and Rhodocyclales were the main orders contributed to nosZ gene in each Group. For norB gene, Rhodocyclales, Burkholderiales and Rhodobacterales showed highest contributions in Group I and III, Group II and IV, and Group V, respectively. The nirK gene in Group I, III and V were largely contributed by Rhodobacterales, while Actinomycetales and Rhizobiales were the main contributors for this gene in Group II and IV. The orders responsible for ammonia oxidation were different from the orders mentioned above. For hao gene, Phycisphaerales was the main contributor in Group I, III, IV and V, and NB1-j was the main contributor in Group II. The relative contributions of PHOS-HE93 and Myxococcales to hao gene were higher in Group IV and V. The amoA gene was predicted to occur in Nitrosomonadales. Overall, the predicted genes and their contributed orders may play important roles in nitrogen cycle. However, the predictive results need further validation based on the actual gene presence and expression using transcriptomic or metabolomics.
4. Conclusions
Our results demonstrated that the temporal dynamics of bacterial community structure, diversity and abundance were not obvious in one-year monitoring in the full-scale WWTP. The intermittent and transient genera showed different dynamics with persistent genera. Dechloromonas, Thauera, Nitrosomonas and Nitrospira were the four dominant persistent genera. WQI had the greatest contribution to bacterial community variation, followed by T and OP. qPCR analysis revealed the occurrence of Comammox. The bacterial functions were relatively stable. Many genes involved in the nitrogen metabolism pathway were predicted to be present with high gene counts. Our data show that the combination of NGS and PICRUSt is a useful method for understanding the information on both the microbial communities and potential functions in complex environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the NSFC (51078007, 51378027 and 51578015), BJNSF (8162010) and National Science and Technology Major Project (2017ZX07103-003) for the financial supports of this study.References
- P. Petropoulos and K. A. Gilbride, Can. J. Microbiol., 2005, 51, 791–799 CrossRef CAS PubMed.
- F. Ju, Y. Xia, F. Guo, Z. P. Wang and T. Zhang, Environ. Microbiol., 2014, 16, 2421–2432 CrossRef CAS PubMed.
- S. Kwon, T. Kim, G. H. Yu, J. Jung and H. Park, J. Microbiol. Biotechnol., 2010, 20, 1717–1723 Search PubMed.
- D. T. Shu, Y. L. He, H. Yue and Q. Y. Wang, Chem. Eng. J., 2016, 290, 21–30 CrossRef CAS.
- G. F. Wells, H. Park, B. Eggleston, C. A. Francis and C. S. Criddle, Water Res., 2011, 45, 5476–5488 CrossRef CAS PubMed.
- R. Hai, Y. Wang, X. Wang, Y. Li and Z. Du, PLoS One, 2014, 9, e901753 Search PubMed.
- X. H. Wang, X. H. Wen, Y. Xia, M. Hu, F. Zhao and K. Ding, PLoS One, 2012, 7, e362724 Search PubMed.
- M. E. Gentile, J. L. Nyman and C. S. Criddle, ISME J., 2007, 1, 714–728 CrossRef CAS PubMed.
- A. Valentin-Vargas, G. Toro-Labrador and A. A. Massol-Deya, PLoS One, 2012, 7, e425248 Search PubMed.
- F. Ju and T. Zhang, ISME J., 2015, 9, 683–695 CrossRef CAS PubMed.
- F. Ju, F. Guo, L. Ye, Y. Xia and T. Zhang, Environ. Microbiol. Rep., 2014, 6, 80–89 CrossRef CAS PubMed.
- M. G. Langille, J. Zaneveld, J. G. Caporaso, D. McDonald, D. Knights, J. A. Reyes, J. C. Clemente, D. E. Burkepile, T. R. Vega, R. Knight, R. G. Beiko and C. Huttenhower, Nat. Biotechnol., 2013, 31, 814–821 CrossRef CAS PubMed.
- C. Lozupone, M. Hamady and R. Knight, BMC Bioinf., 2006, 7, 371 CrossRef PubMed.
- A. R. M. Polonia, D. F. R. Cleary, R. Freitas, N. J. de Voogd and N. C. M. Gomes, Mol. Ecol., 2015, 24, 409–423 CrossRef CAS PubMed.
- W. Ahmed, C. Staley, J. Sidhu, M. Sadowsky and S. Toze, Appl. Microbiol. Biotechnol., 2017, 101, 1253–1266 CrossRef CAS PubMed.
- Q. Kong, X. He, S. S. Ma, Y. Feng and M. S. Miao, Bioresour. Technol., 2017, 244, 872–879 CrossRef CAS PubMed.
- H. Liang, D. Ye and L. Luo, RSC Adv., 2017, 7, 41727–41737 RSC.
- S. Siripong and B. E. Rittmann, Water Res., 2007, 41, 1110–1120 CrossRef CAS PubMed.
- G. Zhu, Y. Peng, S. Wang, S. Wu and B. Ma, Chem. Eng. J., 2007, 131, 319–328 CrossRef CAS.
- J. F. Gao, X. Luo, G. X. Wu, T. Li and Y. Z. Peng, Appl. Microbiol. Biotechnol., 2014, 98, 3339–3354 CrossRef CAS PubMed.
- G. F. Wells, H. Park, C. Yeung, B. Eggleston, C. A. Francis and C. S. Criddle, Environ. Microbiol., 2009, 11, 2310–2328 CrossRef CAS PubMed.
- H. Daims, E. V. Lebedeva, P. Pjevac, P. Han, C. Herbold, M. Albertsen, N. Jehmlich, M. Palatinszky, J. Vierheilig, A. Bulaev, R. H. Kirkegaard, M. von Bergen, T. Rattei, B. Bendinger, P. H. Nielsen and M. Wagner, Nature, 2015, 528, 504–509 CAS.
- M. A. H. J. van Kessel, D. R. Speth, M. Albertsen, P. H. Nielsen, H. J. M. Op Den Camp, B. Kartal, M. S. M. Jetten and S. Lucker, Nature, 2015, 528, 555–559 CAS.
- H. Daims, S. Luecker and M. Wagner, Trends Microbiol., 2016, 24, 699–712 CrossRef CAS PubMed.
- A. Gonzalez-Martinez, A. Rodriguez-Sanchez, M. C. M. van Loosdrecht, J. Gonzalez-Lopez and R. Vahala, Environ. Sci. Pollut. Res. Int., 2016, 23, 25501–25511 CrossRef CAS PubMed.
- Z. Wang, X. X. Zhang, X. Lu, B. Liu, Y. Li, C. Long and A. M. Li, PLoS One, 2014, 9, e11360311 Search PubMed.
- D. H. Eikelboom, Process Control of Activated Sludge Plants by Microscopic Investigation, IWA Publishing, UK, 2000 Search PubMed.
- P. H. Nielsen, C. Kragelund, R. J. Seviour and J. L. Nielsen, FEMS Microbiol. Rev., 2009, 33, 969–998 CrossRef CAS PubMed.
- A. T. Mielczarek, C. Kragelund, P. S. Eriksen and P. H. Nielsen, Water Res., 2012, 46, 3781–3795 CrossRef CAS PubMed.
- C. Y. Cao, I. Lou, C. Huang and M. Y. Lee, Desalin. Water Treat., 2016, 57, 2175–2183 CrossRef CAS.
- F. Guo and T. Zhang, Water Res., 2012, 46, 2772–2782 CrossRef CAS PubMed.
- T. Magoc and S. L. Salzberg, Bioinformatics, 2011, 27, 2957–2963 CrossRef CAS PubMed.
- J. G. Caporaso, J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Pena, J. K. Goodrich, J. I. Gordon, G. A. Huttley, S. T. Kelley, D. Knights, J. E. Koenig, R. E. Ley, C. A. Lozupone, D. McDonald, B. D. Muegge, M. Pirrung, J. Reeder, J. R. Sevinsky, P. J. Turnbaugh, W. A. Walters, J. Widmann, T. Yatsunenko, J. Zaneveld and R. Knight, Nat. Methods, 2010, 7, 335–336 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- M. Hu, X. Wang, X. Wen and Y. Xia, Bioresour. Technol., 2012, 117, 72–79 CrossRef CAS PubMed.
- M. P. Ginige, P. Hugenholtz, H. Daims, M. Wagner, J. Keller and L. L. Blackall, Appl. Environ. Microbiol., 2004, 70, 588–596 CrossRef CAS PubMed.
- Y. P. Mao, Y. Xia and T. Zhang, Bioresour. Technol., 2013, 128, 703–710 CrossRef CAS PubMed.
- M. Albertsen, S. J. McIlroy, M. Stokholm-Bjerregaard, S. M. Karst and P. H. Nielsen, Front. Microbiol., 2016, 7, 1033 Search PubMed.
- L. Ye and T. Zhang, Environ. Sci. Technol., 2011, 45, 7173–7179 CrossRef CAS PubMed.
- P. H. Nielsen, A. T. Mielczarek, C. Kragelund, J. L. Nielsen, A. M. Saunders, Y. Kong, A. A. Hansen and J. Vollertsen, Water Res., 2010, 44, 5070–5088 CrossRef CAS PubMed.
- M. K. de Kreuk, M. Pronk and M. C. M. van Loosdrecht, Water Res., 2005, 39, 4476–4484 CrossRef CAS PubMed.
- J. Wang, Q. Li, R. Qi, V. Tandoi and M. Yang, Process Biochem., 2014, 49, 2258–2265 CrossRef CAS.
- A. M. P. Martins, J. J. Heijnen and M. C. M. van Loosdrecht, Appl. Microbiol. Biotechnol., 2003, 62, 586–593 CrossRef CAS PubMed.
- D. Juang, Bioresour. Technol., 2005, 96, 31–40 CrossRef CAS PubMed.
- D. Frigon, R. Michael Guthrie, G. Timothy Bachman, J. Royer, B. Bailey and L. Raskin, Water Res., 2006, 40, 990–1008 CrossRef CAS PubMed.
- X. Wang, M. Hu, Y. Xia, X. Wen and K. Ding, Appl. Environ. Microbiol., 2012, 78, 7042–7047 CrossRef CAS PubMed.
- Y. Wen, Y. Jin, J. Wang and L. Cai, Pol. J. Environ. Stud., 2015, 24, 1809–1815 CrossRef CAS.
- T. P. Curtis and W. T. Sloan, Water Sci. Technol., 2006, 54, 227–236 CrossRef CAS PubMed.
- V. Kunin, S. He, F. Warnecke, S. B. Peterson, H. G. Martin, M. Haynes, N. Ivanova, L. L. Blackall, M. Breitbart, F. Rohwer, K. D. McMahon and P. Hugenholtz, Genome Res., 2008, 18, 293–297 CrossRef CAS PubMed.
- K. Heylen, D. Gevers, B. Vanparys, L. Wittebolle, J. Geets, N. Boon and P. De Vos, Environ. Microbiol., 2006, 8, 2012–2021 CrossRef CAS PubMed.
- K. Wang, X. Ye, H. Zhang, H. Chen, D. Zhang and L. Liu, Sci. Rep., 2016, 6, 38709 CrossRef CAS PubMed.
- A. Briones and L. Raskin, Curr. Opin. Biotechnol., 2003, 14, 270–276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10704h |
| This journal is © The Royal Society of Chemistry 2017 |