
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase structure and dynamics of polystyrene/poly(vinyl methyl ether) blend studied using solid-state NMR
Yong-jin Peng *a,
Yu-ling Liua,
Jun-hua Haob,
Rong-chun Zhangc and
Ping-chuan Sunc
*a,
Yu-ling Liua,
Jun-hua Haob,
Rong-chun Zhangc and
Ping-chuan Sunc
aTeaching and Research Section of Physics, College of Comprehensive Studies, Jinzhou Medical University, Jinzhou 121001, P. R. China. E-mail: hunterpyj2016@163.com
bDepartment of Physics, Tianjin University Ren'ai College, Tianjin 301636, P. R. China
cCollege of Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 13th December 2017
Abstract
In this work, solid-state 1H NMR experiments were conducted to fully characterize the dynamic characteristics of a polystyrene/poly(vinyl methyl ether) blend with a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (PS/PVME 75/25). About 13% of whole PS was determined to locate in the mobile fraction of the PS/PVME 75/25 blend at room temperature. Increasing the temperature of this blend revealed three transitions. These results are expected to improve our understanding of the glass transition process in polymer systems.
:1 (PS/PVME 75/25). About 13% of whole PS was determined to locate in the mobile fraction of the PS/PVME 75/25 blend at room temperature. Increasing the temperature of this blend revealed three transitions. These results are expected to improve our understanding of the glass transition process in polymer systems.
I. Introduction
A polymer blend (polymer alloy) is a multicomponent system of two or more components with different chemical structures and properties. It can display properties differing from those of its individual components due to the interactions between the components.1–6 Therefore the study of the relationship between the polymer blend structure and properties is very important. There have been many such studies of PS/PVME blends.7–14The PS/PVME blend is a typical lower critical solution temperature (LCST)-type polymer blend, and has garnered much attention, and many studies of its thin film and the properties of this blend doped with materials such as SiO2, carbon nanotubes and so on have been carried out.15–20
Several experiments have shown the weak hydrogen bonds between the oxygens of PVME and aromatic hydrogens of PS to be the source of the compatibility of the PS with PVME in their blend.10–13 This hydrogen bonds interaction explains the dependence of the compatibility of PS with PVME on the solvent of their blend. Wagler et al.21 used wide-line separation (WISE) experiments to reveal the presence of heterogeneities from 3.5 nm to greater than 30 nm within the PS/PVME blends, depending on the temperature of the heat treatment. The relaxation dynamics of PVME in miscible blends with PS at several concentrations under a broad range of pressure and temperature values were studied by Schwartz et al.13 using dielectric spectroscopy. Recently our group investigated the effect of the interaction between PS and PVME on the glass transition process of the blend at the micro-level using Fourier Transform Infrared Spectroscopy (FTIR).22
As a continuation of our previous work,22,23 we utilized the solid state nuclear magnetic resonance (SSNMR) technique combined with Differential Scanning Calorimetry (DSC) to investigate the phase structure and glass transition process of the PS/PVME blend. Owing to the high resolution of the NMR technique, structural and dynamic information of the blend can be obtained at the micro-level. This article is organized as described below.
First, phase structure information at room temperature was successfully obtained by carrying out dipolar filter 1H spin-diffusion experiments. Then, the evolution of the rigid part of the blend with temperature was elucidated by carrying out variable-temperature 1H double quantum filter (DQF) and Combined Rotation and Multiple Pulse Sequences (CRAMPS) experiments. Our experiments provided a molecular picture for understanding the relationships between the structure and properties and glass transition process of the PS/PVME blend.
II. Experiment
Materials and sample preparation
Polystyrene with a molecular weight of about 110 kg mol−1 was purchased from Polymer Source (Canada) and used directly. Poly(vinyl methyl ether) with a molecular weight of 67 kg mol−1 in a 50% methanol solution was purchased from TCI Company (Japan) and used after its methanol was removed. PS/PVME with a 3:1 mass ratio was dissolved in a toluene solution to a concentration of 1% (g ml−1) and was magnetically stirred at room temperature for 24 hours until it completely dissolved. The solution was rotary evaporated to yield less solvent, and then put in a vacuum oven at a temperature of 40 °C for a week and then at 60 °C for more than two days until its mass no longer changed. A transparent blend film was obtained and differential scanning calorimetry (DSC) results showed good compatibility between the PS and PVME in this blend.
DSC
The DSC thermograms were obtained using a Mettler-Toledo DSC1 STARe calorimeter. Before taking the measurements, the temperature and heat flow were calibrated. The procedure and results of the DSC experiment for the PS/PVME 75/25 blend and its corresponding pure original species were reported in detail in our previous work.22Solid-state nuclear magnetic resonance
A frequency of 399.72 MHz for the protons was used in our NMR experiments, which were performed on a Varian Infinitplus-400 wide-bore (89 mm) NMR spectrometer with a CP/MAS T3 probe having a rotor diameter of 4 mm. The 1H 12-pulse dipolar filter (DF) and double quantum filter (DQF) experiments under static conditions were performed for the PS/PVME blend over a wide temperature range.The magic angle spinning (MAS) speed was automatically controlled to be 9.8 kHz ± 1 Hz by using an MAS speed controller for the 1H CRAMPS experiment. The temperature was calibrated with PbNO3 and controlled by using a Varian model-L950 temperature controller. The fractions of rigid and mobile components in the system at a given temperature were derived from the signal intensities of, respectively, 1H DQF and DF spectra at specified filter strengths. For each proton NMR experiment, the number of scans was generally between 24 and 32. The SSNMR pulse sequences are briefly described and shown in Fig. 1.
 | ||
| Fig. 1 (a) 1H spin diffusion measurement with only 1H magnetization of the mobile phases selected by a 12-pulse dipolar filter. (b) 1H double-quantum filter (DQF) experiment. (c) 1H combined rotation and multiple pulse spectroscopy (CRAMPS) experiment. | ||
III. Results and discussion
Compatibility of the PS with PVME
The compatiblity of the PS with PVME was large when dissolved in toluene. The glass transition of the PS/PVME blend showed a wider temperature range than did the homopolymer due to the self-concentration effect (see Fig. 1 in ref. 22 and Fig. 6a in this work). This result indicated the presence of a weak interaction between PS and PVME. Our result was consistent with the previous conclusion published by others.11,12,31,32Phase structure of the PS/PVME 75/25 blend
Use of a proton dipolar filter combined with spin diffusion is an effective NMR method, and was used to characterize the micro-scale phase separation in the polymer system. As shown in Fig. 2, the 1H signal intensity reached an constant value as the diffusion time was increased due to the spin diffusion between the protons at each fixed Ncycle value (the bigger the Ncycle value, the larger was the dipolar filter strength). The ratio of the final 1H signal intensity to the initial selected 1H signal intensity was found to be equal to the ratio of the initial selected 1H signal intensity to the whole 1H signal intensity in the blend. This ratio decreased as the dipolar filter strength was increased, and reached an constant value finally when Ncycle was larger than 14 for PS/PVME 75/25 blend at room temperature as shown in Fig. 3. | ||
| Fig. 2 Static 1H spin-diffusion curve for PS/PVME 75/25 at room temperature. | ||
 | ||
| Fig. 3 Selected 1H fraction as a function of filter strength (Ncycle) for PS/PVME 75/25 at room temperature. The error bars represent the distributions of the experimental data. | ||
The final value of 40%, shown in Fig. 3, indicated the percentage of PS/PVME 75/25 blend at room temperature consisting of mobile components. This proportion exceeded the stoichiometric 1H ratio of the PVME blend by 9%. This result indicated the presence of an interphase in PS/PVME 75/25 and there were some PS components in the interphase at room temperature. This interphase was crucial to the properties of the system. About 13% of whole PS was determined to locate in the mobile fraction of the PS/PVME 75/25 blend at room temperature.
Dynamic characteristics of PS/PVME 75/25
In the calorimetric glass transition temperature range of the blend (about 0–60 °C, see Fig. 6a), the 1H DQF signal intensity of PS/PVME 75/25, which reflected the rigid phase, just showed small variations (as shown in Fig. 4). But near the corresponding PVME glass transition temperature (−25 °C), the 1H DQF signal intensity clearly dropped. This drop was related to the reduction of the dipolar coupling between protons in PVME due to its having become softer as the temperature was increased. However, the whole blend system was still in a glassy state because most of the PS phase was still rigid in this temperature range. So the total 1H DQF signal intensity was still very large.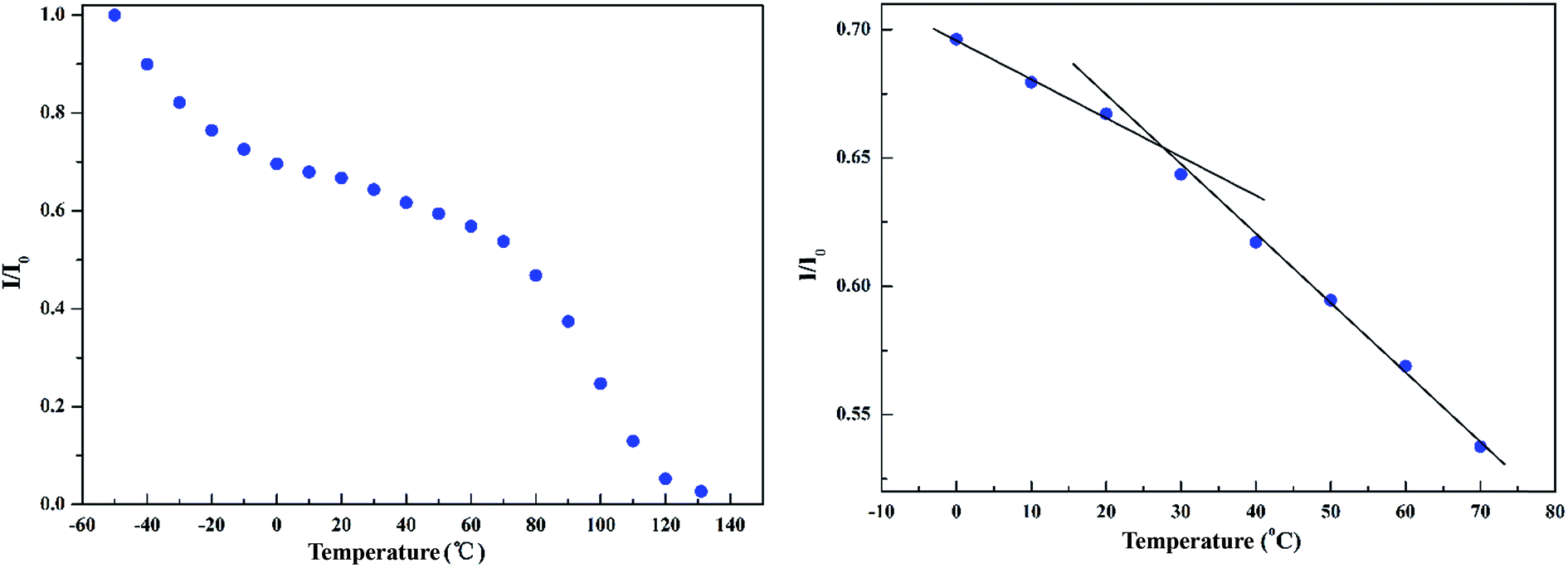 | ||
| Fig. 4 Normalized intensity of 1H DQF of PS/PVME 75/25 as a function of temperature. | ||
The specific heat capacity of the blend showed a steep change during the glass transition temperature range of the system whereas the change of the 1H DQF signal intensity was less in this temperature range than in other temperature ranges. This comparison indicated that the glass transition process evaluated using DSC corresponded to a rigid network collapse process (percolation process) rather than a melting process.23,33,34 In the calorimetric glass transition temperature range, the rigid components of the PS/PVME 75/25 blend were mainly the segments of PS (near 7 ppm corresponding to 1H in the benzene ring) and some PVME segments (near 3.2 ppm corresponding to 1H in the methoxy group) whose mobility was limited by PS as shown in Fig. 5. In this temperature range, most of the connections between the rigid parts of the polymer system were broken but many rigid parts did not yet become soft.
 | ||
| Fig. 5 Double quantum filter 1H CRAMPS of PS/PVME 75/25 at various temperatures, in degrees celsius. | ||
After the percolation process, the system gradually entered into a rubber-like state and the specific heat capacity of the system did not obviously change any longer. Most of the rigid parts of the blend started to become soft when the temperature was increased, which led to the dramatic drop of the 1H DQF signal intensity. The system finally became a liquid, with a very high mobility.
In order to show the dynamic characteristics of the PS/PVME 75/25 blend more clearly, the DSC curve, the intensity and full width at half maximum (FWHM) of the 1H DQF curve with respect to the temperature are shown in Fig. 6.
 | ||
| Fig. 6 Comparison of the SSNMR and DSC experimental results for 75/25 PS/PVME. (a) DSC traces (second heating scan). (b) Temperature dependence of the 1H DQF intensities in arbitrary units. (c) Temperature dependence of 1H DQF FWHM. (d) First derivative of the curve in (c). The three shaded regions in the figures indicate three temperature transition regions. | ||
Note that in both the temperature ranges −40 °C to −20 °C and 80 °C to 110 °C, the 1H DQF intensity of PS/PVME 75/25 dropped obviously due to the softening of PVME and PS, respectively (Fig. 6b). Because the proportion of PVME in this blend was small (25% in mass ratio), the softening of PVME did not lead to an obvious increase in mobility for the whole blend. So the FWHM of the 1H DQF curve showed a small change from −40 °C to −20 °C. In contrast, from 0 °C to 60 °C (calorimetric blend glass transition) and from 80 °C to 110 °C, the FWHM of the 1H DQF curve showed a significant decrease due to the high mobility of the system obtained through the rigid network collapse (calorimetric glass transition process) and the melting of the PS components, respectively (Fig. 6c), which was also indicated by the two local minimums in the first derivative of the 1H DQ-FWHM curve (Fig. 6d).
The above experimental results indicated that the whole glass transition of PS/PVME 75/25 blend included two processes as in the PS:23,33,35 one was the percolation process (calorimetric glass transition process), the other was the softening process of the most rigid component (after the calorimetric glass transition process).
IV. Conclusions
In summary, our 12-pulse dipolar filter 1H spin diffusion experiment results indicated the presence of an interphase in the PS/PVME 75/25 blend, and about 13% of whole PS was determined to locate in the mobile fraction of the PS/PVME 75/25 blend at room temperature.The 1H DQF and CRAMPS experimental results showed the presence of three temperature transition regions for the PS/PVME 75/25 blend: −40 °C to −20 °C (the softening of a portion of PVME), 0 °C to 60 °C (the rigid network collapse process, calorimetric glass transition) and 80 °C to 110 °C (the melting of the rigid PS component).
Our experiment results provided a picture of the dynamics of the PS/PVME 75/25 blend at the micro level and showed similar dynamic characteristics of the glass transition process of the homopolymer and miscible polymer blend. We expect these results to help us understand the glass transition of the polymer system in a deeper way.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars (No. 20825416), and the National Natural Science Foundation of China (No. 21374051), 973 program (No. 2012CB821503), PCSIRT (No. IRT1257) and CERS-1-61.References
- C. C. M. Ma, H. D. Wu and C. T. Lee, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1721–1729 CrossRef CAS.
- A. Alegria, D. Gomez and J. Colmenero, Macromolecules, 2002, 35, 2030–2035 CrossRef CAS.
- S.-C. Chen, S.-W. Kuo, U. S. Jeng, C.-J. Su and F.-C. Chang, Macromolecules, 2010, 43, 1083–1092 CrossRef CAS.
- C. M. Evans, R. W. Sandoval and J. M. Torkelson, Macromolecules, 2011, 44, 6645–6648 CrossRef CAS.
- C. M. Evans and J. M. Torkelson, Macromolecules, 2012, 45, 8319–8327 CrossRef CAS.
- Y. He, B. Zhu and Y. Inoue, Prog. Polym. Sci., 2004, 29, 1021–1051 CrossRef CAS.
- P. Xavier and S. Bose, Phys. Chem. Chem. Phys., 2014, 16, 9309–9316 RSC.
- J. K. Yeganeh, F. Goharpey and R. Foudazi, RSC Adv., 2012, 2, 8116–8127 RSC.
- S. Koizumi, Soft Matter, 2011, 7, 3984–3992 RSC.
- A. A. Bhutto, D. Vesely and B. J. Gabrys, Polymer, 2003, 44, 6627–6631 CrossRef CAS.
- M. M. Green, J. L. White, P. Mirau and M. H. Scheinfeld, Macromolecules, 2006, 39, 5971–5973 CrossRef CAS.
- J. A. Pathak, R. H. Colby, G. Floudas and R. Jerome, Macromolecules, 1999, 32, 2553–2561 CrossRef CAS.
- G. A. Schwartz, J. Colmenero and A. Alegria, Macromolecules, 2007, 40, 3246–3255 CrossRef CAS.
- H. Yin and A. Schoenhals, Polymer, 2013, 54, 2067–2070 CrossRef CAS.
- T. Xia, Y. Qin, Y. Huang, T. Huang, J. Xu and Y. Li, J. Chem. Phys., 2016, 145, 204903 CrossRef PubMed.
- P. Xavier and S. Bose, Phys. Chem. Chem. Phys., 2015, 17, 14972–14985 RSC.
- P. Xavier and S. Bose, Abstracts of Papers of the American Chemical Society, 2014, vol. 248 Search PubMed.
- T. Xia, Y. Huang, X. Jiang, Y. Lv, Q. Yang and G. Li, Macromolecules, 2013, 46, 8323–8333 CrossRef CAS.
- P. Xavier and S. Bose, J. Phys. Chem. B, 2013, 117, 8633–8646 CrossRef CAS PubMed.
- N. Li, Y.-J. Huang, T. Xia, Q. Yang and G.-X. Li, Chem. J. Chin. Univ., 2013, 34, 2896–2902 CAS.
- T. Wagler, P. L. Rinaldi, C. D. Han and H. Chun, Macromolecules, 2000, 33, 1778–1789 CrossRef CAS.
- Y.-j. Peng, Y.-l. Liu, Q. Wu and P.-c. Sun, Anal. Sci., 2017, 33, 1071–1076 CrossRef CAS PubMed.
- Y.-j. Peng, C.-t. Cai, R.-c. Zhang, T.-h. Chen, P.-c. Sun, B.-h. Li, X.-l. Wang, G. Xue and A.-C. Shi, Chin. J. Polym. Sci., 2016, 34, 446–456 CrossRef CAS.
- F. Mellinger, M. Wilhelm and H. W. Spiess, Macromolecules, 1999, 32, 4686–4691 CrossRef CAS.
- X. Wang, Q. Gu, Q. Sun, D. Zhou, P. Sun and G. Xue, Macromolecules, 2007, 40, 9018–9025 CrossRef CAS.
- A. Buda, D. E. Demco, B. Blumich, V. M. Litvinov and J. P. Penning, ChemPhysChem, 2004, 5, 876–883 CrossRef CAS PubMed.
- H. Kimura, S. Kishi, A. Shoji, H. Sugisawa and K. Deguchi, Macromolecules, 2000, 33, 9682–9687 CrossRef CAS.
- R. A. Santos, R. A. Wind and C. E. Bronnimann, J. Magn. Reson., Ser. B, 1994, 105, 183–187 CrossRef CAS PubMed.
- A. L. Webber, B. Elena, J. M. Griffin, J. R. Yates, T. N. Pham, F. Mauri, C. J. Pickard, A. M. Gil, R. Stein, A. Lesage, L. Emsley and S. P. Brown, Phys. Chem. Chem. Phys., 2010, 12, 6970–6983 RSC.
- E. Salager, R. S. Stein, S. Steuernagel, A. Lesage, B. Elena and L. Emsley, Chem. Phys. Lett., 2009, 469, 336–341 CrossRef CAS.
- S. Luo, L. Wei, J. Jiang, Y. Sha, G. Xue, X. Wang and D. Zhou, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1357–1364 CrossRef CAS.
- C. Pellerin, R. E. Prud'homme and M. Pezolet, Macromolecules, 2000, 33, 7009–7015 CrossRef CAS.
- R. P. Wool and A. Campanella, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2578–2590 CrossRef CAS.
- M. I. Ojovan, J. Non-Cryst. Solids, 2013, 382, 79–86 CrossRef CAS.
- R. P. Wool, in 6th International Conference on Times of Polymers, ed. A. Damore, L. Grassia and D. Acierno, 2012, vol. 1459, pp. 5–7 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |