
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surficial nanoporous carbon with high pyridinic/pyrrolic N-Doping from sp3/sp2-N-rich azaacene dye for lithium storage†
Jianfeng Zhaoa,
Kai Chena,
Bing Yanga,
Yanni Zhanga,
Caixia Zhua,
Yinxiang Lic,
Qichun Zhang *b,
Linghai Xie*c and
Wei Huang*acd
*b,
Linghai Xie*c and
Wei Huang*acd
aKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China. E-mail: iamwhuang@njtech.edu.cn
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: qczhang@ntu.edu.sg; Fax: +65-67909081
cKey Laboratory for Organic Electronics & Information Displays (KLOEID), Institute of Advanced Materials, Nanjing University of Posts & Telecommunications, Nanjing 210023, China. E-mail: iamlhxie@njupt.edu.cn
dXi'an Institute of Flexible Electronics (XIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi'an 710072, China
First published on 27th November 2017
Abstract
Rationally designed pyridinic/pyrrolic N-doping of anodic carbonaceous materials is significant but rare for lithium storage materials. Herein, two highly pyridinic/pyrrolic N-doped carbon materials i.e. the pristine material (NC, N content 10.9 wt%) and template (NPC, N content 12.6 wt%) are predictively achieved by the direct pyrolyzation of the azaacene dye with a high sp3/sp2-N content (13.6 wt%). Their total amount of pyridinic and pyrrolic content is as high as 7 at% (NC)/6.98 at% (NPC), which is close to the theoretical value (9.1 at%) with less/no graphitic N. Both of them present higher capacities of 1094.79 mA h g−1 and 411.3 mA h g−1, respectively, by the 3rd cycle at 100 mA g−1. Featured by surficial bumps and hollows of NPC particles, the NPC electrode not only exhibits a first reversible specific discharge capacity as high as 1178.9 mA h g−1 at a 100 mA g−1 current density but is also stabilized at 614 mA h g−1 after 200 cycles at a 200 mA g−1 current density. Our results indicate that the sp3/sp2-N-rich azaacene dye can be a useful highly pyridinic/pyrrolic N-doped carbon source for high performance anodic materials.
Introduction
Carbon-based anode materials have been explored as one of the most key active power components for portable electronic devices due to their high storage capacity, cycling stability, and reversibility in the fields of lithium–ion batteries,1–3 supercapacitors,4–6 oxygen reduction reactions,7 and H2 storage.8 Furthermore, the intrinsic properties, including low theoretical and actual capacities, and limited application ranges4,9 of conventional anodic graphite can no longer meet the requirements for high-efficient and convenient power consumption. Moreover, two main strategies i.e. enlargement of the electrolyte–electrode interfacial area and increasing the redox sites with pre-embedded heteroatoms are applied to optimize the electrochemical properties of carbon-based anode materials. Template precursors and alkaline activators10 that tend to form various well-defined micro/nanostructures11–13 have been applied to enlarge the electrolyte–electrode interfacial area.14–16 Furthermore, sufficient and effective lithiation sites17 between porous electrodes and liquid electrolytes could deeply enhance the chemical stability and electrochemical performance of carbon-based anodes,18–21 and overcome the expansion and other defects of Si and Sn materials.2,22Various approaches including pre-templating,23–26 graphitization,27 pressure-assisted carbonization,28 activating agents,9b,29,30 and alternative carbon sources have been widely applied to improve the specific electrolyte–electrode interface2b,23,31 and especially increase the hetero-doping ratio7d,16,32 in carbon materials to achieve extraordinary electrochemical properties.24,33,34 Since pyridinic/pyrrolic N-doped carbons possess a higher Li atom average adsorption energy than the Li–Li binding energy,17b it is generally acknowledged that a much more higher ratio pyridinic/pyrrolic N-doping would enhance the insertion/de-insertion ability of the loaded Li+ ions in carbonaceous materials.19,24,35,36 Special carbon sources such as biomass,10,37–39 polymers including conjugated polymers,40,41 polyaniline,42 and polyamide,6,43,44 melamine–resorcinol–formaldehyde,45 metal organic frameworks,18,46 and complexes47,48 containing sp2-N and sp3-N could produce versatile N-doped porous carbons.
However, there are few reports17,20 on the rational design of sp3/sp2-N-rich aromatics as a source of high ratio pyridinic/pyrrolic N-doped carbon-based anode materials with N-doping ratios over 10 wt%,3,20 large specific surface areas, and high Li+ ion storage capacity.28,49,50 Therefore, it is highly desirable to explore this type of new carbon source due to its advantages of low-cost, simple synthesis, and high output as high-performing carbon-based anodic lithium storage materials.
Herein, we successfully synthesized two novel amorphous carbon materials, i.e. N-doped porous carbon (NPC) and pristine N-doped carbon (NC), by pyrolyzing the low-cost, easily-synthesized N-rich azaacene dye (mixture of trans isomeric Pigment Orange 43 and cis isomeric Pigment Red 194)51 without/with a micro-SiO2 template under an inert gas atmosphere. NPC possesses the special characteristic of surficial nanopores, whereas NC has no surficial pores. The novel N-doped carbons are systematically characterized via elemental analysis, X-ray photoelectron spectroscopy (XPS), X-ray powder diffraction (XRD), Raman spectroscopy, Brunauer–Emmett–Teller surface area measurements (BET), field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HR-TEM), and electrochemical tests.
Results and discussion
Synthesis
The π-conjugated azaacene dye carbon precursor was synthesized via a facile cyclocondensation route in high yield.52 As shown in Scheme 1, NC was prepared by the direct carbonization of the azaacene dye at high temperatures for 2 h under an Ar atmosphere. Considering that a low temperature will result in incomplete carbonization, whereas a high temperature will lead to a low heteroatom doping content,17d we have finally chosen 700 °C as the ideal temperature. As the temperature increased, the dye powder melted at 497–504 °C (ref. 52e) and decomposed accompanied by swelling. The high melting point led to a high carbon yield. Subsequently, the melted azaacene dye combusted and carbonized into intact, bulky, crisp, and black shiny 3D-like self-supported carbon due to the self-sealed pressure from the released waste gas. For NPC, the π-conjugated azaacene dye was granulated with micro-SiO2 for 15 min and then carbonized under the former conditions. Then, the organic powder melted on the surface of the neighbouring micro-SiO2 particles and then decomposed with waste gas released through the capillary space between micro-SiO2 particles. This led to the formation of an NPC–SiO2 mixture as a dark black powder, which is different from the shiny black 3D-like appearance (Scheme S1†) of NC. Then, the as-prepared NPC–SiO2 mixture was immersed in excess aqueous NH4HF2 and sodium hydroxide in sequence to get rid of SiO2. Finally, NPC was obtained as a black powder with a large surface area.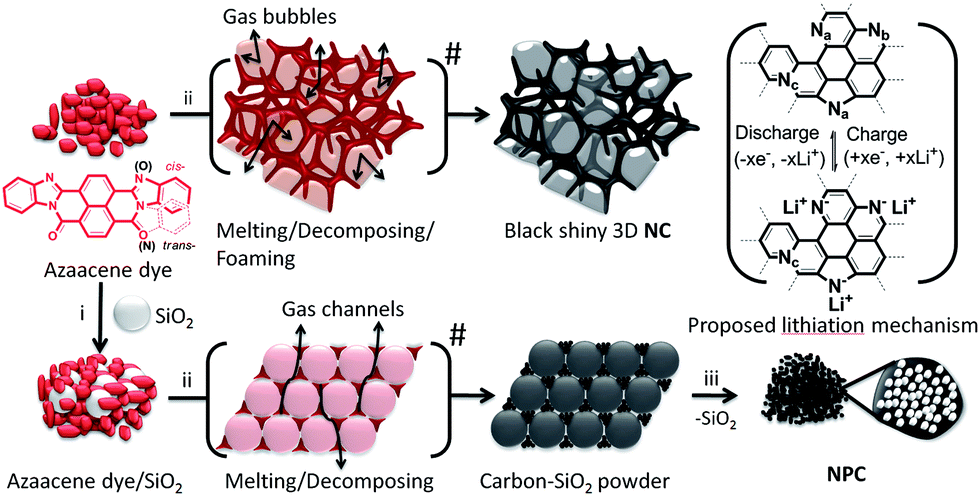 | ||
| Scheme 1 Synthetic route for the preparation of NC and NPC. Na, Nb, and Nc represent pyridinic N, pyrrolic N, and graphitic N (less/none), respectively. Azaacene dye: 1,4,5,8-naphthalenetetracarboxylic dianhydride, o-benzenediamine, DMF, 150 °C, 4 h, 65% yield, red powder. (i) Micro-SiO2, granulation for 15 min; (ii) 700 °C, Ar; and (iii) NH4HF2 (aq.), NaOH (aq.), deionized water. Bracket: proposed lithiation mechanism. | ||
Morphology
The shiny black 3D-like appearance of the self-supported carbon NC (Fig. S1a†) matched well with the smooth surface morphology shown in the FE-SEM images of the ground NC particles. The size of these particles ranged from several to tens of micrometers without well-defined structures, as shown in the magnified images (Fig. 1a inset). Moreover, NPC is presented as unshaped particles (Fig. 1b). From another magnified image (Fig. 1b inset), it was found that the surface of the NPC particles had large quantities of bumps and hollows in the range of tens of nanometers acting as effective Li+ intercalation sites at the liquid–solid interface. The XRD patterns of both NC and NPC are inserted as white curves in Fig. 1a and b, which exhibit broad and low diffraction bands at the 2θ values of around 26–28° and 41–46°, representing the interplanar spacings of (d002) and (d100), respectively. These bands suggest a low graphitization degree for both NC and NPC. According to the Bragg's Law 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ (λ = 1.5405 Å), the values of the weak d002 and d100 bands are 0.34–0.32 nm (26–28°) and 0.22–0.20 nm (41–46°), respectively, indicating the low crystallinity53,54 of these two new carbon materials.
sinθ = nλ (λ = 1.5405 Å), the values of the weak d002 and d100 bands are 0.34–0.32 nm (26–28°) and 0.22–0.20 nm (41–46°), respectively, indicating the low crystallinity53,54 of these two new carbon materials.
 | ||
| Fig. 1 FE-SEM images of NC (a) and NPC (b), TEM (c, d) and HR-TEM (e, f) images of NC (c, e) and NPC (d, f). Inset images: magnified surficial morphologies of NC and NPC. Inset white curves: XRD patterns of NC and NPC. White arrows and bars: distances of the crystal lattices. | ||
TEM and HR-TEM analyses were further conducted to investigate the microstructures of the as-prepared carbon materials. In Fig. 1c and d, NPC displays a similar disordering of its inner crystalline lattice as NC; this indicates that their structural defects are favourable for Li+ ion diffusion and effective contact between active materials and electrolyte.55 The HR-TEM images, as shown in Fig. 1e and f, also reveal that both NC and NPC are amorphous carbon materials without a long range order and straight crystalline lattices. However, a few large and distinguishable short lattice fragments were observed, which were marked by white arrows and bars. The measured width of the lattice fragments of NC was as large as 0.38/0.41 nm, and that for NPC was as large as 0.55/0.64 nm, which was much rougher than NC. It was proposed that the surface pores originated from the interfacial etching reactions among the surficial corrosive matters and groups (–OH, –COOH, etc) on the template. Moreover, for NPC, the micro-SiO2 template interdicted macroscale 3D-like cross-linkage and thermal agglomeration of the melting and combusting azaacene dye during the pyrolysis process.
Therefore, N2 adsorption–desorption isotherms were obtained, as shown in Fig. 2. From the analysis of the BET data shown in Fig. 2a, the specific surface area of NPC was found to be 456.8 m2 g−1, which was almost 264 times that of NC (1.73 m2 g−1). Moreover, NPC possesses both micropores (peaked at 0.415 nm) and mesopores (peaked at 5.10 nm) (Fig. 2b). The pores of NPC supply a number of commodious reservoirs for potential Li+ ion transport and accumulation under electrochemical test conditions.
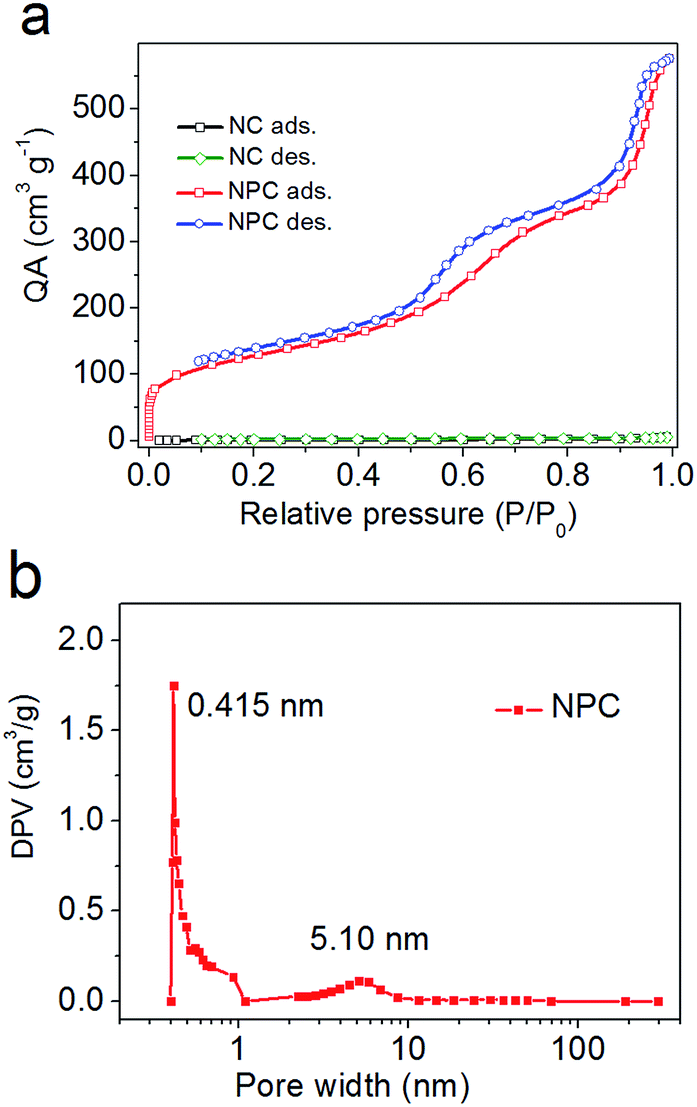 | ||
| Fig. 2 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of NPC. QA: quantity adsorbed and DPV: differential pore volume. | ||
Raman spectroscopy
The Raman spectra in Fig. 3a exhibit two distinct peaks attributed to the D band (ca. 1353/1367 cm−1) and G band (ca. 1600/1571 cm−1) for NC and NPC, respectively. The intensive G bands indicate that the degree of graphitization decreases from NC to NPC after the addition of the micro-SiO2 template. Compared with that of NC, the intensity ratio of the D-band versus G-band (ID/IG) in NPC was calculated to be 1.02, which was larger than 0.86 calculated for NC and suggested much more structural defects and disorder56–58 for NPC. This was further confirmed by its widely distributed surficial pores (Fig. 1b inset).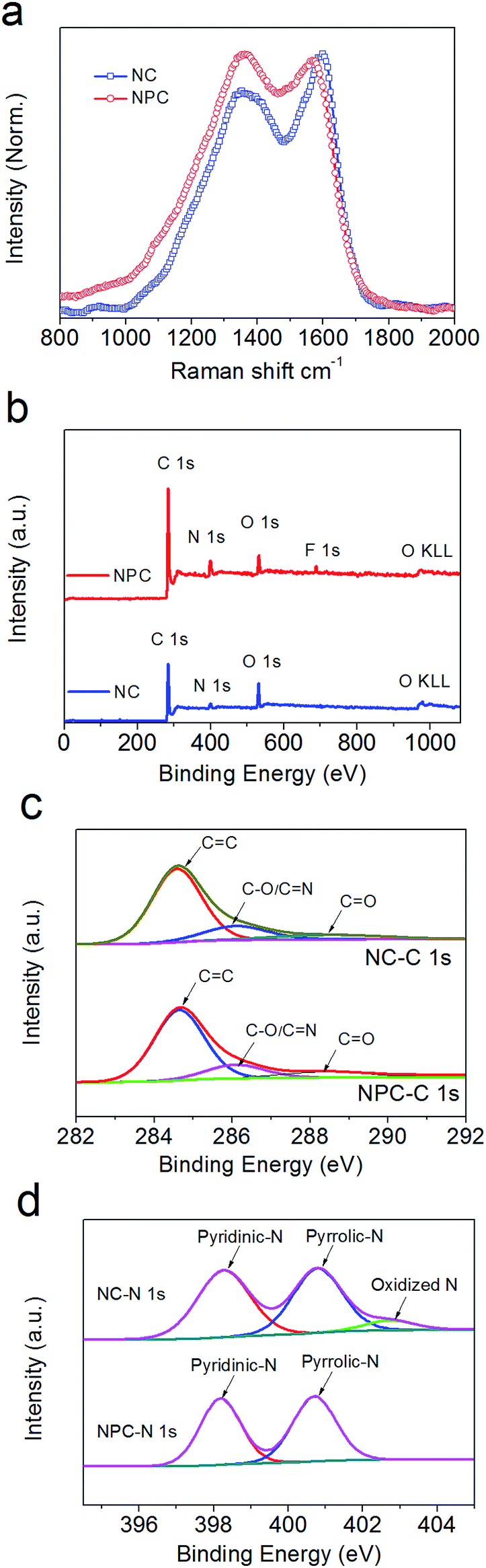 | ||
| Fig. 3 Raman spectra (a) of NC and NPC synthesized from π-conjugated azaacene dye at 700 °C. C 1s (c) and N 1s (d) XPS spectra (b) with the deconvoluted curves of NC and NPC. | ||
X-ray photoelectron spectroscopy (XPS)
XPS was used to analyse the elemental distribution and the related bonding species in NC and NPC. As shown in Fig. 3b, both NC and NPC mainly contain C, N, and O dopants with three characteristic peaks at ∼285 eV, ∼400 eV, and ∼532 ± 1 eV, corresponding to C 1s, N 1s, and O 1s. For NC, the total content of C, N, and O elements was 85.07 at%, 7.48 at%, and 7.45 at%, respectively. For NPC, the content of C, N, and O elements was 85.6 at%, 7 at%, and 5.57 at%, respectively. Moreover, a certain amount of residual fluorine (1.64 at%) and Si (0.17 at%) in NPC was detected, which most likely originated from the F− of NH4HF2 and SiO2 particles, respectively, buried and trapped in the amorphous carbon during the de-SiO2 process.In Fig. 3c, the C 1s spectra for NC and NPC can be deconvoluted into several individual peaks. The fitted peaks at 284.6 eV, 286.1 eV, and 288.5 eV are related to the bonding configurations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN/C–O, and CO, respectively.59 It should be noted that the ratios of sp2-Cs in NC and NPC occupied a large percentage (61.58 at% and 58.99 at%, respectively). The other types of C elements occupied 23.49 at% for NC and 26.61 at% for NPC. In Fig. 3c, the three deconvoluted peaks for N element in NC at 398.3 eV, 400.8 eV, and 402.8 eV belong to pyrrolic-N, pyridinic-N, and graphitic N/oxidized N species, occupying 3.67 at%, 3.31 at%, and 0.5 at%, respectively.29,60,61 However, in Fig. 3d, only pyrrolic-N and pyridinic-N peaks in NPC were detected, which were 3.41 at% and 3.59 at%, and they originated from the pre-embedded sp2-N and sp3-N, respectively. As shown in Fig. S3b,† the spectra of O element in NC were also deconvoluted into two peaks at 531.5 eV and 532.8 eV, occupying 5.11 at% and 2.34 at% from OC and O–C, respectively. For NPC, the corresponding two fitted peaks at 531.5 eV and 532.8 eV of O element occupied 3.55 at% and 2.02 at%, respectively. The FE-SEM mapping images exhibit the homogenous distribution of C, N, and O for both the NC (Fig. S2d–S2f†) and NPC (Fig. S2h–S2j†) particle samples. Elemental analysis disclosed that the N content weight ratios in both NC and NPC were as high as 10.9 wt% and 12.6 wt%, respectively, which were higher than those in other N-contained carbonaceous materials for LIBs.19,62,63
C, CN/C–O, and CO, respectively.59 It should be noted that the ratios of sp2-Cs in NC and NPC occupied a large percentage (61.58 at% and 58.99 at%, respectively). The other types of C elements occupied 23.49 at% for NC and 26.61 at% for NPC. In Fig. 3c, the three deconvoluted peaks for N element in NC at 398.3 eV, 400.8 eV, and 402.8 eV belong to pyrrolic-N, pyridinic-N, and graphitic N/oxidized N species, occupying 3.67 at%, 3.31 at%, and 0.5 at%, respectively.29,60,61 However, in Fig. 3d, only pyrrolic-N and pyridinic-N peaks in NPC were detected, which were 3.41 at% and 3.59 at%, and they originated from the pre-embedded sp2-N and sp3-N, respectively. As shown in Fig. S3b,† the spectra of O element in NC were also deconvoluted into two peaks at 531.5 eV and 532.8 eV, occupying 5.11 at% and 2.34 at% from OC and O–C, respectively. For NPC, the corresponding two fitted peaks at 531.5 eV and 532.8 eV of O element occupied 3.55 at% and 2.02 at%, respectively. The FE-SEM mapping images exhibit the homogenous distribution of C, N, and O for both the NC (Fig. S2d–S2f†) and NPC (Fig. S2h–S2j†) particle samples. Elemental analysis disclosed that the N content weight ratios in both NC and NPC were as high as 10.9 wt% and 12.6 wt%, respectively, which were higher than those in other N-contained carbonaceous materials for LIBs.19,62,63
The investigation on NC and NPC suggested that employment of effective templates and heteroatom pre-embedded precursors is an efficient strategy to produce highly N-doped porous carbon materials,33,64,65 which are expected to display effective Li+ ion insertion/de-insertion behaviour according to the proposed lithiation mechanism (Scheme 1).17
Electrochemical performance
Fig. 4a and S4b† present the first three voltage curves of the NC and NPC electrodes measured through cyclic voltammogram (CV) under ambient conditions between 0.005 and 3.0 V at a scan rate of 0.5 mV s−1. The first discharge semi-cycle did not overlap the subsequent cycles. The strong but broad peaks at 0.4–1 V were mainly contributed by the newly generated solid electrolyte interphase (SEI) film,66–71 especially for the enhanced peak at 0.4 V for NPC with an improved surface area.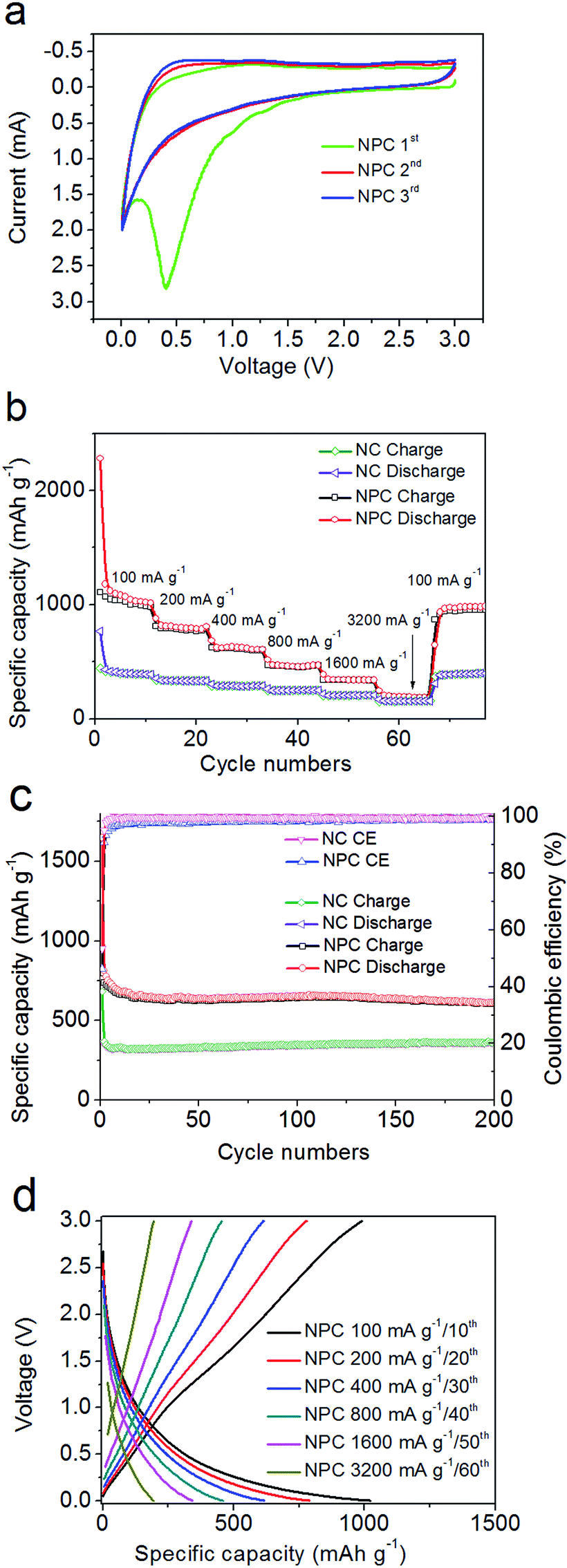 | ||
| Fig. 4 (a) Electrochemical performance of the NPC electrode: the first, second, and third CV profiles at a scan rate of 0.5 mV s−1 over the potential window of 0.005–3 V (vs. Li/Li+). (b) Rate performance at different current densities from 100 to 3200 mA g−1. (c) Cycling performance and CE at 200 mA g−1 d−1) Galvanostatic charge–discharge curves of NPC at different current densities ranging from 100 to 3200 mA g−1 in the 10th–60th cycles. | ||
In Fig. 4b, the NC and NPC electrodes present rate performance curves at different current densities ranging from 100, 200, 400, 800, and 1600 to 3200 mA g−1. At a current density of 100 mA g−1, the initial irreversible discharge capacity of the NPC electrode reached as high as 2283 mA h g−1, which was thrice the corresponding value of 767 mA h g−1 for the NC electrode. It should be noted that these values are almost double of their initial reversible charge capacities of 1107 mA h g−1 and 448 mA h g−1. The irreversibility is most likely due to the SEI effect and unidirectional Li+ ion trapping or consumable reaction. However, in the first reversible cycle, the specific discharge capacity of the NPC electrode reached as high as 1178.9 mA h g−1 and stabilized at 1014 mA h g−1 by the end of the 11th cycle, which were both nearly 2.7 times the corresponding value of 432 mA h g−1, and stabilized at 387 mA h g−1 for the NC electrode. At a higher specific rate of 1.6 A g−1, the capacity was still stabilized at ∼336.0 mA h g−1, which was close to the theoretical value of graphite (372 mA h g−1).69–73 Even at the strongest current density of 3.2 A g−1, the NC and NPC-based electrodes still delivered 155 and 207 mA h g−1 at the 57th cycle. Significantly, when the current density returned to 100 mA g−1 in the 70th cycle, the discharge capacity was still over 976 mA h g−1 and 389 mA h g−1 for NPC and NC, respectively, which recovered to the initial value although slightly lower than that in the first cycle at a current density of 100 mA g−1.
Fig. 4c shows the cyclability and coulombic efficiency (CE) at a current density of 200 mA g−1 for the NC and NPC electrodes. The initial irreversible discharge capacity of the NPC electrode reached as high as 1648 mA h g−1. The NPC electrode exhibited good and stable cyclability with a specific capacity ranging from 700 to 614 mA h g−1 up to the 200th cycle. In contrast, the NC electrode presented a much lower specific capacity ranging from 325 to 364 mA h g−1 up to the 200th cycle. However, both their coulombic efficiencies from the second cycle up to the 200th cycle were almost over 93%. In this situation, the corresponding galvanostatic charge/discharge profiles (Fig. 4d) of the better performing NPC electrode were obtained at six different current densities ranging from 100, 200, 400, 800, 1600, to 3200 mA g−1 at the 10th, 20th, 30th, 40th, 50th, and 60th cycles, respectively, which indicated that the NPC electrode exhibited matching electrochemical properties, almost coinciding with the charge/discharge voltage–capacity curves.
To further understand the charge–transfer ability and illustrate the advantages of the NPC electrode over the NC electrode, EIS tests on NPC and NC electrodes were conducted under the same conditions. The charge transfer resistances are depicted in Fig. S5.† It can be seen that both electrodes present high-frequency semicircles, representing the charge–transfer process. The EIS plots in the low-frequency region suggest that the NPC electrode possesses a lower Li+ ion diffusion resistance than the NC electrode. From the CV measurement and calculation, the charge-transfer resistance of the NPC electrode was calculated to be about 42.5 Ω, smaller than that of NC (55.7 Ω), indicating its better conductivity.
Compared to the NC electrode, the improved high capacity and rate capability of the NPC electrode could be explained by its special porous structure and high-level N-doping. As abovementioned, the carbonaceous NPC electrode plays a key role as conductive channels1,9b,18 for electron transport, whereas its large number of pores possibly act as Li+ ion storage cisterns.71 Furthermore, the enlarged electrode/electrolyte interface of the NPC electrode promotes the rapid absorption and release of Li+ ions with a fast charge-transfer process.
Conclusions
In summary, two novel pyridinic/pyrrolic N-doped carbon materials have been rationally designed and successfully obtained via the pristine and template pyrolyzation of an sp2/sp3-N-rich azaacene dye with a complete π-conjugated framework and high melting point. The anodic NPC electrode exhibited high Li+ ions storage performance due to its high pyridinic/pyrrolic N-content and electrolyte and liquid interface. Its specific capacity was as high as 614 mA h g−1 after 200 cycles even at 200 mA g−1. Our results indicate that the pyrolyzation of azaacene dye rich in sp3/sp2-N is an effective strategy to achieve more highly monospecies or concomitant pyridinic/pyrrolic N-doped carbon materials.Experimental
Materials
Naphthalene-1,4,5,8-tetracarboxylic acid dianhydride was purchased from Zhengzhou Keyulong Chemical Products Co., Ltd. o-Benzene-diamine was purchased from Energy Co., Ltd. Micro-SiO2 (8 ± 2 μm ≥80%) was obtained from high-performance thin layer chromatography plates (200 × 200 mm) purchased from Yantai Jiangyou Guijiao Kaifa Co., Ltd. All other reagents and solvents were purchased from Wanqing Co., Ltd. All solvents were used without further purification.Synthesis of NC and NPC
Azaacene was synthesized, filtered, washed, and dried as a red powder in 64% yield by refluxing DMF in o-benzene-diamine and naphthalene-1,4,5,8-tetracarboxylic acid dianhydride. The mixture of azaacene dye (1.5 g) with micro-SiO2 (3 g) was mixed and ground for 10 min. Azaacene (1 g) and the ground mixture were put into corresponding Al2O3 crucibles, in an OTF-1200X tube furnace with SiO2 glass and protected under an Ar atmosphere and heated to 700 °C at a heating rate of 3 °C min−1 and kept for 0.5 h under a nitrogen atmosphere. The activated mixture was then washed with the NH4HF2 solution (1 mol L−1), NaOH solution (1 mol L−1), and deionized water in sequence until the filtrate became neutral. The sample was finally dried overnight at 100 °C in an oven. The carbonization yields of NC and NPC were 65% and 40%, respectively.Methods
The morphology of the samples was observed via FE-SEM (Hitachi S-4800, Tokyo, Japan). TEM observation was carried out using a JEOL 2100F microscope operating at an accelerating voltage of 100 kV. HR-TEM observation was carried out using an FEI TECAI G2F20 microscope operating at an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns were obtained using a Bruker D8 X-ray diffractometer with Cu Kα radiation (λ = 1.5405 Å). Raman spectroscopy was carried out suing a WITec alpha300M+ micro-Raman confocal microscope. CHN elemental analysis was performed using a Thermo Scientific FLASH 2000 elemental analyzer. XPS measurements were performed using a Thermo Scientific ESCALAB 250XI system with a monochromatic Al Kα X-ray source. Nitrogen adsorption and desorption isotherms were determined by nitrogen physisorption at 77 K using a V-Sorb 2800P BET surface area and pore volume analyser.Electrochemical tests
The electrochemical properties of NC and NPC were measured with 2032 coin cells using pure lithium foil as the counter electrode. The working electrodes were constructed by mixing the as-prepared azaacene dye samples of 80 wt% NC or NPC, 10 wt% polyvinylidene fluoride (PVDF), 10 wt% acetylene black, and N-methyl-2-pyrrolidone as the dispersing solvent to obtain slurries, which were coated homogeneously onto the copper foil and dried at 100 °C overnight in a vacuum oven. Circular electrodes (12 mm in diameter) were prepared using a punching machine and weighed with an electronic balance within 0.01 mg. The electrolyte was a 1.0 mol L−1 LiPF6 in ethylene carbonate and dimethyl carbonate solution (1:1 (w/w)). Celgard 2300 was applied as the separator. Coin cells with both electrodes were assembled in an inert Ar gas-filled glove box. Both the moisture and oxygen contents were kept below 0.1 ppm. The galvanostatic charge/discharge parameters of the cells were measured using a multichannel NEWARE battery-testing system with the voltage ranging from 0.005 to 3.0 V vs. Li+/Li at different rates. The calculated capacity values were determined based on the weight of the loaded carbon materials on the collectors. Galvanostatic cycling was performed using a NEWARE multichannel battery testing system. Alternative current (AC) impedance measurements were conducted using a CHI 760D electrochemical workstation (CH Instruments, Inc.) at room temperature. EIS was performed with an AC voltage of 5 mV amplitude in the frequency range from 100 kHz to 0.1 Hz.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the 973 program (2015CB932200), the National Natural Science Foundation of China (NSFC 21502091, 5161101159), and the Natural Science Foundation of Jiangsu Province (BK20171470, 14KJB430017, BK20150064, BK20130912) for the financial support and Ministry of Education and Synergetic Innovation Center for Organic Electronics and Information Displays.Notes and references
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- (a) M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC; (b) S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- L. Qie, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2013, 6, 2497–2504 Search PubMed.
- M. D. Bhatt and C. O'Dwyer, Phys. Chem. Chem. Phys., 2015, 17, 4799–4844 RSC.
- F. Sun, J. Gao, X. Liu, L. Wang, Y. Yang, X. Pi, S. Wu and Y. Qin, Electrochim. Acta, 2016, 213, 626–632 CrossRef CAS.
- Z. Xu, X. Zhuang, C. Yang, J. Cao, Z. Yao, Y. Tang, J. Jiang, D. Wu and X. Feng, Adv. Mater., 2016, 28, 1981–1987 CrossRef CAS PubMed.
- (a) S. Y. Gao, H. Y. Liu, K. R. Geng and X. J. Wei, Nano Energy, 2015, 12, 785–793 CrossRef CAS; (b) F. P. Pan, Z. Y. Cao, Q. P. Zhao, H. Y. Liang and J. Y. Zhang, J. Power Sources, 2014, 272, 8–15 CrossRef CAS; (c) R. Sharma and K. K. Kar, Electrochim. Acta, 2016, 191, 876–886 CrossRef CAS; (d) S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J.-B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed; (e) Z. Y. Wu, H. W. Liang, L. F. Chen, B. C. Hu and S. H. Yu, Acc. Chem. Res., 2016, 49, 96–105 CrossRef CAS PubMed; (f) C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930–2936 CAS.
- (a) L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed; (b) L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- J. Ou, Y. Zhang, L. Chen, Q. Zhao, Y. Meng, Y. Guo and D. Xiao, J. Mater. Chem. A, 2015, 3, 6534–6541 CAS.
- L. Bhattacharjee, R. Manoharan, K. Mohanta and R. R. Bhattacharjee, J. Mater. Chem. A, 2015, 3, 1580–1586 CAS.
- G. Chen, L. Yan, H. Luo and S. Guo, Adv. Mater., 2016, 28, 7580–7602 CrossRef CAS PubMed.
- C. Chen, D. Yu, G. Zhao, B. Du, W. Tang, L. Sun, Y. Sun, F. Besenbacher and M. Yu, Nano Energy, 2016, 27, 377–389 CrossRef CAS.
- (a) K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS; (b) Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed; (c) J.-H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed; (d) Y. M. Chen, X. Y. Li, K. Park, J. Song, J. H. Hong, L. M. Zhou, Y. W. Mai, H. T. Huang and J. B. Goodenough, J. Am. Chem. Soc., 2013, 135, 16280–16283 CrossRef CAS PubMed.
- J. Yang, J. Xie, X. Zhou, Y. Zou, J. Tang, S. Wang, F. Chen and L. Wang, J. Phys. Chem. C, 2014, 118, 1800–1807 CAS.
- H. Chen, Y. Wei, J. Wang, W. Qiao, L. Ling and D. Long, ACS Appl. Mater. Interfaces, 2015, 7, 21188–21197 CAS.
- (a) T. Hu, X. Sun, H. T. Sun, G. Q. Xin, D. L. Shao, C. S. Liu and J. Lian, Phys. Chem. Chem. Phys., 2014, 16, 1060–1066 RSC; (b) C. C. Ma, X. H. Shao and D. P. Cao, J. Mater. Chem., 2012, 22, 8911–8915 RSC; (c) Z. Q. Tan, K. Ni, G. X. Chen, W. C. Zeng, Z. C. Tao, M. Ikram, Q. B. Zhang, H. J. Wang, T. Sun, X. J. Zhu, X. J. Wu, H. X. Ji, R. S. Ruoff and Y. W. Zhu, Adv. Mater., 2017, 29, 1603414 CrossRef PubMed; (d) W. Ai, Z. M. Luo, J. Jiang, J. H. Zhu, Z. Z. Du, Z. X. Fan, L. H. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- (a) C. Hu, Y. Xiao, Y. Zhao, N. Chen, Z. Zhang, M. Cao and L. Qu, Nanoscale, 2013, 5, 2726–2733 RSC; (b) Z. Q. Luo, S. Lim, Z. Q. Tian, J. Z. Shang, L. F. Lai, B. MacDonald, C. Fu and Z. X. Shen, J. Mater. Chem., 2011, 21, 8038–8044 RSC; (c) B. Zheng, X. L. Cai, Y. Zhou and X. H. Xia, ChemElectroChem, 2016, 3, 2036–2042 CrossRef CAS.
- H. B. Wang, M. S. Xie, L. Thia, A. Fisher and X. Wang, J. Phys. Chem. Lett., 2014, 5, 119–125 CrossRef CAS PubMed.
- Z. Q. Zhu, S. W. Wang, J. Du, Q. Jin, T. R. Zhang, F. Y. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- (a) W. Ai, J. Jiang, J. Zhu, Z. Fan, Y. Wang, H. Zhang, W. Huang and T. Yu, Adv. Energy Mater., 2015, 5, 1500559 CrossRef; (b) D. Dong, D. Fang, H. R. Li, C. X. Zhu, X. H. Zhao, J. W. Li, L. Z. Jin, L. H. Xie, L. Chen, J. F. Zhao, H. M. Zhang and W. Huang, Chem.–Asian J., 2017, 12, 920–926 CrossRef CAS PubMed; (c) J. F. Zhao, R. P. Li, W. Ai, D. Dong, J. W. Li, L. Chen, L. H. Xie, T. Yu and W. Huang, Chem.–Asian J., 2016, 11, 1382–1387 CrossRef CAS PubMed.
- (a) M. Huang, K. Mi, J. H. Zhang, H. L. Liu, T. T. Yu, A. H. Yuan, Q. H. Kong and S. L. Xiong, J. Mater. Chem. A, 2017, 5, 266–274 RSC; (b) J. H. Zhang, Q. H. Kong, L. W. Yang and D. Y. Wang, Green Chem., 2016, 18, 3066–3074 RSC; (c) Z. J. Jiang and Z. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 19082–19091 CrossRef CAS PubMed.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- L. Zhang, M. Zhang, Y. Wang, Z. Zhang, G. Kan, C. Wang, Z. Zhong and F. Su, J. Mater. Chem. A, 2014, 2, 10161–10168 CAS.
- F. D. Han, Y. J. Bai, R. Liu, B. Yao, Y.-X. Qi, N. Lun and J.-X. Zhang, Adv. Energy Mater., 2011, 1, 798–801 CrossRef CAS.
- H. G. Wang, Y. Wang, Y. Li, Y. Wan and Q. Duan, Carbon, 2015, 82, 116–123 CrossRef CAS.
- J. W. F. To, Z. Chen, H. Yao, J. He, K. Kim, H.-H. Chou, L. Pan, J. Wilcox, Y. Cui and Z. Bao, ACS Cent. Sci., 2015, 1, 68–76 CrossRef CAS PubMed.
- (a) S. Zhang, M. Zheng, Z. Lin, N. Li, Y. Liu, B. Zhao, H. Pang, J. Cao, P. He and Y. Shi, J. Mater. Chem. A, 2014, 2, 15889–15896 RSC; (b) X. Xiang, Z. Huang, E. Liu, H. Shen, Y. Tian, H. Xie, Y. Wu and Z. Wu, Electrochim. Acta, 2011, 56, 9350–9356 CrossRef CAS.
- C. Z. Zhang, N. Mahmood, H. Yin, F. Liu and Y. L. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- H. Song, N. Li, H. Cui and C. Wang, Nano Energy, 2014, 4, 81–87 CrossRef CAS.
- (a) Y.-X. Yu, Phys. Chem. Chem. Phys., 2013, 15, 16819–16827 RSC; (b) W. Ai, X. Wang, C. Zou, Z. Du, Z. Fan, H. Zhang, P. Chen, T. Yu and W. Huang, Small, 2017, 13, 1602010 CrossRef PubMed.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 CAS.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- T. Y. Ma, L. Liu and Z.-Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- (a) I. Elizabeth, B. P. Singh, S. Trikha and S. Gopukumar, J. Power Sources, 2016, 329, 412–421 CrossRef CAS; (b) Z. Yang, H. Guo, X. Li, Z. Wang, Z. Yan and Y. Wang, J. Power Sources, 2016, 329, 339–346 CrossRef CAS.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed.
- Z. Zhang and L. Yin, Electrochim. Acta, 2016, 212, 594–602 CrossRef CAS.
- (a) Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. Lou, Nat. Commun., 2015, 6, 8850 CrossRef CAS PubMed; (b) H. W. Song, G. Z. Yang and C. X. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 21661–21668 CrossRef CAS PubMed.
- H. Souri, S. J. Yu, H. Yeo, M. Goh, J. Y. Hwang, S. M. Kim, B.-C. Ku, Y. G. Jeong and N.-H. You, RSC Adv., 2016, 6, 52509–52517 RSC.
- A. Sanchez-Sanchez, F. Suarez-Garcia, A. Martinez-Alonso and J. M. D. Tascon, Carbon, 2014, 70, 119–129 CrossRef CAS.
- X. Liu, S. Li, J. Mei, W.-M. Lau, R. Mi, Y. Li, H. Liu and L. Liu, J. Mater. Chem. A, 2014, 2, 14429–14438 CAS.
- H. X. Zhong, J. Wang, Y. W. Zhang, W. L. Xu, W. Xing, D. Xu, Y. F. Zhang and X. B. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- Z. Xie, Z. He, X. Feng, W. Xu, X. Cui, J. Zhang, C. Yan, M. A. Carreon, Z. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 10324–10333 CAS.
- (a) W. Guo, X. Li, J. T. Xu, H. K. Liu, J. M. Ma and S. X. Dou, Electrochim. Acta, 2016, 188, 414–420 CrossRef CAS; (b) B. Jin, F. Gao, Y. F. Zhu, X. Y. Lang, G. F. Han, W. Gao, Z. Wen, M. Zhao, J. C. Li and Q. Jiang, Sci. Rep., 2016, 6, 19317 CrossRef CAS PubMed.
- D. Deng and J. Y. Lee, Chem. Mater., 2007, 19, 4198–4204 CrossRef CAS.
- V. G. Pol and M. M. Thackeray, Energy Environ. Sci., 2011, 4, 1904–1912 CAS.
- J. Mizuguchi, J. Phys. Chem. B, 2004, 108, 8926–8930 CrossRef CAS.
- (a) G. Li, Y. C. Wu, J. K. Gao, C. Y. Wang, J. B. Li, H. C. Zhang, Y. Zhao, Y. L. Zhao and Q. C. Zhang, J. Am. Chem. Soc., 2012, 134, 20298–20301 CrossRef CAS PubMed; (b) J. Z. Zhao, J. I. Wong, C. Y. Wang, J. K. Gao, V. Z. Y. Ng, H. Y. Yang, S. C. J. Loo and Q. C. Zhang, Chem.–Asian J., 2013, 8, 665–669 CrossRef CAS PubMed; (c) P. Y. Gu, F. Zhou, J. K. Gao, G. Li, C. Y. Wang, Q. F. Xu, Q. C. Zhang and J. M. Lu, J. Am. Chem. Soc., 2013, 135, 14086–14089 CrossRef CAS PubMed; (d) C. Y. Wang, J. Zhang, G. K. Long, N. Aratani, H. Yamada, Y. Zhao and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6292–6296 CrossRef CAS PubMed; (e) M. Mamada, C. S. Pérez-Bolívar and P. Anzenbacher, Org. Lett., 2011, 13, 4882–4885 CrossRef CAS PubMed.
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609–615 CrossRef CAS.
- S. F. Ahmed, S. Das, M. K. Mitra and K. K. Chattopadhyay, Indian J. Pure Appl. Phys., 2006, 44, 700–704 CAS.
- P. M. Panchmatia, A. R. Armstrong, P. G. Bruce and M. S. Islam, Phys. Chem. Chem. Phys., 2014, 16, 21114–21118 RSC.
- R. N. Singh and R. Awasthi, Catal. Sci. Technol., 2011, 1, 778–783 Search PubMed.
- (a) W. Qian, R. Hao, Y. Hou, Y. Tian, C. Shen, H. Gao and X. Liang, Nano Res., 2009, 2, 706–712 CrossRef CAS; (b) Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- D. R. Rolison, Science, 2003, 299, 1698–1701 CrossRef CAS PubMed.
- Y. Meng, X. Zou, X. Huang, A. Goswami, Z. Liu and T. Asefa, Adv. Mater., 2014, 26, 6510–6516 CrossRef CAS PubMed.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed.
- Z. S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- L. G. Bulusheva, A. V. Okotrub, A. G. Kurenya, H. Zhang, H. Zhang, X. Chen and H. Song, Carbon, 2011, 49, 4013–4023 CrossRef CAS.
- C. Li, X. Yin, L. Chen, Q. Li and T. Wang, J. Phys. Chem. C, 2009, 113, 13438–13442 CAS.
- (a) T. Takeichi, Y. Yamazaki, M. Zuo, A. Ito, A. Matsumoto and M. Inagaki, Carbon, 2001, 39, 257–265 CrossRef CAS; (b) W. Ai, W. Zhou, Z. Du, C. Sun, J. Yang, Y. Chen, Z. Sun, S. Feng, J. Zhao, X. Dong, W. Huang and T. Yu, Adv. Funct. Mater., 2017, 27, 1603603 CrossRef.
- L. G. Bulusheva, A. V. Okotrub, A. G. Kurenya, H. K. Zhang, H. J. Zhang, X. H. Chen and H. H. Song, Carbon, 2011, 49, 4013–4023 CrossRef CAS.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim, Y. A. Kim and M. Endo, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. Proietti Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS PubMed.
- J. Wu, X. Rui, C. Wang, W.-B. Pei, R. Lau, Q. Yan and Q. Zhang, Adv. Energy Mater., 2015, 5, 1402189 CrossRef.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 CAS.
- S. B. Yang, X. L. Feng, L. J. Zhi, Q. A. Cao, J. Maier and K. Müllen, Adv. Mater., 2010, 22, 838–842 CrossRef CAS PubMed.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed; N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07850a |
| This journal is © The Royal Society of Chemistry 2017 |