
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalently anchored tertiary amine functionalized ionic liquid on silica coated nano-Fe3O4 as a novel, efficient and magnetically recoverable catalyst for the unsymmetrical Hantzsch reaction and Knoevenagel condensation†
Qiang Zhang *a,
Xiao-Ming Mab,
Huai-Xin Weia,
Xin Zhaoa and
Jun Luoc
*a,
Xiao-Ming Mab,
Huai-Xin Weia,
Xin Zhaoa and
Jun Luoc
aJiangsu Key Laboratory of Environmental Functional Materials, School of Chemistry, Biology and Material Engineering, Suzhou University of Science and Technology, Suzhou 215009, China. E-mail: zhangqiang005@gmail.com
bSchool of Pharmaceutical Engineering & Life Science, Changzhou University, Changzhou 213164, China
cSchool of Chemical Engineering, Nanjing University of Science & Technology, Nanjing 210094, China
First published on 23rd November 2017
Abstract
A novel magnetic nanoparticle supported basic ionic liquid was successfully prepared by covalently anchoring 1-(2′-piperidyl)ethyl-3-(3-propyltriethoxysilane)imidazolium chloride onto the surface of silica-coated Fe3O4 nanoparticles, and characterized by FT-IR, TEM, XRD, TGA, VSM and elemental analysis. The obtained supported ionic liquid was certified as a versatile and robust catalyst for the unsymmetrical Hantzsch reaction and Knoevenagel condensation under solvent-free conditions. Furthermore, the catalyst could be conveniently recovered by an external magnet and reused six times without significant loss of catalytic activity.
Introduction
1,4-Dihydropyridines and polyhydroquinolines are important classes of bioactive molecules and heterocyclic scaffolds in pharmaceutical research.1,2 For instance, nifedipine, nicardipine, amlodipine and felodipine, as well-known calcium antagonists, are often used for the treatment of angina pectoris and hypertension as well.3 Moreover, the 1,4-dihydropyridine and polyhydroquinoline derivatives possess many other therapeutic and pharmacological properties such as antitumor,4 antitubercular,5 antidiabetic,6 antibacterial,7 potassium-channel opening,8 acetylcholinesterase inhibition and neuroprotective agent.9,10 Additionally, these compounds could effectively behave as organic hydride donors for asymmetric transfer hydrogenation.11 Thus, the synthesis of 1,4-dihydropyridine and polyhydroquinoline derivatives is an interesting research challenge to synthetic chemists. A valuable method for the synthesis of 1,4-dihydropyridines was initially established by Arthur Hantzsch in 1881 via a multicomponent reaction of aldehydes with ethyl acetoacetate and ammonia.12 Subsequently, a variety of improved methods for the synthesis of polyhydroquinolines, using metal triflates,13,14 organocatalysts,15 bakers' yeast,16 CAN,17 p-TSA,18 FeF3,19 Hf(NPf2)4,20 Ni or Pd nanoparticles,21,22 KH2PO4,23 microwave and ultrasound irradiation,24,25 have been reported. However, these procedures suffer from one or more drawbacks in terms of using volatile organic solvents, long reaction time, low yields, tedious product separation and catalyst recycling. To overcome these limitations, some attractive heterogeneous catalysts such as In–SiO2,26 cellulose supported sulfamic acid,27 ILOS@Fe/TSPP,28 V–TiO2,29 SPPN polymer,30 PdRuNi@GO,31 boehmite-SSA,32 nano-ZrO2–SO3H,33 Fe3O4 supported Cu and Ni complex34,35 are applied in this regard. Although these reported methods show respective advantages and provide better improvements for the synthesis of the target molecules, there is still scope for improvement, particularly towards developing a milder and cleaner protocol without using precious metals and organic solvents.On the other hand, due to undetectable vapor pressure, high thermal stability, excellent solubility and catalytic activity, ionic liquids (ILs) have attracted considerable attention as green reaction media or catalysts.36,37 Despite of their widespread use in organic reactions, some drawbacks, for instance difficult product separation and catalyst recovery due to their high viscosity, and use of large amounts of ILs in biphasic systems, still exist yet. More recently, a concept of nanoconfined ionic liquids has been proposed,38 which combines the benefits of nano-supports and ILs, such as minimizing the dosage of ILs, high designability and excellent activity. However, nanoparticles are difficult to separate by filtration and expensive ultracentrifugation is the main way to separate the product and catalyst. This limitation can be overcame by using magnetic nanoparticles (MNPs), which can be simply recovered from the reaction mixture by magnetic separation.39 A variety of magnetically retrievable catalysts have been employed in a range of organic transformations,40–42 and some immobilization processes for functional ILs on MNPs supports have been developed.43–45
In order to explore a new protocol for the synthesis of polyhydroquinolines and continue our investigations on the recoverable catalysts for organic transformations,46–50 herein, we demonstrated a novel silica coated nano-Fe3O4 supported basic ionic liquid (BIL@MNP) and its application as a robust and magnetically recoverable catalyst for the one-pot synthesis of polyhydroquinolines via the unsymmetrical Hantzsch reaction. Interestingly, this novel catalyst could act as an efficient and reusable catalyst for Knoevenagel condensation under solvent-free conditions as well (Scheme 1).
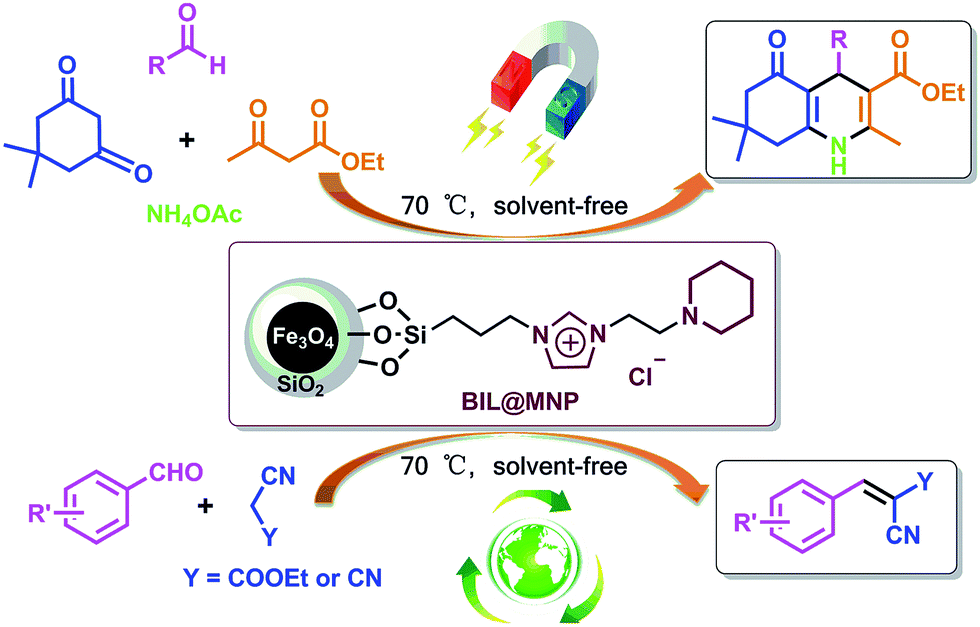 | ||
| Scheme 1 The BIL@MNP catalyzed unsymmetrical Hantzsch reaction and Knoevenagel condensation. | ||
Results and discussion
The magnetic nanoparticle supported basic ionic liquid catalyst (BIL@MNP) was prepared following the procedure shown in Scheme 2. N-(2-Chloroethyl)piperidine was firstly treated with imidazole to give 1-(2-(1H-imidazol-1-yl)ethyl)piperidine 1 under basic conditions. Then, a quaternization of (3-chloropropyl)triethoxysilane and the compound 1 afforded the precursor ionic liquid 2 (BIL). Ultimately, a suspension of SiO2@Fe3O4 with a chloroform solution of the BIL was refluxed for 36 h to undergo a condensation reaction, which provided the desired catalyst 3 (BIL@MNP).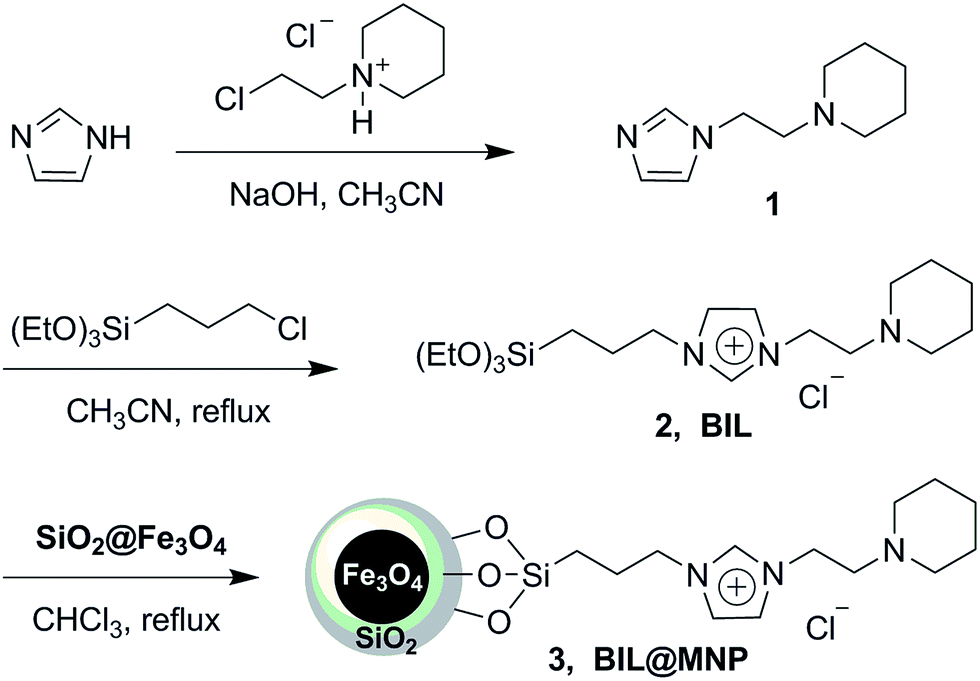 | ||
| Scheme 2 The synthetic route for BIL@MNP. | ||
In order to confirm the successful functionalization of MNPs, FT-IR was employed to give a detailed investigation of the SiO2@Fe3O4, BIL, and BIL@MNP (Fig. 1). The Si–O–Si and Fe–O vibrations of the SiO2@Fe3O4 could be observed at around 1097 and 580 cm−1, respectively. The IR curve of BIL shows typical bands at 1563 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration and CC vibrations of the imidazole ring), 2941, 2866 and 1456 cm−1 (alkyl chain stretching and deformation vibrations). While in the spectrum of BIL@MNP, these characteristic peaks are at the similar wavenumbers, with a slight offset due to the interaction with the support. Nevertheless, all of these significant features cannot be observed in the support SiO2@Fe3O4. Moreover, the nitrogen content of BIL@MNP determined by elemental analysis was 1.51%, which shows that the loading amount of the imino–pyridine ligand was calculated to be 0.36 mmol g−1. The above results indicate that the BIL was successfully grafted onto the SiO2@Fe3O4.
N vibration and CC vibrations of the imidazole ring), 2941, 2866 and 1456 cm−1 (alkyl chain stretching and deformation vibrations). While in the spectrum of BIL@MNP, these characteristic peaks are at the similar wavenumbers, with a slight offset due to the interaction with the support. Nevertheless, all of these significant features cannot be observed in the support SiO2@Fe3O4. Moreover, the nitrogen content of BIL@MNP determined by elemental analysis was 1.51%, which shows that the loading amount of the imino–pyridine ligand was calculated to be 0.36 mmol g−1. The above results indicate that the BIL was successfully grafted onto the SiO2@Fe3O4.
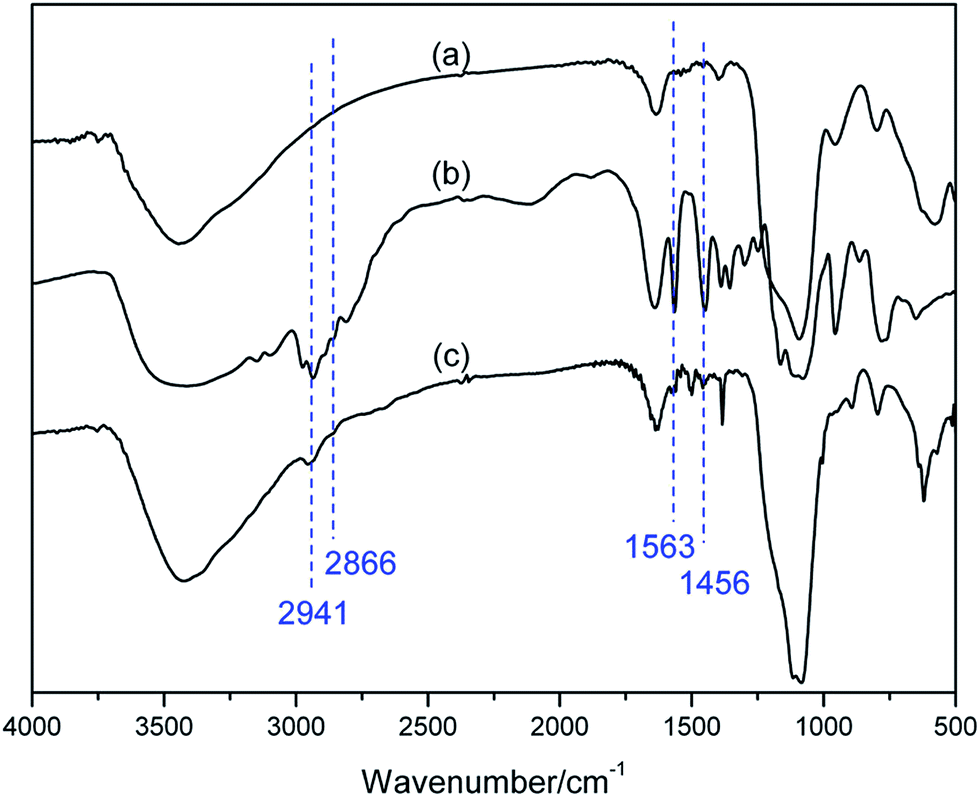 | ||
| Fig. 1 FT-IR spectra of the SiO2@Fe3O4 (a), BIL (b) and BIL@MNP (c). | ||
X-ray reflective diffraction (XRD) and transmission electron microscopy (TEM) were then carried out to obtain detailed information about the structure and morphology of as-prepared catalyst. The high-angle XRD pattern of BIL@MNP (Fig. 2) exhibits diffraction peaks corresponding to the standard Fe3O4 sample (JCPDS file no. 19-0629), which clearly indicates that the surface modification of MNPs do not damage the crystal structure of Fe3O4 core. And the TEM image of BIL@MNP shows that the dark nano-Fe3O4 cores are surrounded by grey silica shell and the average size of BIL@MNP is about 20 nm (Fig. 3a).
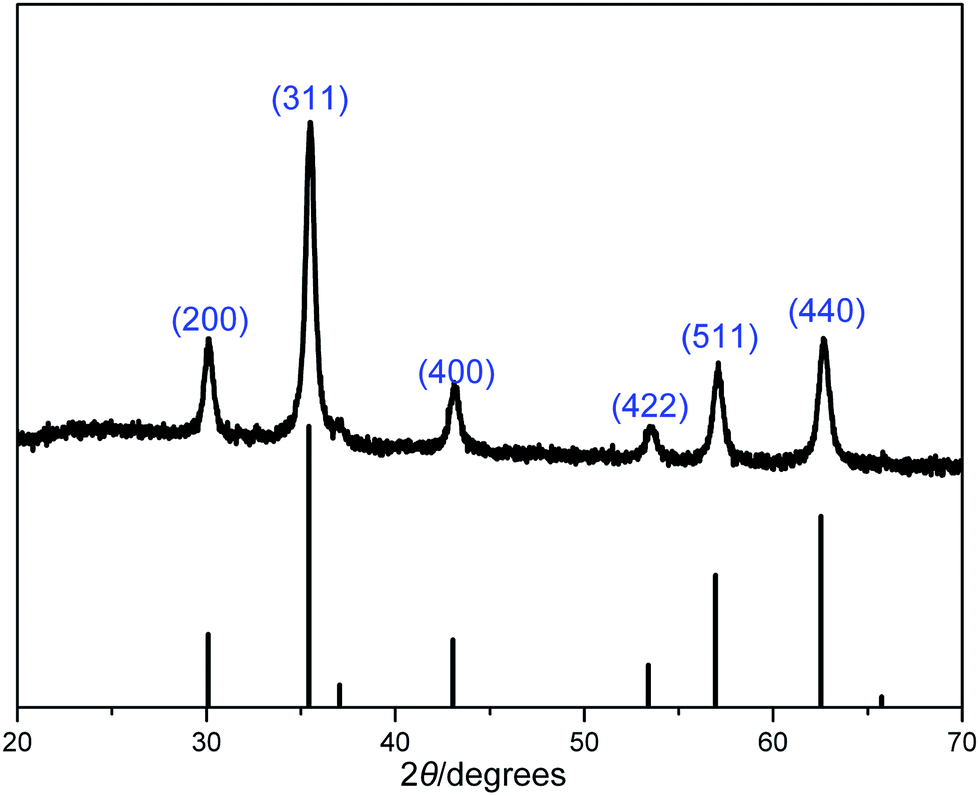 | ||
| Fig. 2 XRD pattern of BIL@MNP. The bottom row of tick marks indicates the reflection positions for a standard magnetite pattern (JCPDS no. 19-0629). | ||
 | ||
| Fig. 3 TEM images of BIL@MNP (a) and six-times reused catalyst (b). | ||
The stability of the BIL@MNP was determined by the thermogravimetric analysis (Fig. 4). The TG curve indicates an initial weight loss of 1.0% up to 150 °C due to the adsorbed water in support. Complete loss of the supported IL was found in the temperature range from 200 °C to 500 °C, and the amount of organic structure was about 17.3% against the total solid catalyst. Meanwhile, the DTG curve shows that the decomposition of ionic moiety mainly occured in one step from 250 °C to 450 °C, which is related to main weight loss of 14.8%. And the peak in the DTG curve shows the fastest loss of the IL occured at 365 °C. Therefore, the BIL@MNP is stable below about 200 °C.
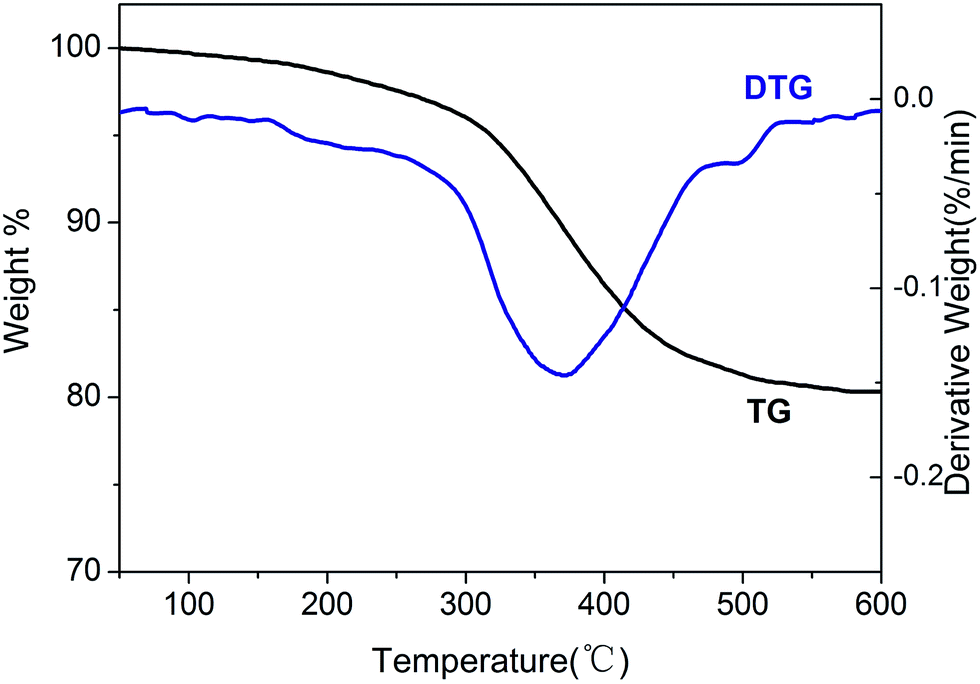 | ||
| Fig. 4 TG-DTG analysis for AIL@MNP. | ||
The magnetic property of BIL@MNP was evaluated using vibrating sample magnetometer (VSM) at room temperature. The VSM magnetization curve goes through the zero point (Fig. 5). The phenomenon of no magnetic hysteresis indicates that the as-prepared catalyst is superparamagnetic. As a result, BIL@MNP can be simply separated from the reaction mixture using an external magnet.
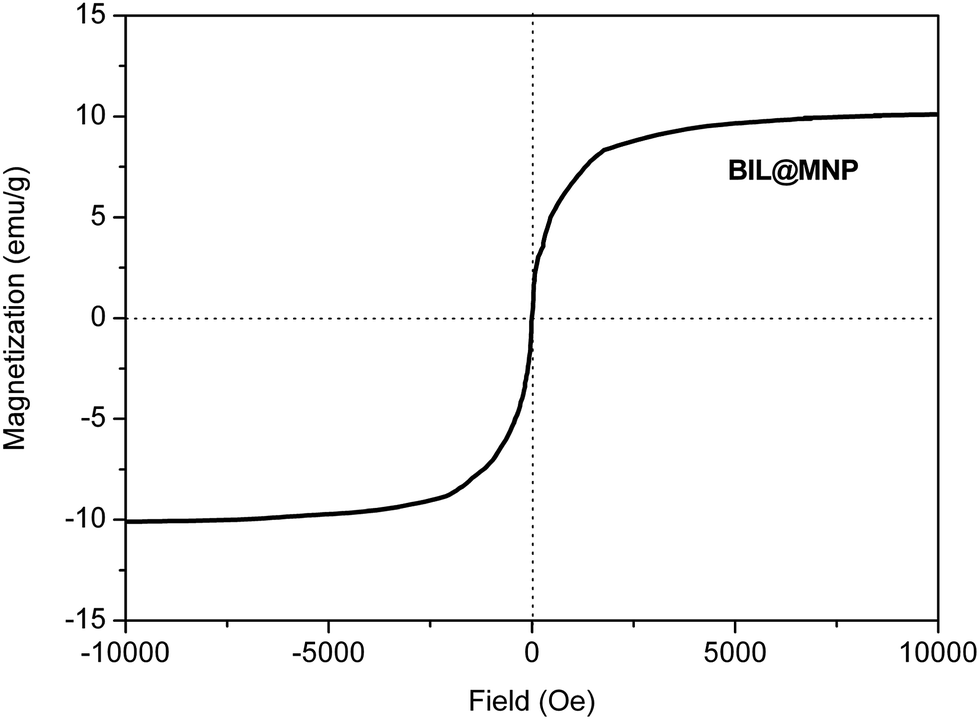 | ||
| Fig. 5 Magnetic curve of BIL@MNP. | ||
To investigate the catalytic activity of the newly designed catalyst, it was initially tested for the one-pot synthesis of polyhydroquinolines. The four-component condensation of benzaldehyde (1 mmol), dimedone (1 mmol), ethyl acetoacetate (1 mmol) and ammonium acetate (1 mmol), as a model reaction, was firstly performed in the presence of BIL@MNP (1.0 mol%) for 30 min at 70 °C under solvent-free conditions. And the corresponding product was obtained in 60% yield. Encouraged by this result, we increased gradually the amount of catalyst from 0 to 3.0 mol% (Table 1, entries 1–6). In the absence of BIL@MNP, only a small amount of product was detected, whereas the yield was improved as the amount of BIL@MNP increased from 0 to 2.5 mol% and became almost steady when the amount of catalyst was further increased beyond this. The influence of reaction temperature was also investigated (Table 1, entries 7–9), which indicated that lower temperatures (50–60 °C) decelerated the reaction rate and led to lower yields and no obvious improvement was achieved by the elevated temperature (80 °C). Hence, the best result was obtained in the presence of 2.5 mol% of BIL@MNP at 70 °C (Table 1, entry 5). In order to show the unique role of BIL@MNP, some control experiments in the presence of the SiO2@Fe3O4 (blank support), BIL (unsupported ionic liquid) and triethylamine (traditional tertiary amine) were further carried out and they could also afford moderate to good yields under the same conditions (Table 1, entries 10–12). However, they were obviously inferior to the BIL@MNP. These results indicated that the high catalytic activity of BIL@MNP for the one-pot synthesis of polyhydroquinolines could be attributed to the synergistic interaction between the surface silanol groups of nano-support and ionic liquid moieties.
| Entry | Catalyst (mol%) | Temperature (°C) | Time (min) | Isolated yield (%) |
|---|---|---|---|---|
| 1 | — | 70 | 30 | 24 |
| 2 | BIL@MNP (1.0) | 70 | 30 | 60 |
| 3 | BIL@MNP (1.5) | 70 | 25 | 73 |
| 4 | BIL@MNP (2.0) | 70 | 20 | 84 |
| 5 | BIL@MNP (2.5) | 70 | 15 | 92 |
| 6 | BIL@MNP (3.0) | 70 | 15 | 92 |
| 7 | BIL@MNP (2.5) | 50 | 15 | 68 |
| 8 | BIL@MNP (2.5) | 60 | 15 | 85 |
| 9 | BIL@MNP (2.5) | 80 | 15 | 93 |
| 10 | SiO2@Fe3O4 (70 mg) | 70 | 15 | 45 |
| 11 | BIL (2.5) | 70 | 15 | 81 |
| 12 | NEt3 (2.5) | 70 | 15 | 70 |
The generality of this method for the four-component condensation of structurally diverse aldehydes, dimedone, ethyl acetoacetate and ammonium acetate under the optimized conditions was then investigated and the results are shown in Table 2. The aromatic aldehydes bearing either electron-withdrawing or electron-donating groups (Table 2, entries 1–13) were converted to the corresponding polyhydroquinolines in good to excellent yields (84–93%). Interestingly, heteroaromatic aldehydes such as thiophene-2-carbaldehyde and furan-2-carbaldehyde (Table 2, entries 14 and 15) also underwent these transformations well to afford the target products in excellent yields (86–90%). Furthermore, the aliphatic aldehyde, which is a usually less reactive substrate,28 was smoothly converted to the corresponding product as well (Table 2, entry 16). These results demonstrate that the designed catalyst BIL@MNP showed an excellent performance for the one-pot synthesis of polyhydroquinolines.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time (min) | Product | Yielda (%) | Mp (°C) [Lit]ref |
| a Isolated yield. | |||||
| 1 | Ph | 15 | 4a | 92 | 202–204 [203–204]29 |
| 2 | 4-Me-C6H4 | 18 | 4b | 90 | 259–261 [261–262]29 |
| 3 | 4-MeO-C6H4 | 20 | 4c | 91 | 254–256 [255–257]45 |
| 4 | 4-OH-C6H4 | 30 | 4d | 87 | 232–233 [230–231]29 |
| 5 | 4-OH-3-MeO-C6H3 | 35 | 4e | 93 | 210–211 [211–212]29 |
| 6 | 3-NO2-C6H4 | 25 | 4f | 84 | 173–175 [174–177]35 |
| 7 | 4-NO2-C6H4 | 20 | 4g | 89 | 241–243 [242–243]45 |
| 8 | 4-F-C6H4 | 20 | 4h | 85 | 191–193 [193–195]45 |
| 9 | 4-Br-C6H4 | 15 | 4i | 92 | 251–253 [253–255]13 |
| 10 | 3-Br-C6H4 | 20 | 4j | 88 | 232–234 [230–232]35 |
| 11 | 2,4-Cl2-C6H3 | 15 | 4k | 90 | 244–245 [241–244]13 |
| 12 | 3-Cl-C6H4 | 18 | 4l | 91 | 203–205 [192–194]51 |
| 13 | 4-Cl-C6H4 | 20 | 4m | 89 | 243–245 [245–247]28 |
| 14 | 2-Thienyl | 15 | 4n | 90 | 238–240 [238–240]13 |
| 15 | 2-Furyl | 18 | 4o | 86 | 246–247 [247–248]29 |
| 16 | n-C3H7 | 20 | 4p | 83 | 178–180 [165–167]52 |
In order to evaluate the merit of the method for the synthesis of polyhydroquinolines, we compared the efficiency of BIL@MNP with some recently reported catalysts for the one-pot four-component synthesis of 4a. As shown in Table 3, BIL@MNP showed a better activity in terms of no precious metals, organic solvents or additional irradiation.
| Entry | Catalyst | Conditions | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | Hf(NPf2)4 (1 mol%) | C10F18/60 °C | 180 | 95 | 20 |
| 2 | Cell-Pr-NHSO3H (50 mg) | EtOH/reflux | 50 | 90 | 27 |
| 3 | PdRuNi@GO (6 mg) | DMF/70 °C | 45 | 93 | 31 |
| 4 | Nano-Pd (4 mol%) | THF/reflux | 240 | 89 | 22 |
| 5 | Boehmite-SSA (30 mg) | EtOH/reflux | 215 | 94 | 32 |
| 6 | V–TiO2 (2.0 mol%) | Solvent-free/80 °C | 10 | 85 | 29 |
| 7 | Ni(NO3)2/Fe3O4@SiO2 (20 mg) | Solvent-free/100 °C | 20 | 96 | 35 |
| 8 | SPPN polymer (6 mg) | EtOH/120 °C/microwave | 5 | 96 | 30 |
| 9 | Co3O4-CNTs (30 mg) | EtOH/50 °C/sonication | 15 | 97 | 53 |
| 10 | GSA@MNPs (50 mg) | EtOH/reflux | 240 | 90 | 54 |
| 11 | Fe3O4@B-MCM-41 (50 mg) | EtOH/reflux | 65 | 90 | 55 |
| 12 | BIL@MNP (2.5 mol%) | Solvent-free/70 °C | 15 | 92 | This work |
A plausible mechanism for the synthesis of polyhydroquinolines catalyzed by BIL@MNP is proposed in Scheme 3. Initially, the enol form of dimedone performs a nucleophilic addition to the activated aldehyde to provide the corresponding Knoevenagel adduct I, assisted by the synergistic interaction between the surface silanol and piperidine sites of BIL@MNP. On the other hand, the β-ketoester reacts with ammonium acetate to afford the enamine intermediate II in the presence of BIL@MNP as well. Then, the Michael addition reaction between these two intermediates generates the intermediate III, which is also promoted by BIL@MNP. Finally, the target products are formed via an intramolecular cyclization and elimination of H2O.
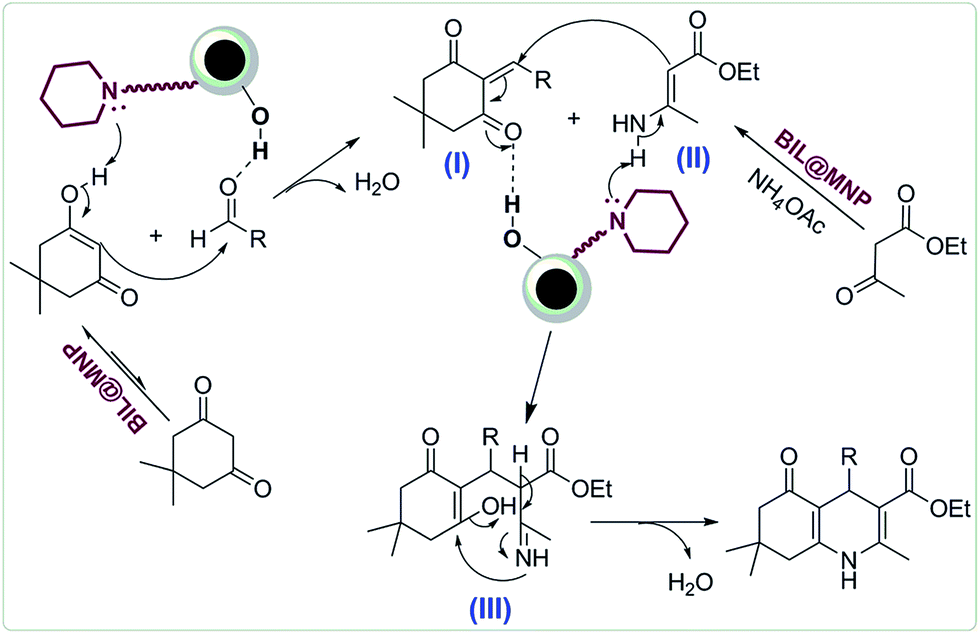 | ||
| Scheme 3 Plausible mechanism for the synthesis of polyhydroquinolines. | ||
The recovery and reuse of catalysts is highly preferable for a sustainable process. Hence, the reusability of BIL@MNP was evaluated by the reaction of benzaldehyde, dimedone, ethyl acetoacetate, and ammonium acetate under the optimized conditions. Upon completion of the reaction, ethyl acetate was added and the catalyst was simply separated by an external magnet and washed with ethyl acetate, and reused for subsequent cycles after drying under vacuum. As depicted in Fig. 6, the catalyst could be reused six times without any significant loss of catalytic activity. The recovered catalyst after six runs had no obvious change in structure, referring to the FT-IR spectra in comparison with fresh one (Fig. 7). And there was also no apparent change in the morphology and size by a TEM observation of the recovered catalyst (Fig. 3b). These results revealed that the catalyst was very stable and could endure the reaction conditions.
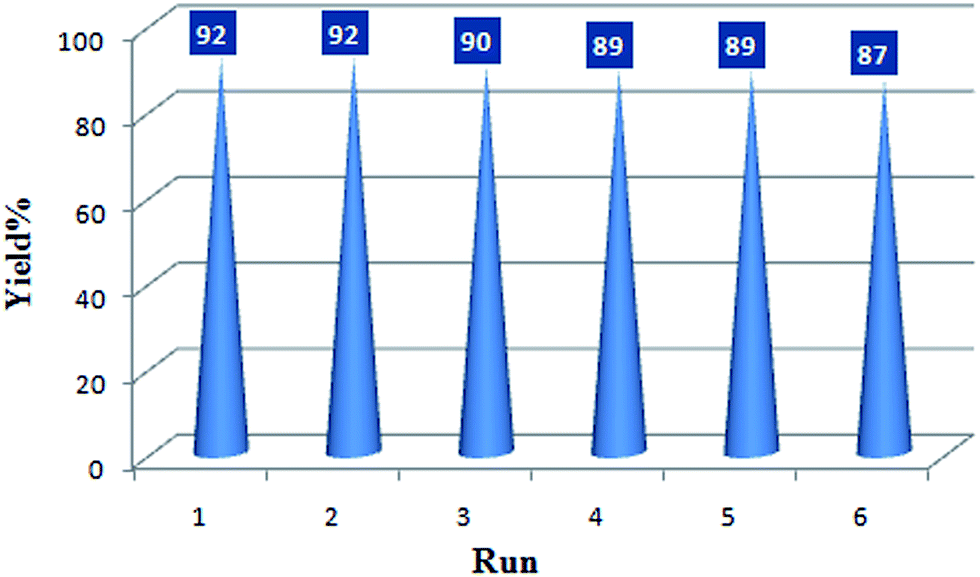 | ||
| Fig. 6 Recycling experiment of BIL@MNP. | ||
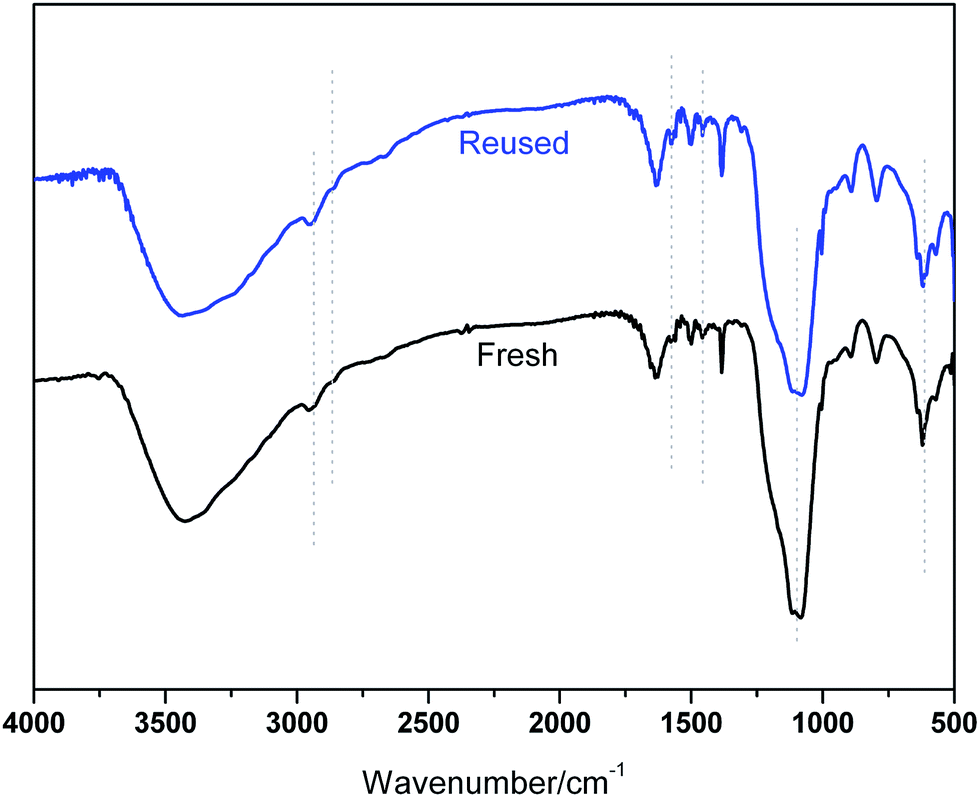 | ||
| Fig. 7 FT-IR spectra of the fresh catalyst and six-times reused catalyst. | ||
Moreover, a hot filtration experiment was carried out to explore the catalyst leaching. The reaction of benzaldehyde (1 mmol), dimedone (1 mmol), ethyl acetoacetate (1 mmol) and ammonium acetate (1 mmol) catalyzed by BIL@MNP (2.5 mol%) was performed at 70 °C under solvent-free conditions. When the reaction time reached 6 min, hot ethyl acetate (10 mL) was added and the BIL@MNP was fleetly separated by magnetic force. The solution was averagely divided into two sections (S1 and S2). The product of S1 was obtained in 47% yield and neat S2 was reacted at 70 °C for another 9 min to afford the product in 49%, which was similar to S1 and less than the normal (92%; Table 2, entry 1). Thus these above results indicated that the leaching of BIL@MNP was negligible in the catalytic process.
Besides the synthesis of polyhydroquinolines, such an environmentally benign catalyst might provide a wider application in traditional organic base catalyzed reactions, and we additionally extended the application scope of the catalytic system to the Knoevenagel condensation. The condensation of various aldehydes and ethyl cyanoacetate or malononitrile were investigated under the above-mentioned optimized conditions. The results are summarized in Table 4. Aromatic aldehydes with electron-donating or electron-withdrawing groups, and even heteroaromatic aldehyde, underwent smoothly transformation in a short time (10–25 min) with good to excellent yields (86–98%). Moreover, the recyclability of BIL@MNP was also evaluated by the Knoevenagel condensation of 3-nitrobenzaldehyde and ethyl cyanoacetate and the yield remained almost constant in a test of six cycles (Table 4, entry 3), which shows that the BIL@MNP still had an excellent recyclability in this traditional base-catalyzed reaction.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Y | Time (min) | Product | Yield (%)a | Mp (°C) [Lit]ref |
| a Isolated yield.b Run 2.c Run 4.d Run 6. | ||||||
| 1 | Ph | COOEt | 15 | 5a | 94 | 46–47 [47–48]56 |
| 2 | 4-NO2-C6H4 | COOEt | 12 | 5b | 95 | 163–165 [165–167]56 |
| 3 | 3-NO2-C6H4 | COOEt | 12 | 5c | 96 (96b, 95c, 93d) | 130–131 [130–131]57 |
| 4 | 4-Cl-C6H4 | COOEt | 20 | 5d | 94 | 87–89 [86–88]57 |
| 5 | 4-F-C6H4 | COOEt | 20 | 5e | 95 | 92–94 [90–92]56 |
| 6 | 4-Br-C6H4 | COOEt | 20 | 5f | 91 | 90–91 [88–89]58 |
| 7 | 4-Me-C6H4 | COOEt | 20 | 5g | 94 | 89–91 [89–90]59 |
| 8 | 4-OH-3-MeO-C6H3 | COOEt | 35 | 5h | 86 | 108–110 [110–111]59 |
| 9 | 2-Furyl | COOEt | 20 | 5i | 97 | 86–88 [85–87]56 |
| 10 | Ph | CN | 10 | 5j | 97 | 79–80 [79–80]56 |
| 11 | 3-NO2-C6H4 | CN | 10 | 5k | 95 | 101–102 [99–101]57 |
| 12 | 4-MeO-C6H4 | CN | 20 | 5l | 98 | 111–113 [111–113]57 |

Conclusions
In conclusion, we have developed a novel silica coated nano-Fe3O4 supported basic ionic liquid (BIL@MNP) as a robust and magnetically recoverable catalyst for the one-pot synthesis of polyhydroquinolines via the unsymmetrical Hantzsch reaction. The notable advantages of this procedure are mild reaction conditions, short reaction time, high yields and no precious metals, organic solvents or additional irradiation, which makes it a better and more practical alternative to the existing methods. Moreover, the catalyst could be simply separated by an external magnet, avoiding the tedious recovery procedure via filtration or centrifugation, and reused without apparent loss of activity in the test of six cycles. Besides the synthesis of polyhydroquinolines, this novel catalyst has been applied as an efficient and reusable catalyst for Knoevenagel condensation under solvent-free conditions as well and the research of application in other base-catalyzed reactions is an ongoing project.Experimental
General
Melting points were determined on a Perkin-Elmer differential scanning calorimeter and uncorrected. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE III spectrometer. The IR spectra were run on a Nicolet spectrometer (KBr). Elemental analysis was performed on Elementar Vario MICRO spectrometer. Mass spectra were obtained with an automated FININIGAN TSQ Advantage mass spectrometer. Thermogravimetric analysis was carried out under nitrogen using a Shimadzu TGA-50 spectrometer. X-Ray diffraction (XRD) images were obtained from a Bruker XRD D8 Advance instrument with Cu Kα radiation. The magnetization curve was obtained by a vibrating sample magnetometer (JDM-13T, China). Transmission electron microscope (TEM) images were obtained from a JEOL JEM-2010 instrument. All the solvents used were strictly dried according to standard operation and stored on 4A molecular sieves. All other chemicals (AR grade) were commercially available and used without further purification.Preparation of the magnetic nanoparticle supported basic ionic liquid catalyst (BIL@MNP)
1H NMR (500 MHz, CDCl3) δ 7.43 (s, 1H), 6.92 (s, 1H), 6.87 (s, 1H), 3.92 (q, J = 6.5 Hz, 2H), 2.52 (t, J = 6.5 Hz, 2H), 2.30 (s, 4H), 1.49–1.45 (m, 4H), 1.35–1.32 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 137.14, 128.78, 119.09, 59.21, 54.45, 44.59, 25.80, 24.00.
3-Chloropropyltriethoxysilane (5.0 mL, 21 mmol) was added dropwise into a solution of 1 (3.6 g, 20 mmol) in acetonitrile (30 mL) under a nitrogen atmosphere and the mixture was refluxed for three days. The solvent was removed by rotatory evaporation under reduced pressure and the obtained residue was washed with absolute ether for several times, then dried under vacuum to give the precursor 2 as a viscous orange liquid in 92% yield.
1H NMR (500 MHz, CDCl3) δ 10.17 (s, 1H), 7.64 (s, 1H), 7.28 (s, 1H), 4.33 (t, J = 5.5 Hz, 2H), 4.17 (t, J = 7.5 Hz, 2H), 3.64 (q, J = 7.0 Hz, 6H), 2.58 (t, J = 5.5 Hz, 2H), 2.27 (s, 1H), 1.87–1.81 (m, 2H), 1.38–1.34 (m, 4H), 1.25 (s, 2H), 1.04 (t, J = 7.0 Hz, 9H), 0.44 (t, J = 8.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 137.26, 122.98, 121.18, 58.41, 57.71, 54.20, 51.51, 46.68, 25.84, 24.25, 23.93, 18.12, 6.97; FT-IR (KBr, cm−1): 3146, 3093, 2986, 2941, 2866, 2809, 1645, 1563, 1456, 1394, 1355, 1172, 1085, 959, 782, 650; ESI-MS: m/z 384.2 (M+–Cl); anal. calc. for C19H38ClN3O3Si: C, 54.33; H, 9.12; N, 10.00. Found: C, 54.49; H, 9.71; N, 9.89.
General procedure for the synthesis of polyhydroquinolines
A mixture of aldehyde (1 mmol), dimedone (1 mmol), ethyl acetoacetate (1 mmol), ammonium acetate (1 mmol) and BIL@MNP (70 mg) was stirred at 70 °C in an oil bath for a certain time, as indicated by TLC for a complete reaction. Ethyl acetate was added and the catalyst was separated magnetically from the product solution, washed with ethyl acetate, and dried under vacuum. Pure polyhydroquinolines were afforded by evaporation of the solvent followed by recrystallization from the ethanol–water mixture. All products were characterized by spectral data and compared with their physical data given in the literature.General procedure for Knoevenagel condensation
A mixture of aromatic aldehyde (1 mmol), activated methylene compounds (1.1 mmol), and BIL@MNP (70 mg) was stirred at 70 °C in an oil bath for a certain time, as indicated by TLC for a complete reaction. Ethyl acetate was added and the catalyst was separated magnetically from the product solution, washed with ethyl acetate, and dried under vacuum. The pure final products were afforded by evaporation of the solvent followed by recrystallization from the ethanol-water mixture. All products were characterized by spectral data and compared with their physical data given in the literature.Selected data for typical compounds
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is financially supported by Natural Science Foundation of Jiangsu Province (China) (No. BK20150282); Innovation and Entrepreneurship Training Program for Undergraduates in Jiangsu Province (201713985010Y, 201710332051X); A project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. The authors would like to thank Excellent Innovation Team in Science and Technology of Education Department of Jiangsu Province for discussions.References
- J. P. Wan and Y. Liu, RSC Adv., 2012, 2, 9763–9777 RSC.
- V. K. Sharma and S. K. Singh, RSC Adv., 2017, 7, 2682–2732 RSC.
- K. C. Yedinak, Am. Pharm., 1993, 33, 49–64 CrossRef.
- M. Kawase, A. Shah, H. Gaveriya, N. Motohashi, H. Sakagami, A. Varga and J. Molnár, Bioorg. Med. Chem., 2002, 10, 1051–1055 CrossRef CAS PubMed.
- P. S. Eharkar, B. Desai, H. Gaveria, B. Varu, R. Loriya, Y. Naliapara, A. Shah and V. M. Kulkarni, J. Med. Chem., 2002, 45, 4858–4867 CrossRef.
- A. K. Ogawa, C. A. Willoughby, R. Bergeron, K. P. Ellsworth, W. M. Geissler, R. W. Myer, J. Yao, H. Harris and K. T. Chapman, Bioorg. Med. Chem. Lett., 2003, 13, 3405–3408 CrossRef CAS PubMed.
- K. Sirisha, D. Bikshapathi, G. Achaiah and V. M. Reddy, Eur. J. Med. Chem., 2011, 46, 1564–1571 CrossRef CAS PubMed.
- P. A. Hopes, A. J. Parker and I. Patel, Org. Process Res. Dev., 2006, 10, 808–813 CrossRef CAS.
- J. Marco-Contelles, R. León, C. de los Rios, A. Guglietta, J. Terencio, M. G. López, A. G. Garcia and M. Villarroya, J. Med. Chem., 2006, 49, 7607–7610 CrossRef CAS PubMed.
- R. León, C. de los Rios, J. Marco-Contelles, O. Huertas, X. Barril, F. J. Luque, M. G. López, A. G. García and M. Villarroya, Bioorg. Med. Chem., 2008, 16, 7759–7769 CrossRef PubMed.
- C. Zheng and S. L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef.
- L. M. Wang, J. Sheng, L. Zhang, J. W. Han, Z. Y. Fan, H. Tian and C. T. Qian, Tetrahedron, 2005, 61, 1539–1543 CrossRef CAS.
- J. L. Donelson, R. A. Gibbs and S. K. De, J. Mol. Catal. A: Chem., 2006, 256, 309–311 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron, 2007, 63, 1946–1952 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron Lett., 2007, 48, 3887–3890 CrossRef CAS.
- S. K. Ko and C. F. Yao, Tetrahedron, 2006, 62, 7293–7299 CrossRef CAS.
- A. Kumar and R. A. Maurya, Synlett, 2008, 883–885 CrossRef CAS.
- R. Surasani, D. Kalita, A. V. D. Rao, K. Yarbagi and K. B. Chandrasekhar, J. Fluorine Chem., 2012, 135, 91–96 CrossRef CAS.
- M. Hong, C. Cai and W. B. Yi, J. Fluorine Chem., 2010, 131, 111–114 CrossRef CAS.
- S. B. Sapkal, K. F. Shelke, B. B. Shingate and M. S. Shingare, Tetrahedron Lett., 2009, 50, 1754–1756 CrossRef CAS.
- M. Saha and A. K. Pal, Tetrahedron Lett., 2011, 52, 4872–4877 CrossRef CAS.
- S. J. Yü, S. Wu, X. M. Zhao and C. W. Lü, Res. Chem. Intermed., 2017, 43, 3121–3130 CrossRef.
- B. D. Bala, K. Balamurugan and S. Perumal, Tetrahedron Lett., 2011, 52, 4562–4566 CrossRef.
- S. Guo and Y. Yuan, Chin. J. Chem., 2010, 28, 811–817 CrossRef CAS.
- R. F. Affeldt, E. V. Benvenutti and D. Russowsky, New J. Chem., 2012, 36, 1502–1511 RSC.
- S. Karhale, C. Bhenki, G. Rashinkar and V. Helavi, New J. Chem., 2017, 41, 5133–5141 RSC.
- D. Elhamifar, P. Badin and G. Karimipoor, J. Colloid Interface Sci., 2017, 499, 120–127 CrossRef CAS PubMed.
- G. B. Dharma Rao, S. Nagakalyan and G. K. Prasad, RSC Adv., 2017, 7, 3611–3616 RSC.
- S. Mondal, B. C. Patra and A. Bhaumik, ChemCatChem, 2017, 9, 1469–1475 CrossRef CAS.
- T. Demirci, B. Çelik, Y. Yıldız, S. Eriş, M. Arslan, F. Sen and B. Kilbas, RSC Adv., 2016, 6, 76948–76956 RSC.
- A. Ghorbani-Choghamarani and B. Tahmasbi, New J. Chem., 2016, 40, 1205–1212 RSC.
- A. Amoozadeh, S. Rahmani, M. Bitaraf, F. B. Abadi and E. Tabrizian, New J. Chem., 2016, 40, 770–780 RSC.
- L. Shiri, A. Ghorbani-Choghamarani and M. Kazemi, Monatsh. Chem., 2017, 148, 1131–1139 CrossRef CAS.
- L. Shiri, L. Heidari and M. Kazemi, Appl. Organomet. Chem., 2017, e3943 CrossRef.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- S. G. Zhang, J. H. Zhang, Y. Zhang and Y. Q. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Coord. Chem. Rev., 2015, 287, 137–156 CrossRef.
- M. B. Gawande, Y. Monga, R. Zboril and R. K. Sharma, Coord. Chem. Rev., 2015, 288, 118–143 CrossRef CAS.
- A. K. Rathi, M. B. Gawande, J. Pechousek, C. Aparicio, M. Petr, K. Cepe, R. Krikavova, Z. Travnicek and R. Zboril, Green Chem., 2016, 18, 2363–2373 RSC.
- H. N. Dadhania, D. K. Raval and A. N. Dadhania, Catal. Sci. Technol., 2015, 5, 4806–4812 CAS.
- N. Candu, C. Rizescu, I. Podolean, M. Tudorache, V. I. Parvulescu and S. M. Coman, Catal. Sci. Technol., 2015, 5, 729–737 CAS.
- M. Yarie, M. A. Zolfigol, Y. Bayat, A. Asgari, D. A. Alonso and A. Khoshnood, RSC Adv., 2016, 6, 82842–82853 RSC.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Green Chem., 2012, 14, 201–208 RSC.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Tetrahedron, 2013, 69, 447–454 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 CAS.
- Q. Zhou, Z. Wan, X. Yuan and J. Luo, Appl. Organomet. Chem., 2016, 30, 215–220 CrossRef CAS.
- Q. Zhang, X. Zhao, H. X. Wei, J. H. Li and J. Luo, Appl. Organomet. Chem., 2017, 31, e3608 CrossRef.
- B. L. Li, A. G. Zhong and A. G. Ying, J. Heterocycl. Chem., 2015, 52, 445–449 CrossRef CAS.
- A. Davoodnia, M. Khashi and N. Tavakoli-Hoseini, Chin. J. Catal., 2013, 34, 1173–1178 CrossRef CAS.
- Z. Zarnegar, J. Safari and Z. M. Kafroudi, New J. Chem., 2015, 39, 1445–1451 RSC.
- M. Hajjami and B. Tahmasbi, RSC Adv., 2015, 5, 59194–59203 RSC.
- M. Abdollahi-Alibeik and A. Rezaeipoor-Anari, J. Magn. Magn. Mater., 2016, 398, 205–214 CrossRef CAS.
- G. W. Li, J. Xiao and W. Q. Zhang, Green Chem., 2012, 14, 2234–2242 RSC.
- R. Khoshnavazi, L. Bahrami and F. Havasi, RSC Adv., 2016, 6, 100962–100975 RSC.
- D. Khan, S. Mukhtar, M. A. Alsharif, M. I. Alahmdi and N. Ahmed, Tetrahedron Lett., 2017, 58, 3183–3187 CrossRef CAS.
- H. Kiyani and F. Ghorbani, Res. Chem. Intermed., 2015, 41, 7847–7882 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10692k |
| This journal is © The Royal Society of Chemistry 2017 |