
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sensitive profiling of trace neurotoxin domoic acid by pressurized capillary electrochromatography with laser-induced fluorescence detection†
Qingai Chena,
Lijun Dengb,
Jinxin Chib,
Min Liub,
Xucong Lin *b and
Zenghong Xieb
*b and
Zenghong Xieb
aDepartment of Tourism, Fujian Business University, P. R. China
bInstitute of Food Safety and Environmental Monitoring, Fuzhou University, P. R. China. E-mail: xulin@fzu.edu.cn
First published on 21st November 2017
Abstract
A stable, sensitive and low-reagent consumption method for the quantification of trace neurotoxin domoic acid (DA) was presented by pressurized capillary electrochromatography (pCEC) with laser-induced fluorescence (LIF) detection. Migration behaviours of DA were studied under different separation modes such as pCEC and capillary HPLC. Separation parameters including the mobile phase, applied voltage, supplementary pressure and sample loading rate were investigated. Double modes including electrophoretic migration and chromatographic retention were employed and an efficient separation of DA was achieved with a flow of 1.2 μL min−1 in a capillary column. Sensitive analysis of DA was achieved with a detection limit of DA in shellfish tissue extracts as low as 10 ng mL−1 (equivalent to 15 ng DA per g, wet weight). The stability and repeatability of pCEC-LIF was evaluated and a good result was gained with RSDs (n = 3) for the retention time and peak area of DA less than 0.5% and 2.0%, respectively. Applied to shellfish samples, the recoveries were satisfactory and the variation of the quantification of DA was acceptable when compared with the HPLC-MS/MS method.
1 Introduction
Domoic acid (DA), the principal causative toxin of amnesic shellfish poisoning (ASP), is produced by the algae pseudonitzschia and could accumulate in the edible tissue of shellfish.1,2 Consuming the contaminated shellfish seafoods could cause severe neurological symptoms such as memory loss, headaches, loss of balance, nausea, etc.3 Marine animals and even human beings will exhibit acute intoxication after consumption of these contaminated foods. Fabricating a facile and sensitive method to monitor trace DA is important and is attracting more and more attention.Commonly, HPLC with UV detection has been widely utilized for quantifying DA in shellfish.4 By means of fluorescence detection (FLD) with pre-column or post-column derivatization, the HPLC-FLD method is often used with a low detection limit for trace analysis of DA.5–7 HPLC with MS detection including LC-MS/MS, LC-laser ablation ESI-MS and SPE-LC-MS/MS, have also been developed for the detection of DA.8–10 These methods are stable, efficient and sensitive, however, a considerable amount of organic elution solution is consumed and consequently these methodologies might be unfavourable for economical and environment-friendly applications to a certain degree.
As the effective microfluid separation technology, capillary electrophoresis (CE) and capillary electrochromatography (CEC) have been proposed. Fast analysis of DA using microchip electrophoresis with laser-induced fluorescence (LIF) detection has been proposed.11 CEC coupling a high voltage to induce electroosmotic flow (EOF) with a capillary LC column has been formed for the analysis of ASP toxins.12,13a Leaó Martins et al. firstly developed a CEC method for the analysis of DA.12 A. Gago-Martínez et al. reported the detection of DA with the combination of CEC and photodiode array detection (PAD).13 CE and CEC have been studied and applied to algae toxins. However, the primary problem in CE or CEC was associated with bubble formation produced by Joule heating as a result of the electric current passing through the column.13 The bubbles generate baseline noise and make it difficult to obtain stable flow conditions. Besides, by using electrokinetic injection, quantitative sample introduction was achieved in CE or CEC for the DA analysis, but it might be influenced by the conductivity of sample matrix. The main drawback associated with CE or CEC is the lack of adequate robustness and reproducibility.13,14
To address these problems mentioned above, pressurized capillary electrochromatography (pCEC) has been proposed by coupling an HPLC pump onto the inlet end of the capillary to provide auxiliary pressure to flush capillary column with solvent and inhibit bubble formation.13a,b Quantitative sample injection could be facilely achieved through a reliable rotary-type injector in pCEC system. In pCEC, both high efficiency of CEC and high selectivity of HPLC were successfully combined.15–17 The mobile phase is driven by both hydraulic pressure and electroosmotic flow (EOF), and resulted in an efficient elution and the questions such as bubble formation and unstable quantitation could be effectively solved.18–20 By superimposing a hydraulic pressure onto the electrically generated flow in capillary column, double modes that are electronic interaction and chromatographic retention could be employed for the efficient separation with a high resolution.18,19 Furthermore, the mobile solution as low as nano-litre flow level in capillary column could be carried out facilely in pCEC. It is desirable and become an attractive alternative to common HPLC and pure CEC for the robust and effective analysis.
To date, with the popular UV detection, pCEC-UV method has been used to rapid quantify of DA in shellfish seafood with a determination limit equivalent to 2.0 μg DA per g.21 It always suffers from low detection sensitivity due to a rather short optical path length in capillary column for on-line photometric detection. To gain a high sensitivity, laser induced fluorescence detection (LIF) that is well suitable for capillary micro-separation with a strong laser to overcome the limit of optical path length in capillary column could be utilized. Ultratrace levels of detection limit (10−11 mol L−1 levels for fluorescein) were achieved with LIF and presented an obvious advantage over common UV detection.22 Since introduced firstly by Yan et al. in 1995,23 capillary electrochromatography with LIF methods have attracted close attentions in trace detection of polycyclic aromatic hydrocarbons,23 aflatoxins,24 flavins in plasma25 or proteins.26 So far, pCEC-LIF method for the trace detection of non-protein amino acids toxin DA has still not reported and its utilization will provide an attractive way for the low-reagent consumption and sensitive analysis of ASP.
Here, we present the development and application of the pCEC-LIF method for the quantification of trace DA in shellfish tissue extracts. Migration behaviours of DA were studied under different separation modes including pCEC and capillary HPLC. A series of parameters including mobile phase, applied voltage, hydraulic pressure and sample loading rate were investigated for optimum electrochromatography behaviour. The stability and reproducibility of pCEC-LIF method were evaluated, as well as the detection limit of DA. The recoveries of shellfishes samples spiked with DA were analyzed, and the method validation in detection results was finally evaluated by comparing with the authoritative HPLC-MS/MS method.
2 Experimental
2.1 Instrumentation
pCEC was performed on pCEC system (Trisep™ 2100 GV, Unimicro Technologies, USA), which consisted of a solvent delivery gradient module, a micro-fluid manipulation module (including a 0.8 μL six-port injection valve, a four-port split valve, and a back-pressure regulator), a high voltage power supply (0–±30 kV), a data acquisition module, and a LD-DPSS LIF detector (10 mW, λex = 473 nm; λem = 530 nm). Samples are injected to sample loop and then carried to four-port split valve by mobile phase. Under a constant pressure, the total flow from four-port valve was split and a nano-flow was driven into capillary column with a split device. Applied voltage was imposed at the outlet end of column, and the inlet end was connected to the four-port split valve and grounded. HPLC-MS/MS system and the program were shown in ESI.†2.2 Reagents and materials
Domoic acid standard (≥97%, HPLC-grade) was purchased from Sigma-Aldrich (USA), 4-fluoro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-F) (≥98%, HPLC-grade) was purchased from TCI (Japan), HPLC-grade acetonitrile and methanol were purchased from Chemical Reagent Corp (Shanghai, China), the other chemicals were analytical grade. A packed capillary column (100 μm/id. 375 μm/od., total length 55 cm, of which 20 cm was packed with 3 μm octadecyl silica (ODS) particles) was obtained from Global Chromatography (SuZhou, China). Sep-Pak SAX (3 mL, 200 mg) SPE columns were purchased from Waters (Milford, MA, USA). Deionized water was prepared with a Milli-Q water purification system (Milford, MA, USA).2.3 Sample preparation
The procedure for handling shellfish tissue was based on the previous ref. 6 and 21. Approximately 50 g of shellfish tissue was homogenized with the blender. A 4.0 g homogenated tissue was used and extracted with 15.0 mL extraction solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–water) and homogenized again for 3 min at 13000 rpm. The resulting slurry was then centrifuged at 4000 rpm for 10 min, and the centrifuged sample was then filtered through a 0.22 μm membrane and was purified further using the following SPE procedure according to the work reported by M. Maroulis et al.6
:1 methanol–water) and homogenized again for 3 min at 13000 rpm. The resulting slurry was then centrifuged at 4000 rpm for 10 min, and the centrifuged sample was then filtered through a 0.22 μm membrane and was purified further using the following SPE procedure according to the work reported by M. Maroulis et al.6
A 5.0 mL fraction of the filtered crude extract was loaded onto the cartridge in a flow rate of about one drop per second. The cartridge was washed at about one drop per second with 5 mL wash solution (acetonitrile–water = 10:90, v/v). Then the effluent was discarded and 0.5 mL of 0.1 mol L−1 formic buffer eluent (pH 2.92 ± 0.01, adjusted with sodium hydroxide) was added. The effluent was discarded again. The formic buffer (0.1 mol L−1) were loaded onto the cartridge and allowed to flow at a rate of one drop per second. Exactly 2.0 mL of the final eluted DA extract were collected for further analysis.
2.4 Derivatization of domoic acid with NBD-F
According the ref. 8, NBD-F in acetonitrile (1.0 mg mL−1, 50 μL) and 0.10 mol L−1 sodium borate (50 μL) were mixed with sample extract or standard solution (50 μL) in an amber vial (2 mL), at 20 °C and the reaction was terminated after 3 min by adding 1.0 mol L−1 HCl (50 μL) to the mixture. An aliquot of this derivatised mixture was analysed directly by pCEC-LIF.2.5 pCEC-LIF procedure
Mobile phase was composed of phosphate buffer solution and ACN with the appropriate concentration and desired pH value. The pH values and salt concentrations mentioned in this work were that of phosphate buffer solution.By using acetonitrile–phosphate buffer solution (60:40, v/v) as mobile phase, isocratic pressurized electrochromatography was performed with a octadecyl silica (ODS) packed capillary column mentioned above. A supplementary pressure (7.2 MPa) was applied to the column inlet and the nano-flow was driven by the pump with a split ratio of 1/15. Before conducting pCEC experiments, capillary column was conditioned with mobile phase for 30 min and the column was kept at 25 °C. The applied voltage was ramped from 0 to +8 kV at first and then maintained at +6 kV. Fluorimetric detection was used (λex = 473 nm; λem = 530 nm).
2.6 Method validation
The method was evaluated and validated through linearity, limit of detection (LOD), fortified recoveries and precision, and the program was shown in ESI.† Precision of the method was evaluated by the intra-day and inter-day RSD. The intra-day RSD was obtained from analysing five replicates of the sample spiked with 0.25 μg mL−1 of DA on the same day, and the inter-day RSDs were determined by analysing the sample based on five replicates on three consecutive days. The comparison with the authoritative HPLC-MS/MS method was carried out to further evaluate the feasibility of this method.3 Results and discussion
3.1 pCEC mode for DA detection
In pCEC, an HPLC pump to superimpose a hydraulic pressure at the inlet of capillary column onto an electrically generated flow. Mobile phase is driven by both EOF and pressurized flow, and the linear velocity of a charged solute in mobile phase (um) is the sum of the velocities contributed from electrophoretic migration velocity (uem), pressurized flow (up), and EOF (ueo). As shown in Fig. 1, double driving forces including a hydraulic pressure and an electroosmotic flow (EOF) could be employed and double modes including electrophoretic migration (EM) between the electrode and charged solutes and capillary micro-liquid chromatographic (μ-HPLC) retention of packed stationary phase could be employed for the separation of DA derivatization products.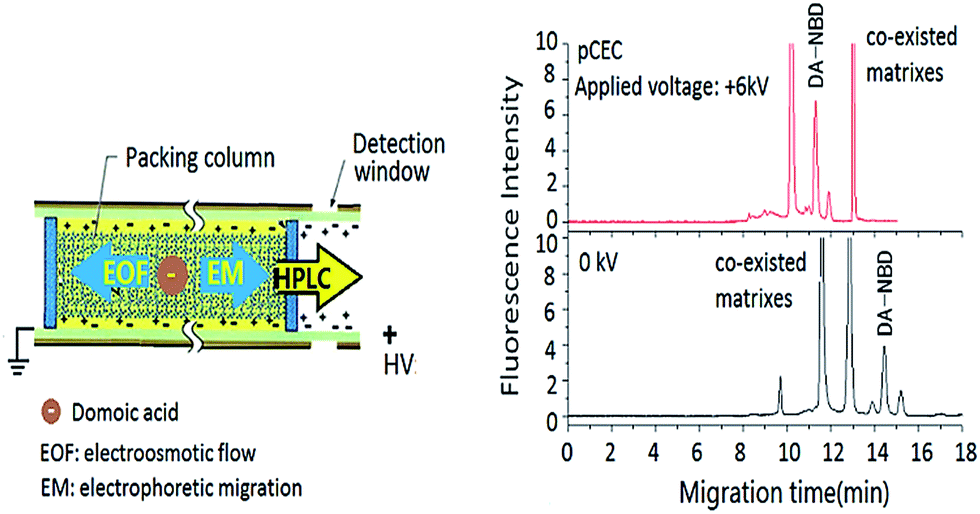 | ||
| Fig. 1 Scheme of multiple separation interactions in pCEC for DA detection. | ||
To evaluate the chromatographic behaviour of DA derivatization product DA-NBD, DA standard solutions and shellfish tissue extracts spiked with DA were used and the results were shown in Fig. 2 and 3. In Fig. 2, the migration time of DA-NBD was shortened to 12 min in pCEC with a positive high voltage of +6 kV, while prolonged to more than 16 min when a negative high voltage −6 kV was applied. It indicated that DA-NBD was of negatively charge and the electrophoresis action could enhance the migration rate and the fluorescence response of DA-NBD remarkably, which was in favour of the rapid and sensitive analysis of trace DA. By using a positive high voltage, the migration rate of DA-NBD was accelerated obviously benefitting from electrophoretic migration with the same direction of hydraulic pressure though the cathode EOF was in the oppose direction. In pCEC, the electrophoretic migration and electroosmotic flow were of importance for the migration and effective separation of DA.
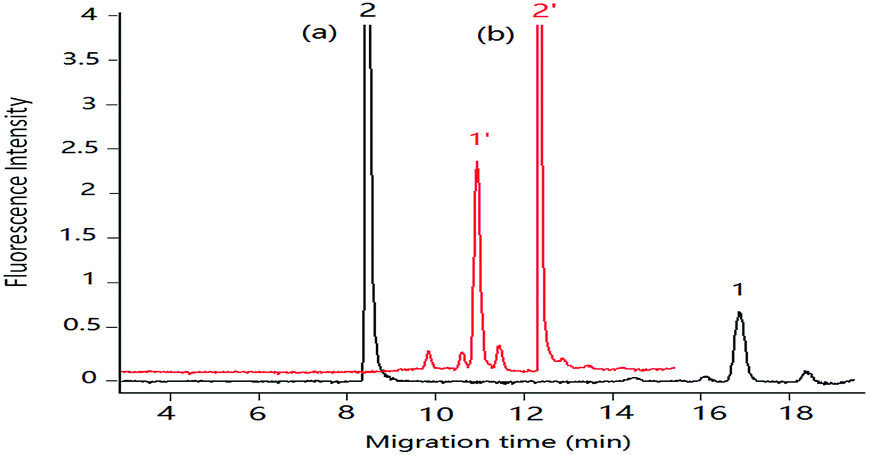 | ||
| Fig. 2 Migration of DA in pCEC with negative and positive applied voltage respectively. Mobile phase, 60% (v/v) acetonitrile, 40% (v/v) of 5.0 mmol L−1 phosphate buffer (pH 2.5), supplementary pressure 1000 psi, flow rate in column: 3 μm L min−1; DA standard solution: 0.50 μg mL−1. Applied voltage: (a) −6 kV, (b) +6 kV. Peak 1 and 1′: DA-NBD, 2 and 2′: NBD-F hydrolysis products. | ||
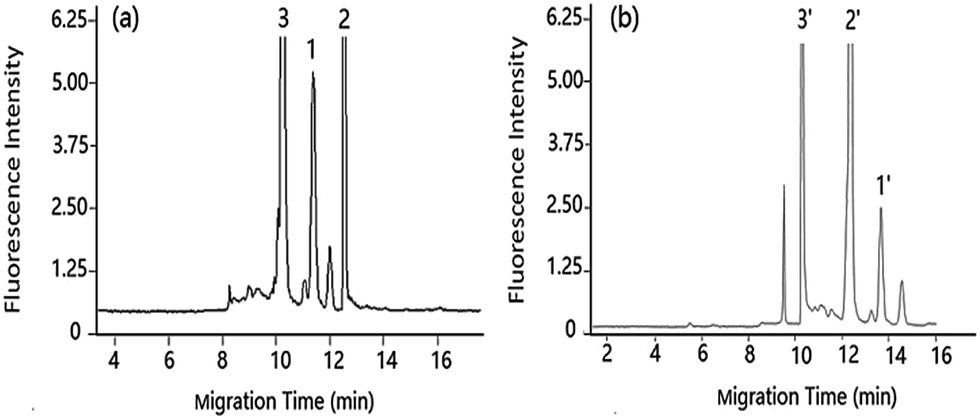 | ||
| Fig. 3 Separation of DA under pCEC and cLC in shellfish tissue extracts shellfish tissue extracts spiked with DA: 1.00 μg mL−1. Applied voltage: (a) +6 kV, (b) 0 kV. Other conditions as Fig. 2. Peak 1 and 1′: DA-NBD, 2 and 2′: NBD-F hydrolysis products, 3 and 3′: co-eluted compounds. | ||
Compared with the μ-HPLC method which achieved under the same experimental conditions as that of pCEC method except applied voltage, a higher sensitivity and a rapid elution of DA were obtained (shown in Fig. 3). Applied to shellfish tissue extracts, the pCEC-LIF method was suitable for DA detection. The separation of DA-NBD and co-eluted matrixes were successfully achieved with a high sensitivity.
3.2 Optimization of separation parameters
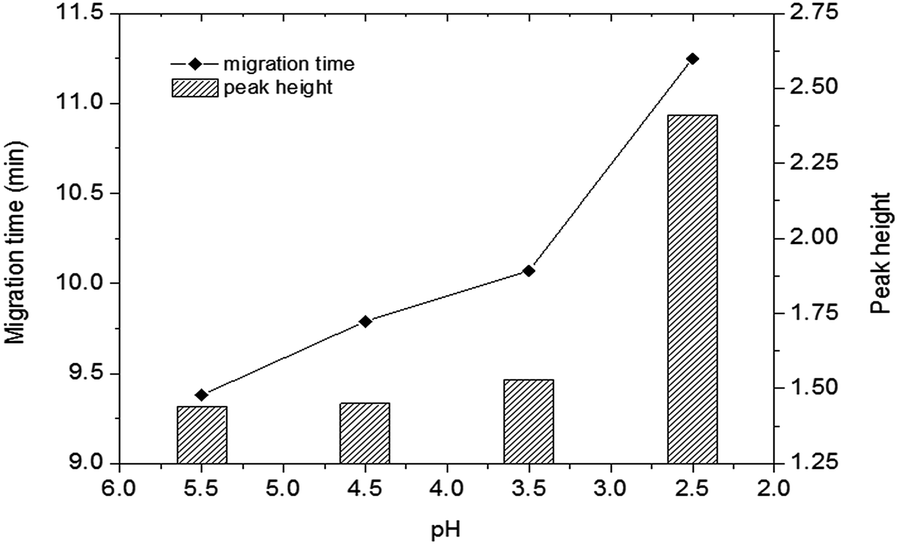 | ||
| Fig. 4 Effect of pH on the detection of DA in shellfish tissue extracts. Mobile phase, 60% (v/v) acetonitrile, 40% (v/v) of 5 mmol L−1 phosphate buffer, applied voltage: +6 kV; supplementary pressure: 1000 psi. Other conditions as Fig. 2. DA: 0.50 μg mL−1. | ||
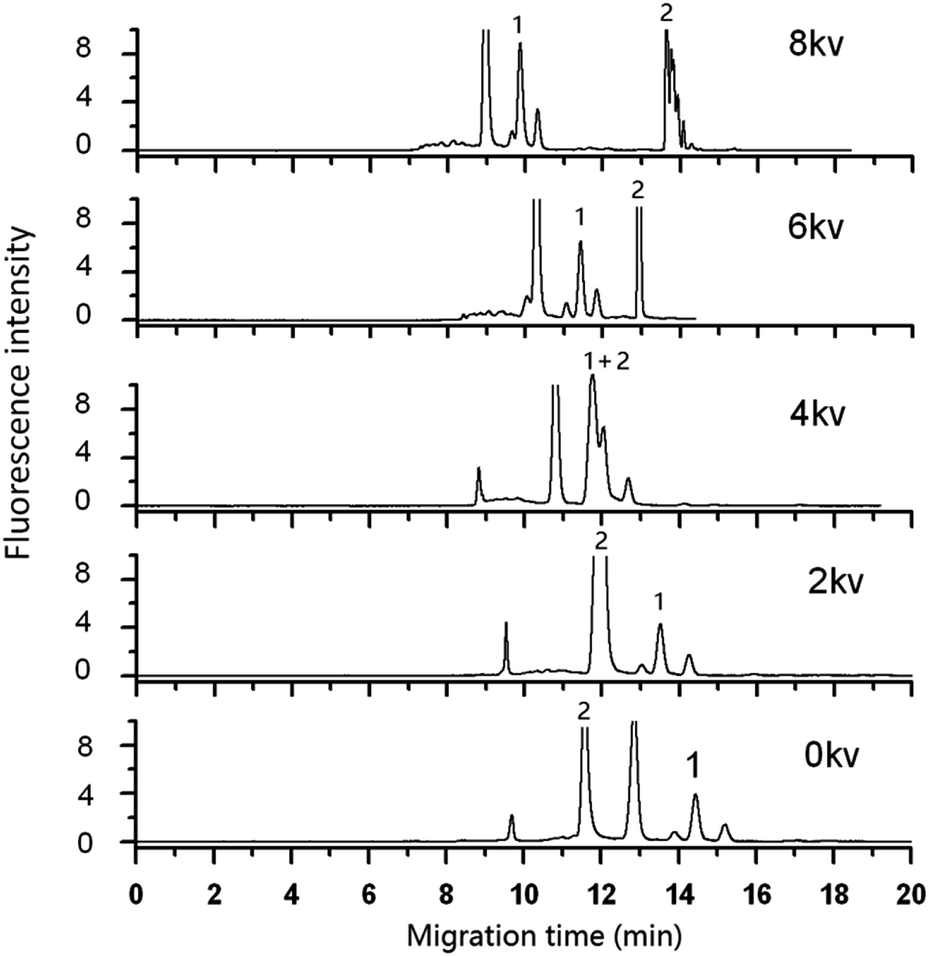 | ||
| Fig. 5 Effect of applied voltage on the separation of DA in shellfish tissue extracts. Mobile phase, 60% (v/v) acetonitrile, 40% (v/v) of 5.0 mmol L−1 phosphate buffer solution (pH 2.5); other as in figure. Peak 1: DA-NBD, 2: NBD-F hydrolysis compounds. | ||
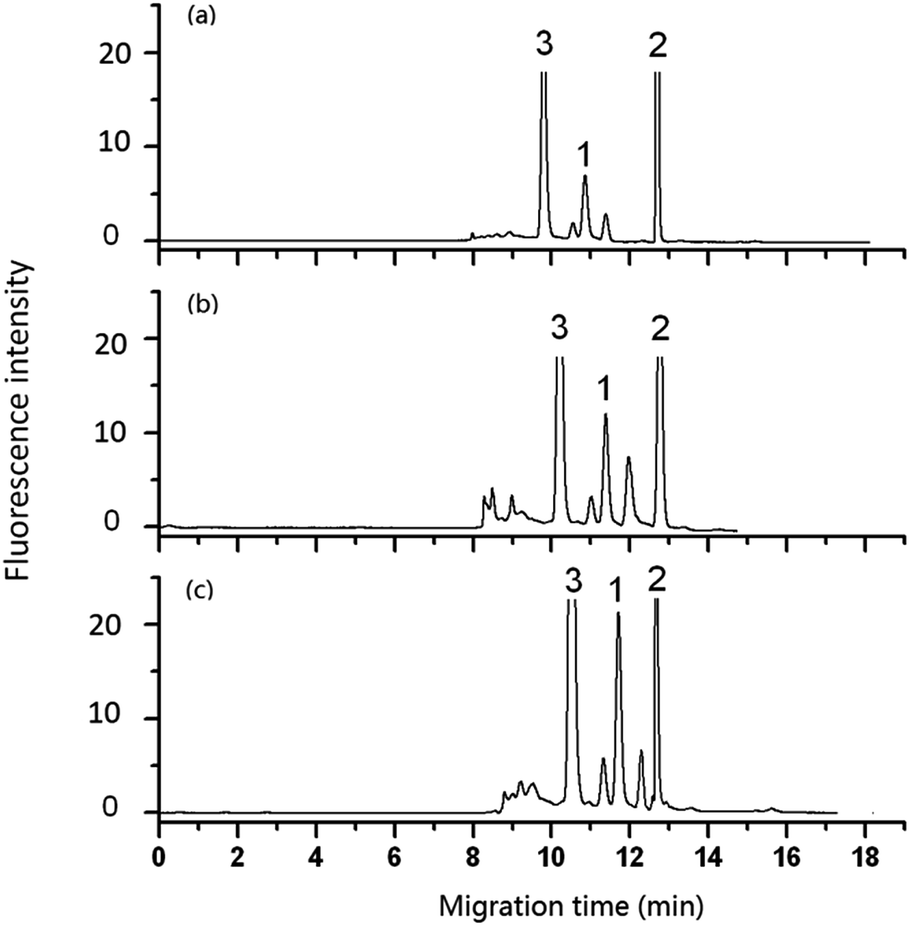 | ||
| Fig. 6 Effect of flow rate of pump on the analysis of DA in shellfish tissue extracts. Mobile phase, 60% (v/v) acetonitrile, 40% (v/v) of 5 mmol L−1 phosphate buffer solution (pH 2.5); other conditions as in Fig. 5. Flow rate in column: (a) 3 μL min−1, (b) 2 μL min−1, (c) 1.2 μL min−1. Peak 1: DA-NBD, 2 and 3: co-eluted compounds. | ||
3.3 Analytical performance
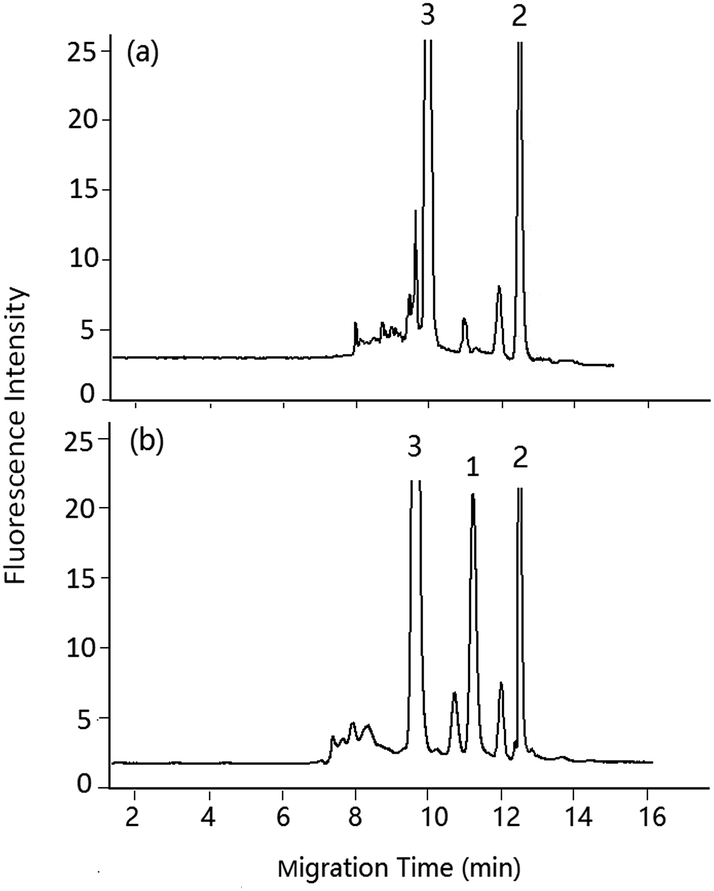 | ||
| Fig. 7 pCEC analysis of DA in shellfish tissue extracts. Mobile phase, 60% (v/v) acetonitrile, 40% (v/v) of 5 mmol L−1 PBS buffer (pH 2.5), applied voltage: +6 kV, supplementary pressure: 1000 psi, spit-flow rate: 1.2 μL min−1. (a) Blank sample, (b) spiked with DA standard solution 1.00 μg mL−1, peaks: 1: DA-NBD, 2: NBD-F hydrolysis products, 3: co-eluted compounds. | ||
| Analytes | Linear range (μg mL−1) | Regression equation | Correlation coefficient | Detection limit (μg mL−1) | Intra-day RSD (%) (n = 5) | Inter-day RSD (%) (n = 15)a | ||
|---|---|---|---|---|---|---|---|---|
| Retention time | Peak area | Retention time | Peak area | |||||
| a The inter-day RSDs were determined by analyzing the sample based on five replicates on three consecutive days. | ||||||||
| DA | 0.05–4.00 | y = 1.516 × 105x + 4.627 × 103 | 0.9903 | 0.01 | 0.5 | 2.1 | 2.0 | 4.0 |
| Shellfish sample | Added amount (μg DA per g) | pCEC-LIF | HPLC-MS/MS | ||
|---|---|---|---|---|---|
| Recovery (%) (n = 3) | RSD (%) | Recovery (%) (n = 3) | RSD (%) | ||
| a According to the ref. 27. | |||||
| Oyster | 0.30 | 95.4 | 4.4 | 95.8 | 2.2 |
| Mytilus edulis | 0.30 | 90.9 | 2.8 | 89.6 | 2.5 |
| Ruditapes | 0.30 | 100.1 | 2.4 | 93.3 | 2.0 |
| Oyster | 0.60 | 110.0 | 3.4 | 104.1 | 1.5 |
| Oyster | 3.00 | 105.5 | 2.5 | 98.7 | 2.3 |
4 Conclusions
A stable and sensitive pCEC-LIF hyphenated method for the analysis of trace DA in shellfishes was formed with low-reagent consumption, and the quantification for trace DA in shellfish seafoods was successfully achieved. Under the optimal conditions, a baseline separation with a high resolution of DA from shellfish matrices was gained within 12 min. The pCEC-LIF method was robust and sensitive with the LOD of DA as low as 10 ng mL−1 (S/N = 3) (equivalent to 15 ng DA per g), which was more sensitive than most method fabricated with CEC-PAD and pCEC-UV. An extremely little amount of mobile phase was consumed economically with a flow of 1.2 μL min−1 in capillary column, which was only 1/800 of that used in normal HPLC. A good repeatability was also obtained and RSD values (intraday, n = 3) for retention time and peak area were less than 0.5% and 2.0%, respectively. Applied to shellfish seafoods, the recoveries were obtained with satisfactory values in the range of 90.9–110%. Compared with the accepted authoritative HPLC-MS/MS method, the detection results were similar and in a good level. The results indicated that pCEC-LIF hyphenated technology was robust and sensitive enough for the quantitative analysis of trace DA toxin in shellfish seafoods and hold promise for the application to food safety monitoring.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (21377024), National Key Technologies R&D Program of China (2014BAD13B01), and Scientific Research and develop project of Fujian province (2015Y0052, 2016Y4009 and 2017Y0056).References
- Y. He, A. Fekete and G. Chen, et al., J. Agric. Food Chem., 2010, 58, 11525 CrossRef CAS PubMed.
- V. L. Trainer, B. M. Hickey and R. A. Horner, Limnol. Oceanogr., 2002, 47(5), 1438 CrossRef CAS.
- P. Ajani, D. T. Harwood and S. Murray, Mar. Drugs, 2017, 15, 33 CrossRef PubMed.
- J. Regueiro, G. Álvarez and A. Mauriz, et al., Food Chem., 2011, 127, 1884 CrossRef CAS.
- I. O. M. Chan, V. W. H. Tsang and K. K. Chu, et al., Anal. Chim. Acta, 2007, 583, 111 CrossRef CAS PubMed.
- M. Maroulis, I. Monemvasios and E. Vardaka, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 876, 245 CrossRef CAS PubMed.
- K. J. James, M. Gillman and M. Lehane, et al., J. Chromatogr. A, 2000, 871, 1 CrossRef CAS PubMed.
- T. Jiang, L. Liu and Y. Li, et al., Chemosphere, 2017, 183, 80 CrossRef CAS PubMed.
- D. G. Beach, C. M. Walsh and P. Cantrell, et al., Rapid Commun. Mass Spectrom., 2016, 30, 2379 CrossRef CAS PubMed.
- S. K. Zervou, C. Christophoridis and T. Kaloudis, et al., J. Hazard. Mater., 2017, 323, 56 CrossRef CAS PubMed.
- Y. Cheng, C. Guo and B. Zhao, et al., J. Sep. Sci., 2017, 40, 1583 CrossRef CAS PubMed.
- J. M. Leaó Martins, A. Gago-Martinez and E. Dabek-Zlotorzynska, et al., J. Sep. Sci., 2002, 25, 342 CrossRef.
- (a) A. Gago-Martínez, N. Pinéiro and E. C. Aguete, et al., J. Chromatogr., A, 2003, 992, 159 CrossRef; (b) T. Eimer, K. K. Unger and T. Frensenius Tsuda, Anal. Chem., 1995, 352, 649 CrossRef CAS.
- (a) Q. Ru, G. Luo and Y. Fu, et al., J. Chromatogr. A, 2001, 924, 331 CrossRef CAS PubMed; (b) F. Ye, Z. Xie and X. Wu, et al., Talanta, 2006, 69, 97 CrossRef CAS PubMed.
- J. Wang, F. Wu and R. Xia, J. Chromatogr. A, 2016, 1449, 100 CrossRef CAS PubMed.
- Y. Huo and W. T. Kok, Electrophoresis, 2008, 29, 80 CrossRef CAS PubMed.
- X. Lin, Y. Li and D. Xu, et al., Analyst, 2013, 138, 635 RSC.
- Z. Liang, J. C. Duan and L. H. Zhang, et al., Anal. Chem., 2004, 76, 6935 CrossRef CAS PubMed.
- J. Wang, F. Wu and R. Xia, et al., J. Chromatogr. A, 2016, 1449, 100 CrossRef CAS PubMed.
- X. Wang, Y. Zheng and C. Zhang, et al., J. Chromatogr. A, 2012, 1239, 56 CrossRef CAS PubMed.
- W. Wu, X. Wu and X. Lin, et al., J. Sep. Sci., 2009, 32, 2117 CrossRef CAS PubMed.
- C. Liu, Q. Deng and G. Fang, et al., Anal. Biochem., 2017, 530, 50 CrossRef CAS PubMed.
- C. Yan, R. Dadoo and H. Zhao, et al., Anal. Chem., 1995, 67(13), 2026 CrossRef CAS.
- Q. Wan, X. Ru and X. Wang, et al., Chin. J. Anal. Chem., 2015, 43(7), 1063 CAS.
- Y. Wu, J. Lin and Q. Wu, et al., J. Pharm. Biomed. Anal., 2010, 53, 1324 CrossRef CAS PubMed.
- S. Štěpánová and V. Kašička, Anal. Chim. Acta, 2016, 933, 23 CrossRef PubMed.
- W. Zhang, M. Lin and P. Tong, et al., J. Chromatogr. A, 2016, 1443, 54 CrossRef CAS PubMed.
- C. Bignardi, A. Cavazza and C. Corradini, Electrophoresis, 2012, 33, 2382 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10662a |
| This journal is © The Royal Society of Chemistry 2017 |