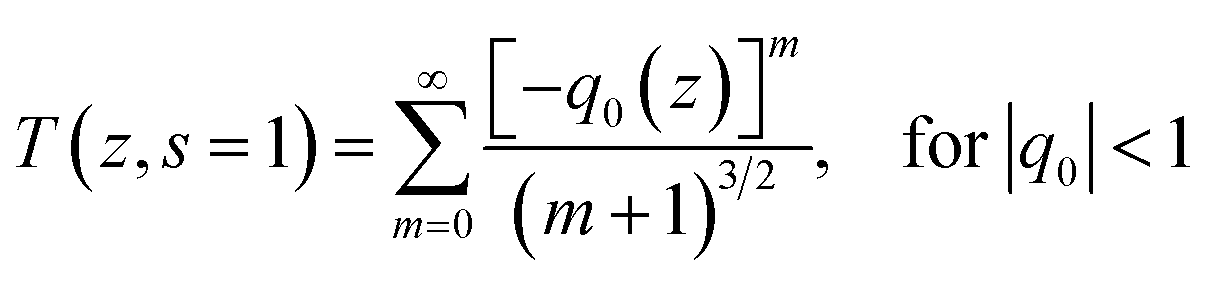DOI:
10.1039/C7RA08499D
(Paper)
RSC Adv., 2017,
7, 53785-53796
Polyfluorenylacetylene for near-infrared laser protection: polymer synthesis, optical limiting mechanism and relationship between molecular structure and properties†
Received
1st August 2017
, Accepted 2nd November 2017
First published on 22nd November 2017
Abstract
A series of functional polyacetylenes bearing the fluorene moiety with different conjugated lengths and terminal substituents, poly[2-ethynyl-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene] (P1), poly[2-ethynyl-7-(4-(4-nitrostyryl)styryl)-9,9-dioctyl-9H-fluorene] (P2), poly[2-ethynyl-7-(4-(4-methoxystyryl)styryl)-9,9-dioctyl-9H-fluorene] (P3), poly[2-ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-dioctyl-9H-fluorene] (P4), and poly[2-ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-didodecyl-9H-fluorene] (P5), were designed and prepared using [Rh(nbd)Cl]2 as the catalyst. Their structures and properties were characterized and evaluated by FTIR, NMR, UV, FL, GPC and TGA analyses. The optical limiting properties were investigated using 450 fs laser pulses at 780 nm. These results show that the incorporation of styryl/stilbene functionalized polyfluorenylacetylenes has endowed resultant polyacetylenes with novel near-infrared laser protection properties and enhanced thermal stability. The optical limiting mechanism for laser protection was studied. It was found that the optical limiting properties mainly originated from two-photon absorption (TPA) of molecules in the resulting polyacetylenes. Additionally, it was also found that the functionalized polyacetylene with a longer fluorene-based conjugated chromophore and a stronger terminal electron acceptor group exhibits better optical limiting properties because of the larger π-electron delocalization and dipolar effect.
1. Introduction
With the rapid development of new laser technology, laser protection in recent years is believed to be promising for both civilians and the military.1 These works have mainly focused on the visible range, where the nonlinear transmission phenomena are mainly dependent on multiphoton absorption (MPA) or reverse saturable absorption (RSA), nonlinear scattering. The materials designed for laser protection involve graphene families,2–4 single-walled and multi-walled carbon nanotubes,5–9 organic dyes,10 inorganic materials,11,12 π-electron conjugated systems such as fullerene13,14 and porphyrins, phthalocyanines,15–18 and organometallic complexes19–21 as optical limiting materials for protecting human eyes and sensitive optical sensors from laser damage. Accordingly, to obtain an ideal optical limiting implement, some major factors should be taken into account such as fast response speed, low transmittance at high intensities and high transmittance at low intensities, temporal response and broad spectral characteristics.22 For the classical benchmark, porphyrins, phthalocyanines and fullerenes can also provide excellent optical limiting properties. However, their narrow excited-state absorption (ESA) spectral regions and poor optical transparency characteristics restricted practical applications in broadband optical limiting properties. Up to now, a considerable crowd of papers have reported that some materials have broad spectral optical limiting properties under nanosecond photoexcitation.2,23–32 Specifically, metal-based materials are premeditated as novel generation optical limiting materials displaying a high transparent characteristic in the visible spectral region.33,34 However, to the best of our knowledge, only a few papers reported the laser protection on the near-infrared spectral region has been investigated. The relationship between molecular structure and near-infrared laser protection properties has not been understood.11,35–38 It is well known that substituted polyacetylenes (PAs) are considerable very promising laser protection materials owing to their large third order nonlinear optical susceptibilities, fast time response, good conductivity,39 well photophysical stability and processability for optical devices.40–42 In our previous work, many functional polyacetylene materials for laser protection in the visible optical wavelength region were designed.43–52 It was found that the incorporation of nonlinear optical chromophore as a pendant group into polyacetylene backbone has endowed polyacetylenes with novel optical limiting properties. The optical limiting properties were mainly influenced by the molecular structure of substituted chromophore group and electron interaction between polyacetylene backbone and substituted chromophore. The interaction generally increases with the decrease of space length between substituted chromophore group and polyacetylene backbone.45 The functional polyacetylene containing long conjugated chromophore with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond as π electron conjugation bridge exhibited better optical limiting property than that with CN or NN double bond as π-electron conjugation bridge under the same linear transmittance.47,51 Their optical limiting mechanism was attributed to the reverse saturated absorption (RSA) of molecules.
C double bond as π electron conjugation bridge exhibited better optical limiting property than that with CN or NN double bond as π-electron conjugation bridge under the same linear transmittance.47,51 Their optical limiting mechanism was attributed to the reverse saturated absorption (RSA) of molecules.
Thus, it was reported that polyacetylenes may be a potential near-infrared laser protection materials.53 However, to our knowledge, the polyacetylenes with optical limiting properties for near-infrared optical region have seldom been reported. In particular, the relationship between the structure and near-infrared optical limiting properties of the molecules, so far, has not been understood. It is known that fluorene is a well near-infrared TPA chromophore.54,55 It is expected that the incorporation of fluorene-based chromophores as the pendant groups into polyacetylene could endow the polyacetylenes with novel optical properties. Therefore, in the work, we designed and synthesized a series of novel polyacetylenes that contain fluorene pendant with different conjugation length and terminal substituent such as electron withdrawing or electron donating groups. At the same time, the alkyl chains at nine positions extending away from the conjugated π-system of fluorene was introduced for improving the solubility of resultant functional polyacetylenes in common organic solvents and provide good compatibility with other matrices.56 The relationship between near-infrared optical limiting properties and molecular structure and the optical limiting mechanism were investigated in detail.
2. Experimental section
2.1 Materials
Unless otherwise noted, all commercial reagents were used as received. 4-Vinylbenzyl chloride, triethyl phosphite, 2-methyl-3-butyn-2-ol, p-nitrostyrene, palladium(II) acetate, bis(triphenylphosphine) palladium(II) chloride [PdCl2(PPh3)2], norbornadiene rhodium(I) chloride dimer ([Rh(nbd)Cl]2), WCl6, TaCl5, MoCl5 and tetraphenyltin (Ph4Sn) were all purchased from Aldrich, and used as received without further purification. 2,7-Dibromofluorene was obtained from TEIKO International LTD. 1-Bromooctane, 1-bromododecane, tetrabutyl ammonium bromide, 4-methoxybenzaldehyde, 4-methylbenzaldehyde, 4-nitrobenzaldehyde, NaH, NaOH, KOH, MgSO4, triphenylphosphine, tetrahydrofuran (THF), dimethylsulfoxide (DMSO), dimethylformamide (DMF), triethylamine (Et3N), dioxane, dichloromethane, petroleum ether and ethyl acetate were all purchased from Shanghai Chemical Reagent Company. Dioxane and THF were distilled from sodium benzophenone ketyl immediately before use. DMF was dried over anhydrous MgSO4 and freshly distilled prior to use. Et3N was distilled from KOH before use. Technical grade methanol was used to precipitate the polymers.
2.2 Instrumentation
FTIR spectra of KBr disks were recorded with a Thermo Nicolet 5700 spectrometer at room temperature, 32 scans were collected at a resolution of 1 cm−1. 1H NMR (400 MHz) and 13C NMR (100 MHz) were recorded on a Bruker DMX-400 spectrometer utilizing tetramethylsilane (TMS, 0.00 ppm) as the internal standard in chloroform-d (CDCl3) at room temperature. Elementary analyses were conducted on Vario EL-III elementary analysis apparatus. UV-vis spectra were recorded on a Shimadzu UV-265 spectrometer using a 1 cm square quartz cell. Fluorescence spectra were obtained on a Shimadzu RF-5301PC spectrofluorimeter equipped with a 450 W Xe lamp and a time-correlated single-photon counting card. Weight-average (Mw) and number-average (Mn) molecular weights, and polydispersity index (PDI, Mw/Mn) were determined by gel permeation chromatograph (GPC) using a Waters 510 HPLC equipped with a Rheodyne 7725i injector with a stand kit, a set of styragel columns (HT3, HT4, and HT6; molecular weight range 102–107), a column temperature controller, a Waters 486 wavelength tunable UV-vis detector, a Waters 410 differential refractometer, and a system DMM/scanner possessing an 8-channel scanner option. All polymer solutions were prepared in THF (ca. 2 mg mL−1) and filtered through 0.45 μm PTFE syringe-type filters before injected into the GPC system. THF was used as the eluent at a flow rate of 1.0 mL min−1. The column temperature was maintained at 30 °C and the working wavelength of the UV detector was set at 254 nm. A set of mono-disperse polystyrene standards (waters) was used for calibration purposes. Differential scanning calorimetry (DSC) was performed on a TA Instruments Q2000 equipped with a liquid nitrogen cooling accessory (LNCA) unit under a continuous nitrogen purge (50 mL min−1). The scan rate was 10 °C min−1 within the temperature range 30–250 °C. Samples (4–6 mg) were weighed and sealed in aluminum pans. Thermogravimetric analysis (TGA) was carried out using a NETZSCH Instruments 449F3 thermogravimetric analyzer with a heating rate of 20 °C min−1 from 20 to 800 °C under a continuous nitrogen purge (100 mL min−1). Samples (15–25 mg) were loaded in alumina pans. The thermal degradation temperature (Td) was defined as the temperature of 5% mass loss.
The molecular two-photon absorption (TPA) cross sections were measured by a Z-scan technique. The laser pulses were produced by a mode-locked Ti:sapphire laser (Quantronix, IMRA), which seeded a Ti:sapphire regenerative amplifier, and focused onto a 1 mm-thick quartz cuvette containing the solutions of these molecules with the minimum beam waist of 25 ± 5 μm. The incident and transmitted laser pulse energy were monitored by moving the cuvette along the propagation direction of the laser pulses. The maximum on-axis irradiance of the laser pulses was ∼120 GW cm−2 at the focus point. The Z-scan experimental system was calibrated using a piece of cadmium sulfide (CdS) bulk crystal as a reference because it possesses large TPA at the wavelength of 780 nm and has been well investigated in our laboratory. The TPA coefficient of CdS was determined to be 6.4 ± 0.6 cm GW−1, which was in good agreement with theoretical values within the experiment uncertainty. The investigation of the optical limiting properties of the samples was carried out by using the same laser system as in the Z-scan experiment. The experimental arrangement was similar with that in the literature.57 The samples were contained in quartz cells with a thickness of 1 mm and positioned at the focal point. Two-photon fluorescence (TPF) measurements of the polymers in solid form were carried out using the same laser system described above. The details of the two-photon technique experimental setup are described elsewhere.58
2.3 Monomer synthesis (cf. Scheme 1)
4-Ethenyl-4′-nitro-1,2-stilbene (1a). 3.9 mL 4-vinylbenzyl chloride (25 mmol) and 5.5 mL triethyl-phosphite (30 mmol) were heated at 110 °C for 6 h in a solvent-free system. Excess triethylphosphite was removed by vacuum distillation, and the resultant phosphonate was obtained as the thick oil, which was used for the Wittig–Horner reaction without further purification. After the crude phosphonate in 50 mL of dry THF was cooled to 0 °C, 1.2 g NaH (50 mmol) were added slowly and reacted for 30 min. 3.78 g 4-nitrobenzaldehyde (25 mmol) in 10 mL THF was then added dropwise, and the contents were stirred under nitrogen for 12 h. The mixture was added slowly to 150 mL of ice-chilled water, and the red brown solid was filtered and rinsed twice with water and ethanol. The crude product was recrystallized from THF/ethanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) to provide red brown crystal powder. Yield: 53%. IR (KBr), ν (cm−1): 3021, 997, 927, 972 (C–H), 1629 (CC), 1589, 850 (Ar), 1514, 1338 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.24 (d, J = 8.8 Hz, 2H, Ar-H ortho to NO2), 7.65 (d, J = 8.8 Hz, 2H, Ar-H meta to NO2), 7.54 (d, J = 8.4 Hz, 2H, Ar-H meta to vinyl), 7.46 (d, J = 8.4 Hz, 2H, Ar-H ortho to vinyl), 7.30–7.14 (q, J = 18.7 Hz, 2H, –CHCH–), 6.79–6.72 (q, J = 9.5 Hz, 1H, –CHCH2), 5.82 (d, J = 17.6 Hz, 1H, –CHCH2), 5.33 (d, J = 11.2 Hz, 1H, –CHCH2).
:1 v/v) to provide red brown crystal powder. Yield: 53%. IR (KBr), ν (cm−1): 3021, 997, 927, 972 (C–H), 1629 (CC), 1589, 850 (Ar), 1514, 1338 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.24 (d, J = 8.8 Hz, 2H, Ar-H ortho to NO2), 7.65 (d, J = 8.8 Hz, 2H, Ar-H meta to NO2), 7.54 (d, J = 8.4 Hz, 2H, Ar-H meta to vinyl), 7.46 (d, J = 8.4 Hz, 2H, Ar-H ortho to vinyl), 7.30–7.14 (q, J = 18.7 Hz, 2H, –CHCH–), 6.79–6.72 (q, J = 9.5 Hz, 1H, –CHCH2), 5.82 (d, J = 17.6 Hz, 1H, –CHCH2), 5.33 (d, J = 11.2 Hz, 1H, –CHCH2).
4-Ethenyl-4′-methoxy-1,2-stilbene (1b). The synthesis process is similar to compound 1a by using 4-methoxybenzaldehyde. The product was gray crystal. Yield: 39%. IR (KBr), ν (cm−1): 3001, 970, 992, 905 (C–H), 2954, 2834 (CH3), 1624 (CC), 1601, 1514, 1462, 840 (Ar), 1254, 1036 (C–O–C). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.48 (d, J = 8.8 Hz, 4H, Ar-H–CHCH2) 7.42 (d, J = 8.4 Hz, 2H, Ar-H meta to OCH3), 7.11–6.96 (q, J = 13.6 Hz, 2H, Ar-H ortho to OCH3), 6.92 (d, J = 8.8 Hz, 2H, –CHCH–), 6.74 (q, J = 17.6 Hz, 1H, –CHCH2), 5.77 (d, J = 17.6 Hz, 1H, –CHCH2), 5.26 (d, J = 12.0 Hz, 1H, –CHCH2), 3.86 (s, 3H, Ar-OCH3).
4-Ethenyl-4′-methyl-1,2-stilbene (1c). The synthesis process is similar to compound 1a by using 4-methylbenzaldehyde. The product was white crystal. Yield: 44%. IR (KBr), ν (cm−1): 3021, 970, 992, 898 (C–H), 2915, 2855 (CH3), 1624 (CC), 1513, 835 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.48 (d, J = 8.4 Hz, 2H, Ar-H meta to CH3), 7.43 (t, J = 13.6 Hz, 4H, Ar-H–CHCH2) 7.19 (d, J = 8.0 Hz, 2H, Ar-H ortho to CH3), 7.08 (d, J = 6.0 Hz, 2H, –CHCH–), 6.74 (q, J = 18.2 Hz, 1H, –CHCH2), 5.77 (d, J = 17.6 Hz, 1H, –CHCH2), 5.26 (d, J = 11.2 Hz, 1H, –CHCH2), 2.39 (s, 3H, Ar-CH3).
2,7-Dibromo-9,9-dioctylfluorene (2a). 1-Bromooctane (5.60 g, 29.0 mmol) was added by using a syringe to a mixture of 2,7-dibromofluorene (3.88 g, 12.0 mmol), tetrabutyl ammonium bromide (0.0225 g, 0.0975 mmol) and 3.75 mL of 50% aqueous NaOH in DMSO (50 mL). The reaction mixture was stirred at room temperature for 3 h. The mixture was poured into H2O (500 mL), and then was extracted three times with dichloromethane. The combined organic layers were dried over anhydrous MgSO4 and decolor by active carbon. The solvent was removed under reduced pressure. The crude product was purified by recrystallization by hexane as the solvent to yield a colorless crystal. Yield: 89%. IR (KBr), ν (cm−1): 2955, 2920, 2851 (CH2, CH3); 1467, 1448, 1416, 812 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.51 (d, 2H, J = 7.7 Hz), 7.45 (m, 4H), 1.90 (m, 4H, –CH2(CH2)6CH3), 1.25–0.86 (m, 20H, –CH2(CH2)5C2H5), 0.83 (t, 6H, J = 7.2 Hz, –(CH2)7CH3), 0.58 (s, 4H, –(CH2)6CH2CH3).
2,7-Dibromo-9,9-1-didodecylfluorene (2b). The synthesis process is similar to compound 2a by using 1-bromododecane. Yield: 78%. IR (KBr), ν (cm−1): 2935, 2923, 2850 (CH2, CH3); 1464, 1449, 1416, 808 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.51 (d, 2H, J = 7.7 Hz), 7.45 (m, 4H), 1.90 (m, 4H, –CH2(CH2)10CH3), 1.30–1.00 (m, 36H, –CH2(CH2)9C2H5), 0.87 (t, J = 7.2 Hz, 6H, –(CH2)11CH3), 0.59 (s, 4H, –(CH2)10CH2CH3).
2-Bromo-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene (3). 2,7-Dibromo-9,9-dioctylfluorene (1.096 g, 2.0 mmol), Pd(OAc)2 (4.48 mg, 2.0 × 10−2 mmol), triphenylphosphine (10.50 mg, 4.0 × 10−2 mmol) and p-nitrostyrene (0.3 mL, 2.0 mmol) were dissolved in 20 mL DMF and 4 mL Et3N. The resultant mixture was combined in a screw cap vial and heated for 48 h at 110 °C under a nitrogen atmosphere. After cooling to room temperature, the precipitate was filtered and the filtrate was concentrated. The mixture was extracted with CH2Cl2 and dried over MgSO4. The crude product was purified by silica gel column chromatography using petroleum ether/dichloromethane (6:1 v/v) as the eluent. Product was obtained as a yellow solid. Yield: 62%. IR (KBr), ν (cm−1): 2975, 2922 (CH2, CH3), 1604, 1512, 856 (Ar), 1517, 1346 (NO2), 1062 (Ar-Br). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.23 (d, J = 8.8 Hz, 2H), 7.70–7.42 (m, 8H), 7.35 (d, J = 16.0 Hz, 1H), 7.21 (d, J = 16.0 Hz, 1H), 1.91 (m, 4H, –CH2(CH2)6CH3), 1.25–0.86 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.58 (m, 4H, –(CH2)6CH2CH3).
2-Bromo-7-(4-(4-nitrostyryl)styryl)-9,9-dioctyl-9H-fluorene (4a). The synthesis process is similar to compound 3 by using 4-ethenyl-4′-nitro-1,2-stilbene (1a). The crude product was purified by silica gel column chromatography using petroleum ether/dichloromethane (5:1 v/v) as the eluent. Product was obtained as a yellowish-red solid. Yield: 79%. IR (KBr), ν (cm−1): 3023, 962 (C–H), 2955, 2924, 2852 (CH2, CH3), 1588, 1460, 816 (Ar), 1514, 1338 (NO2), 1063 (Ar-Br). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.25 (d, J = 8.4 Hz, 2H) 7.68–7.46 (m, 12H), 7.33–7.17 (q, 4H), 1.98 (m, 4H, –CH2(CH2)6CH3), 1.25–1.09 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 6.8 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3).
2-Bromo-7-(4-(4-methoxystyryl)styryl)-9,9-dioctyl-9H-fluorene (4b). The synthesis process is similar to compound 3 by using 4-ethenyl-4′-methoxy-1,2-stilbene (1b). The crude product was purified by silica gel column chromatography using petroleum ether/dichloromethane (10:1 v/v) as the eluent. Product was obtained as a yellow solid. Yield: 68%. IR (KBr), ν (cm−1): 3022, 963 (C–H), 2953, 2926, 2852 (CH2, CH3), 1603, 1512, 1458, 825 (Ar), 1250, 1033 (C–O–C), 1061 (Ar-Br). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.66 (d, J = 7.6 Hz, 1H) 7.57–7.46 (m, 11H), 7.21–6.92 (m, 6H), 3.87 (s, 3H, Ar-OCH3), 1.98 (m, 4H, –CH2(CH2)6CH3), 1.23–1.08 (m, 20H, –CH2(CH2)5C2H5), 0.83 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3).
2-Bromo-7-(4-(4-methylstyryl)styryl)-9,9-dioctyl-9H-fluorene (4c). The synthesis process is similar to compound 3 by using 4-ethenyl-4′-methyl-1,2-stilbene (1c). The crude product was purified by silica gel column chromatography using petroleum ether/dichloromethane (20:1 v/v) as the eluent. Product was obtained as a yellow solid. Yield: 65%. IR (KBr), ν (cm−1): 3023, 962 (C–H), 2953, 2926, 2852 (CH2, CH3), 1514, 1458, 820 (Ar), 1062 (Ar-Br). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.66 (d, J = 7.6 Hz, 1H) 7.57–7.44 (m, 11H), 7.20 (m, 4H), 7.11 (d, 2H), 2.39 (s, 3H, Ar-CH3), 1.98 (m, 4H, –CH2(CH2)6CH3), 1.25–1.09 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 6.8 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3).
2-Bromo-7-(4-(4-methylstyryl)styryl)-9,9-didodecyl-9H-fluorene (4d). The synthesis process is similar to compound 3 by using 4-ethenyl-4′-methyl-1,2-stilbene (1c) and 2,7-dibromo-9,9-didodecylfluorene (2b). The crude product was purified by silica gel column chromatography using petroleum ether/dichloromethane (25:1 v/v) as the eluent. Product was obtained as a yellow solid. Yield: 54%. IR (KBr), ν (cm−1): 3023, 965 (C–H); 2926, 2852 (CH2, CH3); 1514, 1457, 821 (Ar), 1061 (Ar-Br). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.66 (d, J = 8.0 Hz, 1H) 7.57–7.44 (m, 11H), 7.26–7.07 (m, 6H), 2.39 (s, 3H, Ar-CH3), 1.98 (m, 4H, –CH2(CH2)10CH3), 1.23–1.08 (m, 36H, –CH2(CH2)9C2H5), 0.88 (t, J = 7.2 Hz, 6H, –(CH2)11CH3), 0.66 (m, 4H, –(CH2)10CH2CH3).
2-Ethynyl-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene (M1). 2-Methyl-3-butyn-2-ol (0.8 mL, 8.0 mmol) was added to a solution of PdCl2(PPh3)2 (28.00 mg, 4.0 × 10−2 mmol), CuI (2.00 mg, 1.0 × 10−2 mmol), and 3 (0.38 g, 0.5 mmol) in 20 mL of triethylamine under a nitrogen atmosphere. The mixture was reacted at 80 °C for 48 h. After cooling to room temperature, the precipitate was filtered and the filtrate was concentrated. The crude product was purified by aluminum oxide column using ethyl acetate/petroleum ether mixture (1:6 v/v) as eluent. The resulting solid 2-(2-methyl-3-yn-2-ol)-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene, was a yellow powder in 60% yield. Sodium hydroxide (144 mg, 3.6 mmol) was added with stirring to a solution of 2-(2-methyl-3-yn-2-ol)-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene (0.39 g, 0.5 mmol) in dry dioxane (20.0 mL) under nitrogen. The reaction mixture was then heated at 80 °C under reflux for 4 h. After cooling to room temperature and the removal of the solvent with a rotary evaporator, the raw product was extracted with dichloromethane three times, dried over MgSO4 and recrystallized from dichloromethane/ethanol mixture (2:1 v/v). Product was obtained as a yellow powder. Yield: 85%. Mp: 120.0 °C. IR (KBr), ν (cm−1): 3310 (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 2974, 2922 (CH2, CH3), 2103 (CC), 1602, 1512, 850 (Ar), 1516, 1343 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.26 (d, J = 8.0 Hz, 2H), 7.61–7.53 (m, 4H), 7.51–7.42 (m, 4H), 7.28 (d, J = 16.0 Hz, 1H), 7.12 (d, J = 16.0 Hz, 1H), 3.18 (s, 1H, –CCH), 1.96 (m, 4H, –CH2(CH2)6CH3), 1.04 (m, 20H, –CH2(CH2)5C2H5), 0.61 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.56 (m, 4H, –(CH2)6CH2CH3). Anal. calcd for C39H37NO2: C 84.90, H 6.75, N 2.53. Found: C 84.89, H: 6.77, N 2.51.
C–H), 2974, 2922 (CH2, CH3), 2103 (CC), 1602, 1512, 850 (Ar), 1516, 1343 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.26 (d, J = 8.0 Hz, 2H), 7.61–7.53 (m, 4H), 7.51–7.42 (m, 4H), 7.28 (d, J = 16.0 Hz, 1H), 7.12 (d, J = 16.0 Hz, 1H), 3.18 (s, 1H, –CCH), 1.96 (m, 4H, –CH2(CH2)6CH3), 1.04 (m, 20H, –CH2(CH2)5C2H5), 0.61 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.56 (m, 4H, –(CH2)6CH2CH3). Anal. calcd for C39H37NO2: C 84.90, H 6.75, N 2.53. Found: C 84.89, H: 6.77, N 2.51.
2-Ethynyl-7-(4-(4-nitrostyryl)styryl)-9,9-dioctyl-9H-fluorene (M2). This compound was prepared as the similar approach of M1, using 4a instead of 3, as the starting material. Product was recrystallized from dichloromethane/ethanol mixture (2:1 v/v), and obtained as an orange-red powder. Yield: 65%. Mp: 136.1 °C. IR (KBr), ν (cm−1): 3318 (C–H), 3022, 961 (C–H), 2923, 2851 (CH2, CH3), 2104 (CC), 1584, 1466, 821 (Ar), 1511, 1339 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.25 (d, J = 8.4 Hz, 2H), 7.70–7.50 (m, 12H), 7.32–7.17 (q, 4H), 3.18 (s, 1H, –CCH), 2.00 (m, 4H, –CH2(CH2)6CH3), 1.24–1.08 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151.7, 151.0, 146.8, 143.9, 141.5, 140.4, 138.1, 136.7, 135.4, 132.9, 131.2, 129.9, 127.6, 127.5, 127.0, 126.8, 126.5, 126.0, 125.8, 124.2, 120.9, 120.4, 120.3, 119.6 (Ar and –CHCH–), 84.7 (–CCH), 77.2 (–CCH), 55.1, 40.4, 31.8, 30.0, 29.2, 23.7, 22.6, 14.1 (–(CH2)7CH3). Anal. calcd for C47H53NO2: C 85.02, H 8.05, N 2.11. Found: C 85.09, H: 8.01, N 2.07.
2-Ethynyl-7-(4-(4-methoxystyryl)styryl)-9,9-dioctyl-9H-fluorene (M3). This compound was prepared as the similar approach of M1, using 4b instead of 3, as the starting material. Product was recrystallized from dichloromethane/ethanol mixture (1:1 v/v), and obtained as a yellow powder. Yield: 73%. Mp: 135.8 °C. IR (KBr), ν (cm−1): 3310 (C–H), 3021, 968 (C–H), 2926, 2852 (CH2, CH3), 2104 (CC), 1605, 1512, 1462, 830 (Ar), 1255, 1038 (C–O–C). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.70–7.64 (q, J = 7.8 Hz, 2H) 7.57–7.49 (m, 10H), 7.26–6.92 (m, 6H), 3.87 (s, 3H, Ar-OCH3), 3.17 (s, 1H, –CCH), 2.00 (m, 4H, –CH2(CH2)6CH3), 1.23–1.08 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 7.2 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 159.4, 151.7, 151.0, 141.6, 140.0, 137.0, 136.9, 136.4, 131.2, 130.2, 128.8, 128.2, 128.0, 127.7, 126.8, 126.6, 126.5, 126.2, 125.7, 120.7, 120.3, 120.1, 119.6, 114.2 (Ar and –CHCH–), 84.8 (–CCH); 77.2 (–CCH), 55.3 (Ar-OCH3), 55.1, 40.4, 31.8, 30.0, 29.2, 23.7, 22.6, 14.1 (–(CH2)7CH3). Anal. calcd for C48H56O: C 88.84, H 8.70. Found: C 88.80, H 8.64.
2-Ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-dioctyl-9H-fluorene (M4). This compound was prepared as the similar approach of M1, using 4c instead of 3, as the starting material. Product was recrystallized from dichloromethane/ethanol mixture (1:1 v/v), and obtained as a yellow powder. Yield: 67%. Mp: 144.2 °C. IR (KBr), ν (cm−1): 3316 (C–H), 3022, 963 (C–H), 2926, 2852 (CH2, CH3), 2106 (CC), 1514, 1464, 825 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.70–7.64 (q, J = 8.0 Hz, 2H), 7.58–7.44 (m, 10H), 7.26–7.07 (m, 6H), 3.17 (s, 1H, –CCH), 2.39 (s, 3H, Ar-CH3), 2.01 (m, 4H, –CH2(CH2)6CH3), 1.23–1.08 (m, 20H, –CH2(CH2)5C2H5), 0.84 (t, J = 6.8 Hz, 6H, –(CH2)7CH3), 0.65 (m, 4H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151.7, 151.0, 141.6, 140.1, 137.6, 137.0, 136.9, 136.6, 134.6, 131.2, 129.4, 128.9, 128.6, 128.0, 127.3, 126.8, 126.7, 126.5, 126.4, 125.8, 120.8, 120.3, 120.2, 119.6 (Ar and –CHCH–), 84.8 (–CCH), 77.2 (–CCH), 55.1, 40.4, 31.8, 30.0, 29.2, 23.7, 22.6, 14.1 (–(CH2)7CH3), 21.3 (Ar-CH3). Anal. calcd for C48H56: C 91.08, H 8.92. Found: C 91.11, H 8.90.
2-Ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-didodecyl-9H-fluorene (M5). This compound was prepared as the similar approach of M1, using 4d instead of 3, as the starting material. Product was recrystallized from dichloromethane/ethanol mixture (1:1 v/v), and obtained as a yellow powder. Yield: 65%. Mp: 104.8 °C. IR (KBr), ν (cm−1): 3308 (C–H); 3023, 960 (C–H), 2926, 2851 (CH2, CH3), 2104 (CC), 1514, 1463, 826 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.70–7.64 (q, J = 8.0 Hz, 2H) 7.57–7.44 (m, 10H), 7.22–7.11 (m, 6H), 3.17 (s, 1H, –CCH), 2.39 (s, 3H, Ar-CH3), 2.00 (m, 4H, –CH2(CH2)10CH3), 1.33–1.01 (m, 36H, –CH2(CH2)9C2H5), 0.88 (t, J = 6.8 Hz, 6H, –(CH2)11CH3), 0.65 (m, 4H, –(CH2)10CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151.7, 151.0, 141.6, 140.1, 137.6, 137.0, 136.9, 136.6, 134.6, 131.2, 129.4, 128.9, 128.6, 128.0, 127.3, 126.8, 126.7, 126.5, 126.4, 125.8, 120.8, 120.3, 120.2, 119.6 (Ar and –CHCH–), 84.8 (–CCH), 77.2 (–CCH), 55.1, 40.4, 31.9, 30.0, 29.6, 29.5, 29.3, 29.2, 23.7, 22.6, 14.1 (–(CH2)11CH3), 21.3 (Ar-CH3). Anal. calcd for C56H72: C 90.26, H 9.74. Found: C 90.19, H 9.80.
2.4 Polymerization
Scheme 2 all the polymerization reactions and manipulations were performed under purified nitrogen using Schlenk techniques either in vacuum-line system or an inert-atmosphere glove box, except for the purification of the polymers, which were done in open air. Typical procedure is given below: 1.0 mmol of the monomer was added into a baked 20 mL Schlenk tube with a side arm. The tube was evacuated under vacuum and then flushed with dry nitrogen three times through the side arm. Three milliliters of dioxane was injected into the tube to dissolve the monomer. The catalyst solution was prepared in another tube by dissolving 4.6 mg (0.01 mmol) [Rh(nbd)Cl]2 and 2.02 mg (0.02 mmol) Et3N in 2 mL of dioxane, which was transferred to the monomer solution using a hypodermic syringe. The reaction mixture was stirred at 60 °C under nitrogen for 4 h. The mixture was then diluted with 5 mL of dioxane and added dropwise to 200 mL of methanol under stirring. The precipitate was centrifuged and redissolved in THF. The THF solution was added dropwise into 200 mL of methanol to precipitate the polymer. The dissolution–precipitation process was repeated three times, and the finally isolated precipitant was dried under vacuum at 30 °C to a constant weight.
Poly[2-ethynyl-7-(4-nitrostyryl)-9,9-dioctyl-9H-fluorene] (P1). Deep red solid. Yield: 60%. GPC: Mw = 10100, PDI = 1.384 (polystyrene calibration, Table 1, no. 1). IR (KBr), ν (cm−1): 2927, 2857 (CH2, CH3), 1610, 856 (Ar), 1513, 1340 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.32 (d, 2H), 8.07–6.92 (br, 10H), 5.43 (s, 1H), 1.93 (br, 4H), 1.05 (br, 22H), 0.81 (br, 8H).
Table 1 Polymerization summary of P1–P5a
| Entry |
Polymer |
Yield (%) |
Mwb |
Mw/Mnc |
| Unless otherwise specified, the polymerization was carried out in dioxane under nitrogen with [Rh(ncb)Cl]2-Et3N as a catalyst for 4 h at 60 °C. Estimated by GPC in term of polystyrene calibration. Polymerized in THF. |
| 1 |
P1 |
60 |
10100 |
1.384 |
| 2 |
P1′ |
67 |
12760 |
2.121 |
| 3 |
P2 |
38 |
4910 |
1.259 |
| 4 |
P2′ |
54 |
9650 |
2.080 |
| 5 |
P3 |
65 |
96200 |
1.585 |
| 6 |
P4 |
67 |
152000 |
2.269 |
| 7 |
P5 |
84 |
257600 |
1.882 |
Poly[2-ethynyl-7-(4-(4-nitrostyryl)styryl)-9,9-dioctyl-9H-fluorene] (P2). Deep red solid. Yield: 38%. GPC: Mw = 4910, PDI = 1.259 (polystyrene calibration, Table 1, no. 3). IR (KBr), ν (cm−1): 3022, 961 (C–H); 2923, 2851 (CH2, CH3), 1584, 1466, 821 (Ar), 1511, 1339 (NO2). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 8.27 (d, J = 8.4 Hz, 2H) 7.68–7.54 (br, 12H), 7.32–7.17 (br, 4H), 7.02 (s, 1H), 2.07 (br, 4H, –CH2(CH2)6CH3), 1.28–1.08 (br, 20H, –CH2(CH2)5C2H5), 0.91–0.81 (br, 10H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151.7, 151.0, 146.8, 143.9, 141.5–138.1, 136.7, 135.4, 132.9, 131.2, 129.9, 127.6–124.2, 120.9–119.6 (Ar and –CHCH–), 55.1, 40.4, 31.8, 30.0–29.2, 23.7, 22.6, 14.1 (–(CH2)7CH3).
Poly[2-ethynyl-7-(4-(4-methoxystyryl)styryl)-9,9-dioctyl-9H-fluorene] (P3). Deep red solid. Yield: 65%. GPC: Mw = 96200, PDI = 1.585 (polystyrene calibration, Table 1, no. 5). IR (KBr), ν (cm−1): 3021, 958 (C–H), 2926, 2852 (CH2, CH3), 1605, 1512, 1462, 824 (Ar), 1251, 1036 (C–O–C). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.57–7.49 (br, 12H), 7.26–6.92 (br, 6H), 7.02 (s, 1H), 3.87 (s, 3H, Ar-OCH3), 2.07 (br, 4H, –CH2(CH2)6CH3), 1.28–1.08 (m, 20H, –CH2(CH2)5C2H5), 0.91–0.81 (br, 10H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 159.4, 151.7, 151.0, 141.6, 140.0, 137.0–136.4, 131.2, 130.2, 128.8–125.7, 120.7, 120.3, 120.1, 119.6, 114.2 (Ar and –CHCH–), 55.3 (Ar-OCH3); 55.1, 40.4, 31.8, 30.0, 29.2, 23.7, 22.6, 14.1 (–(CH2)7CH3).
Poly[2-ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-dioctyl-9H-fluorene] (P4). Deep red solid. Yield: 67%. GPC: Mw = 152000, PDI = 2.269 (polystyrene calibration, Table 1, no. 6) IR (KBr), ν (cm−1): 3022, 963 (C–H), 2926, 2852 (CH2, CH3), 1514, 1464, 825 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.55–7.41 (br, 12H), 7.21–7.12 (br, 6H), 7.02 (s, 1H), 2.39 (s, 3H, Ar-CH3), 2.07 (br, 4H, –CH2(CH2)6CH3), 1.28–1.10 (br, 20H, –CH2(CH2)5C2H5), 0.91–0.81 (br, 10H, –(CH2)6CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151.0, 141.0, 137.5–134.6, 129.4–126.4, 120.6 (Ar and –CHCH–), 55.0, 40.4, 31.9–29.2, 23.9–22.6, 14.1 (–(CH2)7CH3), 21.3 (Ar-CH3).
Poly[2-ethynyl-7-(4-(4-methylstyryl)styryl)-9,9-didodecyl-9H-fluorene] (P5). Deep red solid. Yield: 84%. GPC: Mw = 257600, PDI = 1.882 (polystyrene calibration, Table 1, no. 7). IR (KBr), ν (cm−1): 3023, 960 (C–H); 2926, 2851 (CH2, CH3), 1514, 1463, 826 (Ar). 1H NMR (400 MHz, CDCl3), δ (TMS, ppm): 7.56–7.38 (br, 12H), 7.22–7.11 (br, 6H), 7.02 (s, 1H), 2.39 (s, 3H, Ar-CH3), 2.07 (br, 4H, –CH2(CH2)10CH3), 1.28–1.11 (br, 36H, –CH2(CH2)9C2H5), 0.91–0.81 (br, 10H, –(CH2)10CH2CH3). 13C NMR (75 MHz, CDCl3), δ (ppm): 151, 141, 137.5–134.6, 129.4–126.4, 120.8–119.6 (Ar and –CHCH–), 55.1, 40.4, 31.9–29.2, 23.7, 22.6, 14.1 (–(CH2)11CH3), 21.3 (Ar-CH3).
3. Results and discussion
3.1 Synthesis and polymerization
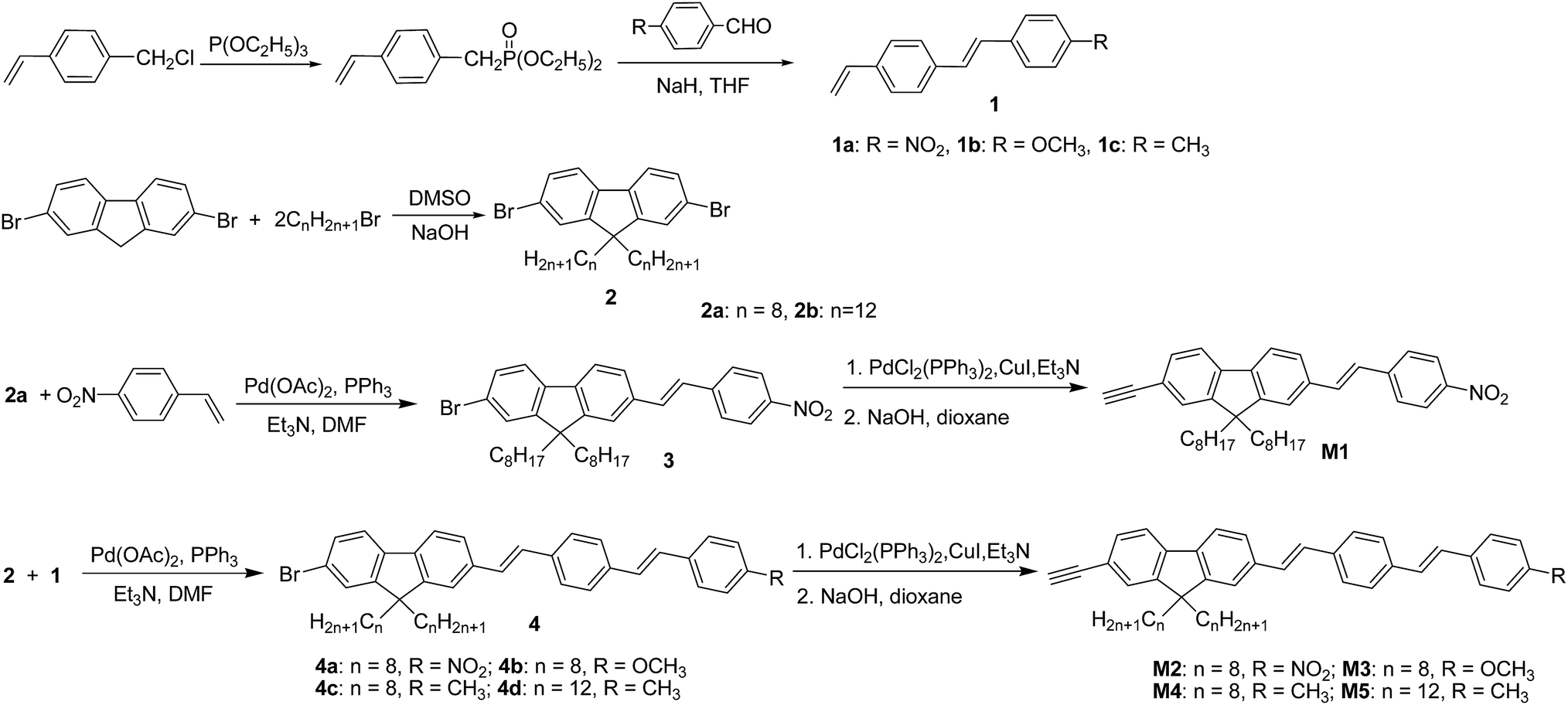
We designed and synthesized one styryl-substituted fluorene derivate M1 and four different stilbene-substituted fluorene derivates M2–M5 with different terminal substituted group (NO2, OCH3 and CH3) and side alkyl chain (Scheme 1). The Wittig Horner reaction of these different benzaldehyde derivatives with 4-vinylbenzyl chloride gave stilbene derivatives 1a–1c. 2,7-Dibromofluorene were alkylated at the 9 position by 1-bromooctane or 1-bromododecane in strongly basic conditions to form 2a–2b. 3 was prepared from 2a and p-nitrostyrene by Heck reaction, and then 4a–4d were made from 2a–2b and 1a–1c by the same reaction. The resultant products (3 and 4a–4d) were then reacted with 2-methyl-3-butyn-2-ol using Pd(PPh3)2Cl2 as the catalyst, the obtained semifinished products with 2-propyl-2-ol group were cleaved in dioxane/NaOH solution to give the desirable monomers M1–M5 in high yields, respectively.
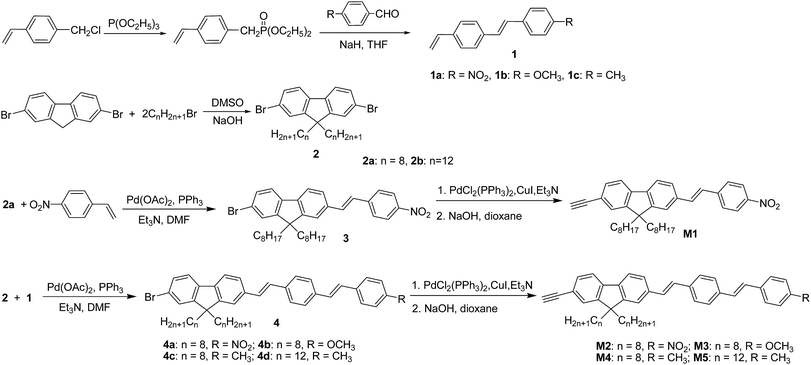 |
| | Scheme 1 Synthetic routes of monomer molecules M1–M5. | |
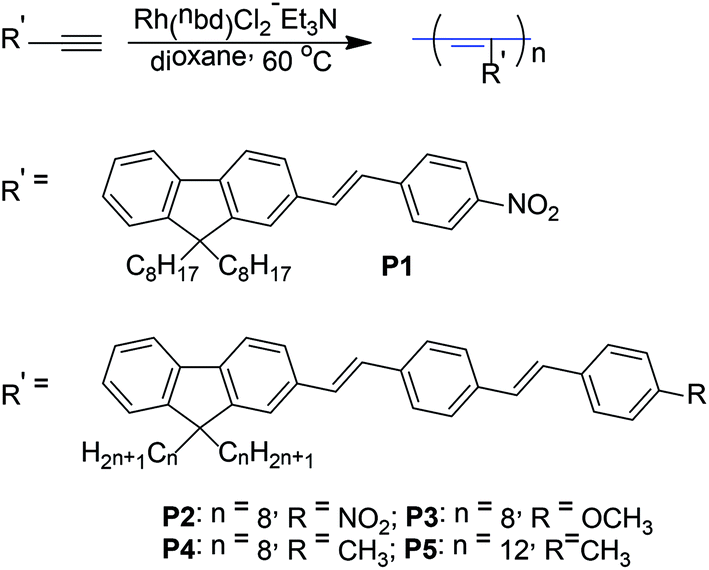
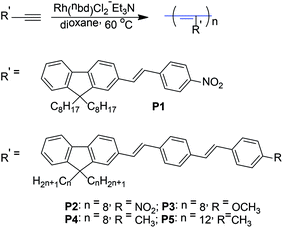 |
| | Scheme 2 Synthetic routes to the polymers P1–P5. | |
All polymerization reactions were carried out under nitrogen atmosphere using a standard Schlenk vacuum-line system. We first attempted to polymerize these monomers (M1–M5) using the classical metathesis catalysts, such as WCl6–Ph4Sn, TaCl5–Ph4Sn and MoCl5–Ph4Sn. However, only trace polymeric products were obtained. On the contrary, [Rh(nbd)Cl]2/Et3N catalyst effectively works for these monomers and the results are listed in Table 1. It was found that P1 and P2 were partly insoluble in dioxane, only giving molecular weight of the soluble part of the polymers (Table 1, entry 1 and 3). However, the yield of P1′ and P2′ was increased by using THF as the polymerization solvent (Table 1, entry 2 and 4). P1 and P2 exhibited the low polymerization yield in dioxane, which may be mainly originated from the enwrapped effect of insoluble polymer to prevent further polymerization of monomers. Moreover, the polymerization yield of P2 in dioxane or P2′ in THF was almost lower than that of corresponding P1 or P1′, hinting that polymerization activity of monomer M2 may be lower than that of M1. The result exhibited that polymerization activity of functionalized acetylene monomer decreased with the increase of conjugated chain length of substituted chromophore group. P3, P4, and P5 were soluble in dioxane and gave dark red polymers with high molecular weight (Mw = 9 × 104–2.5 × 105) in high polymerization yield (Table 1, entry 5, 6, 7), and P5 obtained the highest molecular weight and yield, hinting that the longer flexible alkyl side chain on fluorene was beneficial to polymerization of monomer.
3.2 Solubility
It's reported that PAs have weak solubilities in common solvents.25 However, when the styryl/stilbene–fluorene pendant with flexible alkyl group as side tailor was incorporated into the PA backbone chain, all the polymers were readily soluble in various common organic solvents such as chloroform, toluene, THF, and 1,2-dichloroethane.
3.3 Structural characterization of the polymers
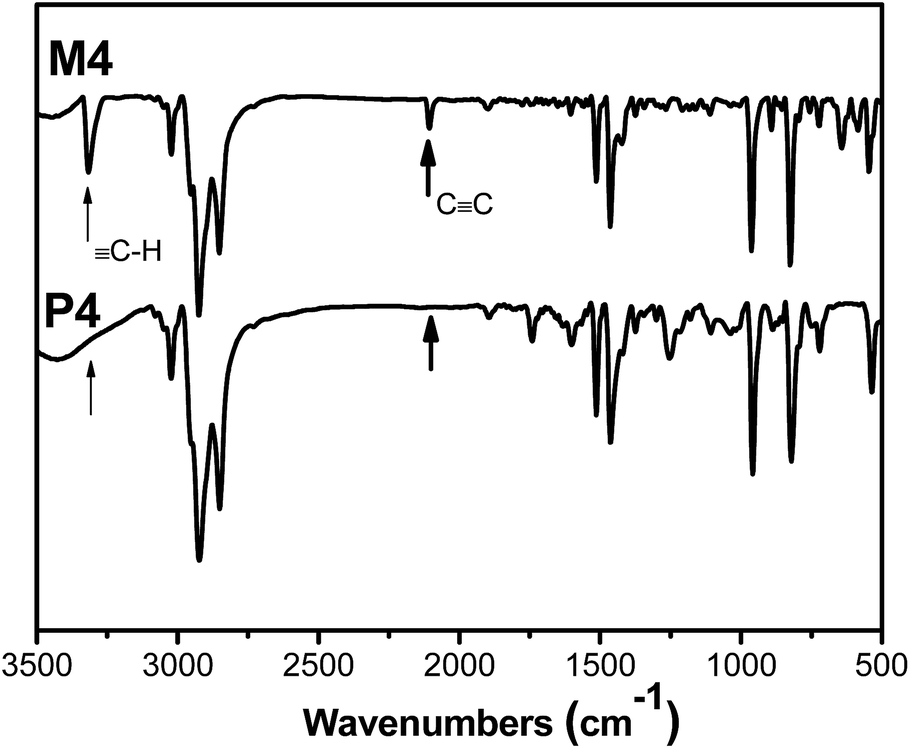
All the polymers and corresponding monomers were characterized by standard spectroscopic methods, from which satisfactory analysis data were obtained (see the Experimental section for details). An example of the FTIR spectrum of P4 is given in Fig. 1. For comparison, the FTIR spectrum of its corresponding monomer M4 is also given in the same figure. As can be seen from Fig. 1, M4 clearly exhibits two characteristic absorptions at 3319 and 2104 cm−1, respectively, corresponding to the C–H and CC stretching vibrations in the mono-substituted acetylene. The acetylenic absorption bands disappear in the spectrum of P4 and the characteristic vibration intensity of aromatic ring is significantly enhanced, suggesting that the triple bond in the monomer has changed to double bond in the polymer. Similar results (Fig. S1–S4†) were found in other monomers (M1–M3 and M5) and their polymers (P1–P3 and P5), suggesting that the triple bonds of these monomers have been transferred to double bond polyacetylene backbone.
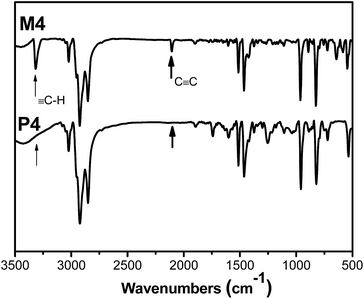 |
| | Fig. 1 FTIR spectra of M4 and P4. | |
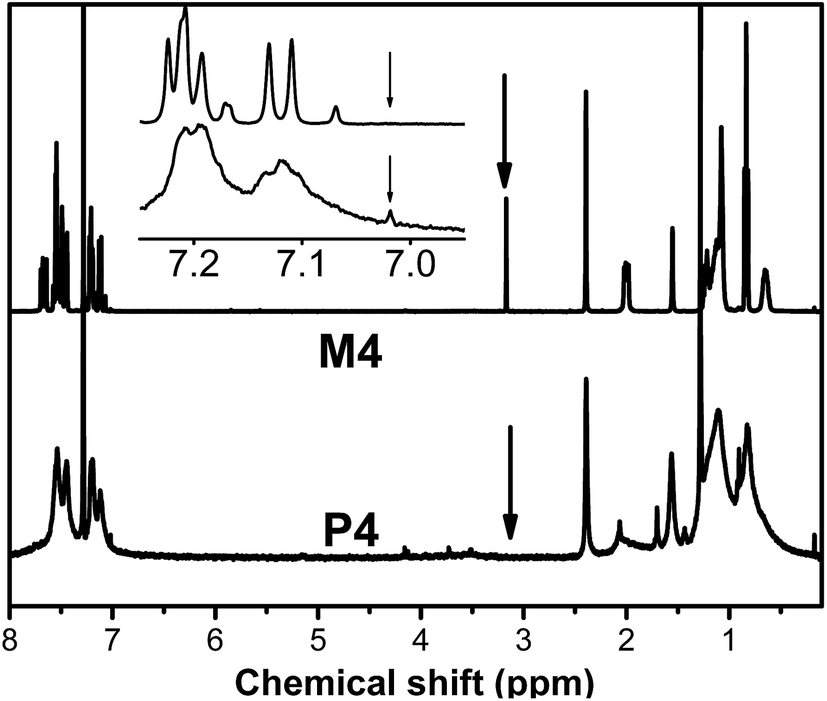
Fig. 2 shows the 1H NMR spectra of M4 and P4 in chloroform-d, respectively. The absorption peak of the acetylene proton in M4 at 3.17 ppm as a singlet peak, completely disappears in the spectrum of its polymer P4. Meanwhile, all the corresponding characteristic absorption peaks of monomer M4 significantly were widened in the spectra of P4 with a widened and enhanced peak at 7.02 ppm corresponding to the olefin protons and aromatic protons absorption, further proving that the M4 has been polymerized to yield the functional polyacetylene P4. The results are consistent with that of FTIR spectra. Alternatively, no absorptions are found in the cis-olefin absorption region of 5.20–6.20 ppm, suggesting that these polymers possess a predominantly trans conformation.59,60 The high trans content may result from the strong steric hindrance of the pendants.61
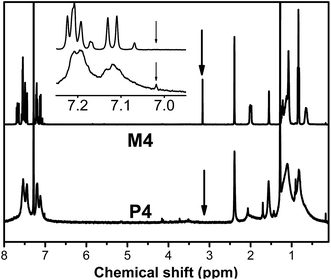 |
| | Fig. 2 1H NMR spectra of M4 and P4. | |
Compared to that of M4, the signals at 77.2 and 84.8 ppm corresponding to the acetylenic carbons disappear in the 13C NMR spectrum of P4 (Fig. S9†), and the relevant absorption peak intensity of the carbon atoms of the aromatic pendants in polymers is significantly enhanced because of the absorption overlapping effect of the olefin carbon atoms of the polyacetylene backbone. Thus, the 1H NMR and 13C NMR spectra data further confirm that the acetylene polymerization has taken place. The similar results were found in polymerization of other monomers. The satisfactory spectral data are given in the ESI (Fig. S10–S12†).
3.4 Linear optical properties
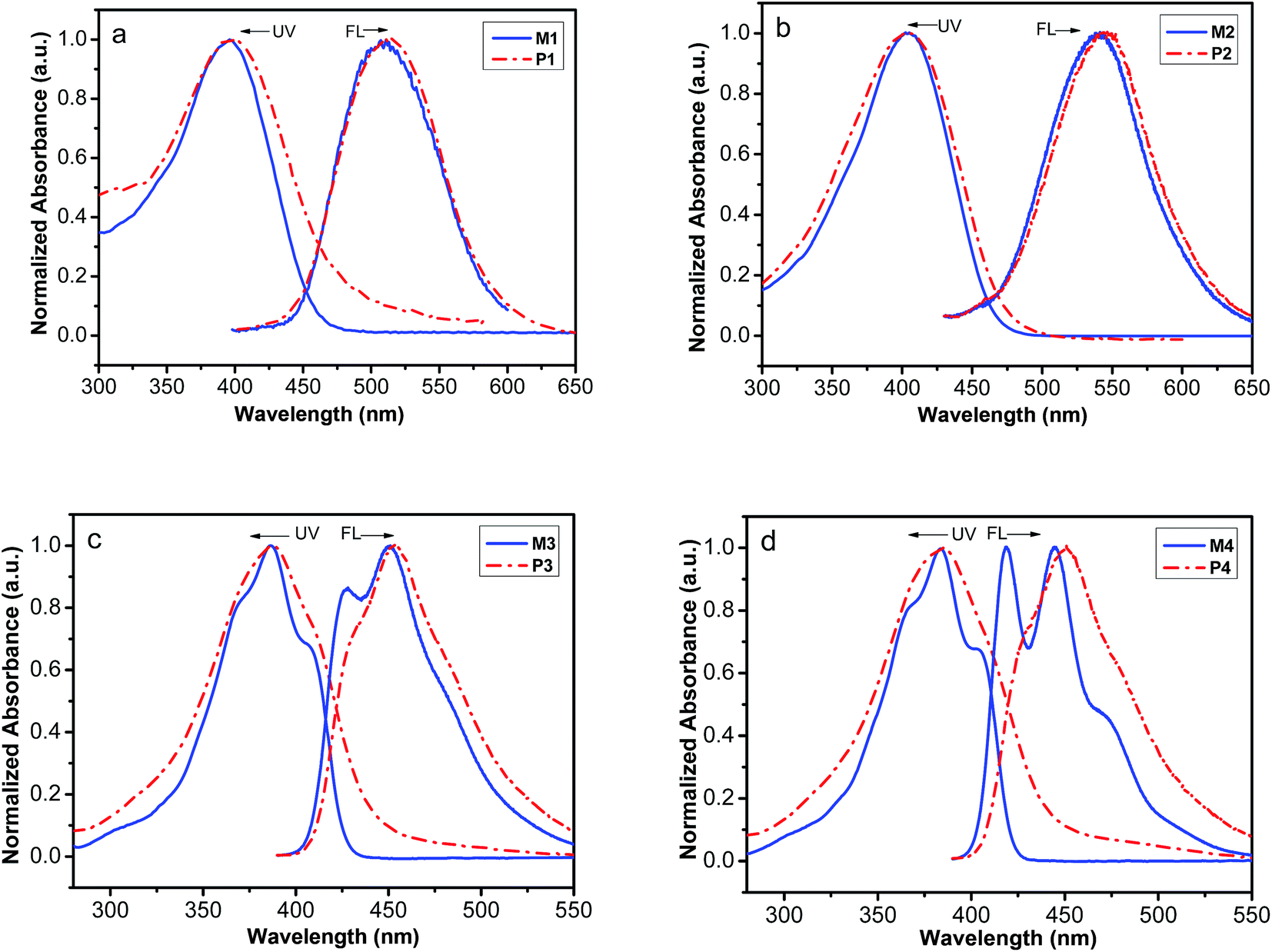
The linear optical properties of the polymers and corresponding monomers were investigated by UV-vis absorption and fluorescence in THF solutions at 20 °C in extremely diluted solution (2 × 10−5 M) and these spectra are depicted in Fig. 3. It can be seen from Fig. 3 that the maximum absorption wavelengths of P1–P5 are located at 400, 406, 388, 385 and 385 nm, respectively, which are assigned to the π–π* electronic transitions of the corresponding conjugated styryl/stilbene–fluorene chromophores. Obviously, the maximum absorption wavelengths of the polymers changes at the different terminal substituents or the length of conjugated systems, but aren't influenced by the length of alkyl flexible chain. The absorption band of the nitro-substituted polymers P1 and P2 shows significantly red-shifts, which results from the larger π-electron delocalization and stronger dipolar effect of NO2 group. P2 with longer conjugated chromophore substituent shows larger red-shift at maximum absorption wavelength than P1. The absorption band of the methyl-substituted polymers P4 and P5 with the different alkyl flexible chain exhibit the almost same maximum absorption wavelength. Although the polymers and their corresponding monomers show nearly the same λmax, the polymers show significant broad backbone absorption at longer wavelength than 420 nm, which result from the double contribution of conjugated double-bond backbones of polyacetylene and enhancement conjugation effect between the double-bond backbones of the polyacetylenes and styryl/stilbene–fluorene pendant groups. These results further support that the monomers have been converted to polymers.
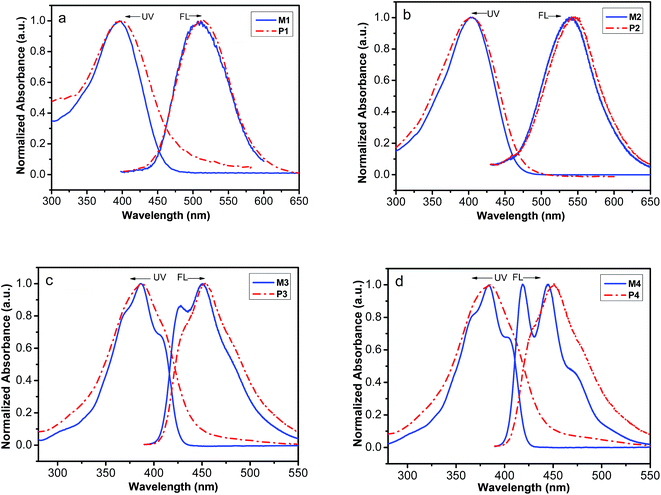 |
| | Fig. 3 Normalized UV-vis absorption and FL emission spectra of the monomers (solid line) and corresponding polymers (dash dot) in THF solutions (spectra of M5 and P5 were showed in Fig. S13†). | |
An interesting feature for the polymers P1–P5 is that they all possess luminescent properties upon photo irradiation while PA itself is nonluminescent.62 When excited at relative maximum absorption wavelength in THF solution, P1–P5 emit fluorescence with maximum emitting peak at around 515 nm, 541 nm, 453 nm, 450 nm and 447 nm respectively, which are higher than that of the corresponding monomers (Fig. 3). The red-shift of fluorescence emitting peak probably results from a strong pendant–pendant intermolecular interaction and/or intramolecular interaction between styryl/stilbene–fluorene pendants and the polyacetylene conjugation main chain. Similar phenomena are found by Balcar.63
3.5 Optical limiting properties and mechanism
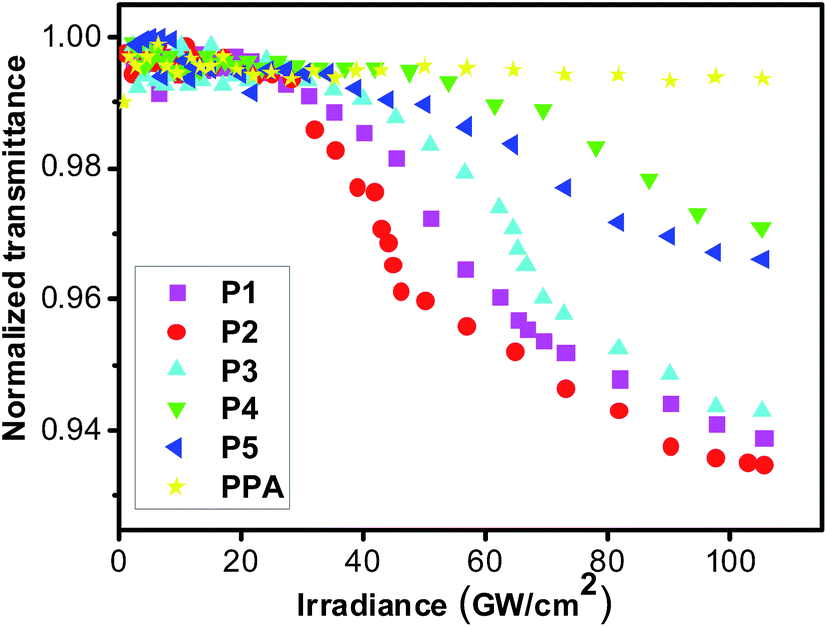
Many optical limiting materials for visible optical region have been investigated. However, the optical limiting materials for the near-infrared optical region were rarely reported. Thus, we investigated the near infrared optical limiting properties of resultant polymers by a mode-locked Ti:sapphire laser (Quantronix, IMRA) at 780 nm using 450 femtosecond laser pulses at 1 kHz repetition rate. For avoiding the influence from thermally induced scattering effects, the optical limiting properties of the polymers in THF solution were measured at low unit concentration (1 mM). Fig. 4 shows the optical limiting properties of P1–P5 in THF and the results are summarized in Table 1S.† For comparison, the optical limiting property of polyphenylacetylene (PPA) at the same condition is also made. The results were shown in Fig. 4. At low incident irradiance, the transmittances of all the polymer solutions remain unchanged with the incident irradiance obeying the Beer–Lambert law. However, the transmittances of the solutions start to deviate from the linear transmission with a further increase in the incident irradiance, and a nonlinear relationship is observed between the output and input irradiance, while that of PPA still rises linearly because of the laser-induced photolysis of the polyacetylene chains,68 suggesting that the incorporation of conjugated styryl/stilbene–fluorene group into polyacetylene backbone has endowed the resultant polyacetylenes with novel laser protection characteristics at 780 nm for 450 femtosecond pulses laser. It is also found that the limiting thresholds of the resultant polymers, which are defined as the incident irradiance fluence where the transmittance starts to deviate from linearity, are significantly influenced by molecular structures. As seen in Table 1S,† polymer P5 (34.9 GW cm−2) with longer tail alkyl chain length in side position of fluorene pendant exhibits better optical limiting properties than P4 (47.6 GW cm−2). Moreover, polymer P3 shows the better optical property when the terminal methyl group of stilbene–fluorene pendant of P4 is replaced by methoxy group. The limiting threshold of P2 (16.7 GW cm−2) is better than P1 (21.4 GW cm−2), which should be attributed to the larger electron conjugation of chromophoric moiety in P2. It can be found that P2 shows the best optical limiting properties (lowest optical limiting thresholds) among all the polymers, owing to the longer conjugated chromophore substituent and larger dipolar effect of strong electron acceptor NO2.
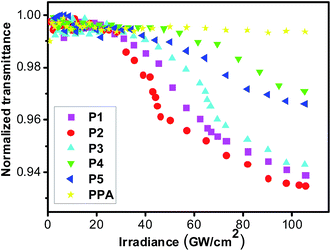 |
| | Fig. 4 Optical limiting properties of P1–P5, and polyphenylacetylene (PPA) for femtosecond laser pulse. | |
The optical limiting mechanisms of organic compounds are often based on TPA or RSA. Generally, TPA based optical limiting effect can be yielded in principle under the laser irradiation of picosecond or shorter pulses, while RSA is achieved on a nanosecond or longer time scale, owing to the different excited state lifetimes involved in a multilevel energy process.64 In this work, the polymers are excited by 450 femtosecond laser pulses at 780 nm. Therefore, we consider that the optical limiting properties of the polymers may mainly originate from TPA.
For investigation of optical limiting mechanism, we performed open-aperture Z-scans with the same laser. The Z-scan experiments of the polymer solution were carried out at 780 nm using femtosecond laser pulses (450 fs, 1 kHZ). In theory, the normalized transmittance for the open aperture can be written as65
| |
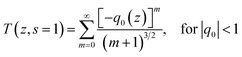 | (1) |
where
q0(
z) =
α2I00(
t)
Leff/(1 +
z2/
z02) with
β is the TPA coefficient,
I0 is the intensity of laser beam at focus (
z = 0),
Leff = [1 − exp(−
α0L)]/
α0 is the effective thickness (
α is the linear absorption coefficient,
L is the sample thickness,
z0 is the Rayleigh range of the laser beam, and
z is the sample position). The TPA cross-section
σ2 is calculated by
55where
N is the number of molecules per cm
3 and
hν is the excitation photon energy. The TPA cross-section
σ2 is expressed in Göppert Mayer (GM) units, in which 1 GM = 1 × 10
−50 cm
4 s per photon. The Z-scan data (or the transmittance as a function of
z position) were normalized to the linear (small-signal) transmittance. The nonlinear-optical signal from the solvent was negligible, compared to the signals from the fullerene derivatives. The Z-scan results confirmed that the samples had surprisingly good photostability, which was verified by indifference between the linear absorption spectra measured before and after intense laser irradiation in the Z-scans. In addition, the TPA coefficients
β, could be extracted from the best fitting between the Z-scan theory and the data, and the TPA cross sections were then calculated from the definition
(2).
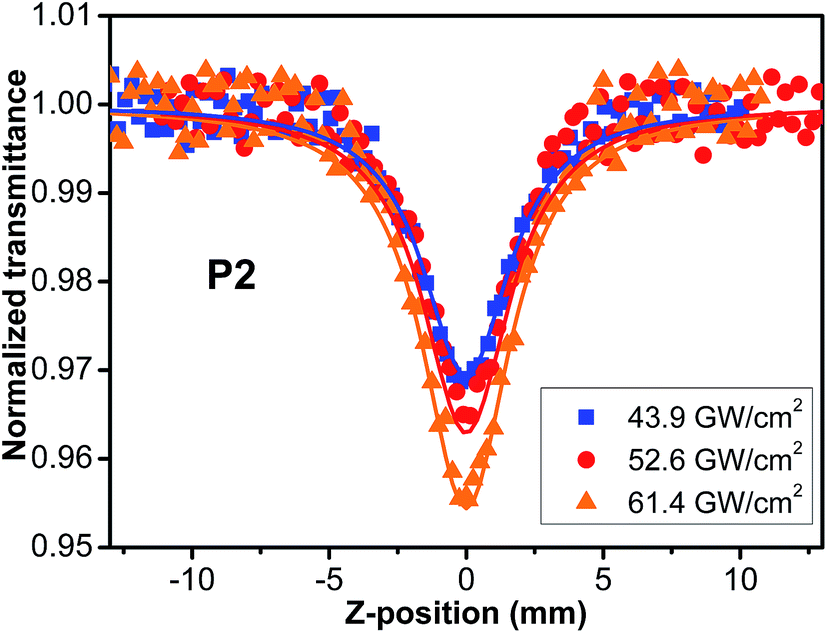
As thermally induced scattering effects and exited-states absorption caused by high irradiance may influence the accuracy of overall TPA cross sections measured, we undertook the Z-scans of P1–P5 in THF with different irradiances. As an example, the results of P2 are shown in Fig. 5. After calculation from definition (2), it is found that the TPA cross sections (σ2) of the polymers are independent of laser irradiance and remain nearly constant across the irradiance range of interest, indicating the observed nonlinear absorption is induced from a pure third-order nonlinear process (Table 1S†). Other nonlinear mechanisms such as thermally induced scattering and excited-state absorption, in particular singlet excited-state absorption, are negligible in our experiments. The similar phenomena (Fig. S14–S17†) were also observed in other polymers. Furthermore, it is found that TPA cross sections of the polymers P1–P5 are obviously larger than those of PPA.42 Thus, the attachment of styryl/stilbene–fluorene moieties into the PA main chain has endowed the resultant polyacetylenes with enhanced the nonlinear optical properties significantly, which may be attributed to the enhanced conjugation effect between substituent fluorene pendants and conjugated backbone of polyacetylene. The similar results were also found in oxadiazole-based or azobenzene-based chromophore functionalized polyacetylenes.45,49
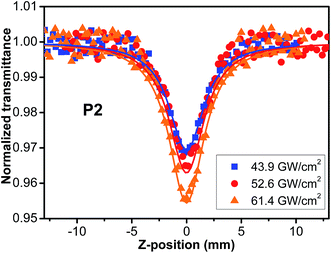 |
| | Fig. 5 Open-aperture Z-scans of P2 at the different pulse energy in THF solution. Solid lines are the theoretical fit for two-photon absorption. | |
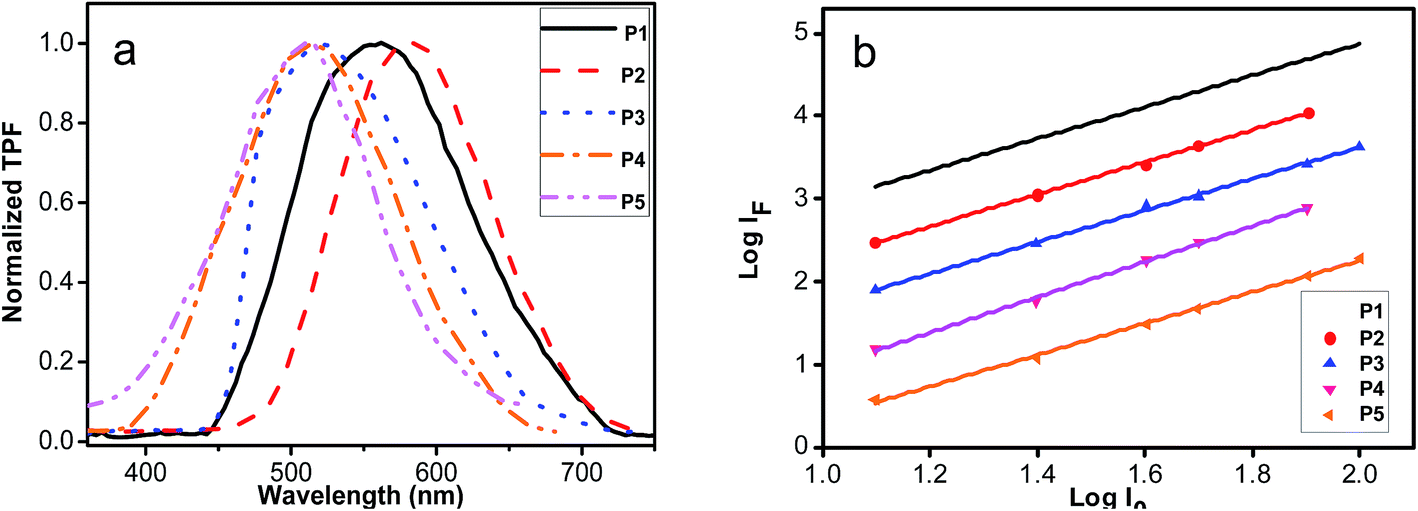
Fig. 6 shows the TPF emission spectra of all polymers (P1–P5) in solid form. The maximum TPF peaks of P1–P5 located at 562 nm, 584 nm, 512 nm, 506 nm and 506 nm, respectively. Comparing the TPF curves of P1–P5 with those of one-photon fluorescence of P1–P5 in THF (Fig. 3), red shifts can be identified, supporting that the re-absorption effects are involved in the nonlinear process in the solid polymers. It is well known that the fluorescence emission intensity presented a square dependence of the fluorescence on the input intensity to prove the TPA nature. When the sample is represented by TPA without stimulated emission, self-quenching, or any dark states influencing the emission intensity, the signal intensity (Ifluor) of the detected fluorescence from the sample cell is given by55
here
I0 is the input light intensity,
l0 and
β are the optical path length and TPA coefficient of the medium, respectively. Therefore,
Fig. 6(a) and (b) showed the two-photon fluorescence (TPF) spectra at different input light intensities and the log–log plots of the signal intensity (
Ifluor) of the detected fluorescence
vs. the input light intensity (
I0) of
P1–P5, respectively. The results displayed that the log–log plots of the detected TPF intensity are linear with the input light intensity, and the slopes are approximately 2, further confirming that the optical limiting properties are mainly attributed to the molecular TPA absorption of resultant polyacetylenes.
66
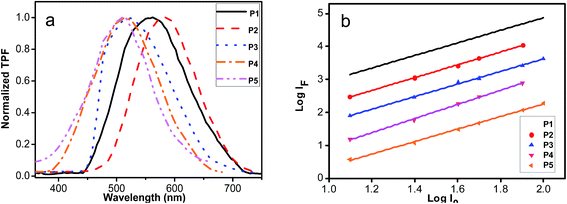 |
| | Fig. 6 (a) Two-photon fluorescence (TPF) spectra of P1–P5; (b) the log–log plots of the signal intensity (Ifluor) of the detected fluorescence vs. the input light intensity (I0) of P1–P5. | |
3.6 Thermal property
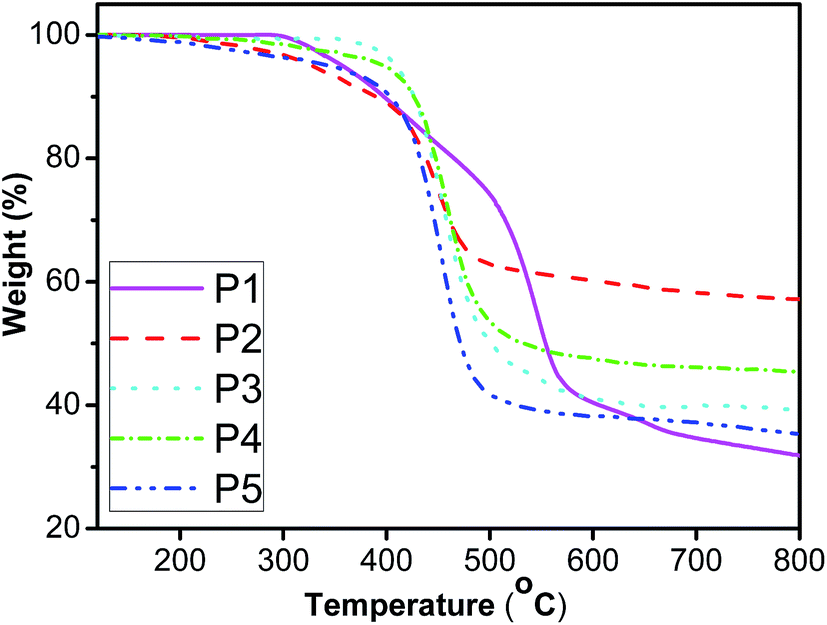
The thermal stability of the resulting polymers was evaluated by thermogravimetric analysis (TGA) under nitrogen atmosphere. It is well known that poly(1-alkyne)s such as poly(1-butyne) and poly(1-hexyne), are so unstable that even the isolation process of the polymer products from the polymerization reactions leads to degradation.59,62 However, as shown in Fig. 7, all the fluorene-containing polyacetylenes exhibit good thermal stability. Td (defined as the temperature of 5% weight loss) for P1 is as high as 357 °C, while Td for P2 with longer conjugated pendant is at 327 °C. When para-position of stilbene–fluorene substituted by different groups, the resulting polymers showed Td at 407, 397 and 345 °C for P3, P4 and P5, respectively, which are much higher than that for PPA (225 °C).55 Thus, the incorporation of the rigid styryl/stilbene–fluorene groups into polyacetylenes significantly improves the thermal stability of resultant polyacetylenes. The enhancement of the thermal stability may be due to the “jacket effect” of the styryl/stilbene–fluorene pendants. Namely, the alternating-double-bond backbone of the polymers may be surrounded by a rigid “jacket” formed through the strong intra- and interchain molecular electronic interaction of the styryl/stilbene–fluorene groups, shielding the polymer main chains from the thermal attack. Similar phenomena were also found by Tang,59,62 Masuda67 and our previous work.45–48,68,69
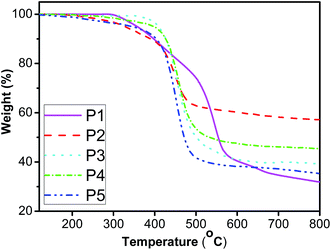 |
| | Fig. 7 TGA thermograms of P1–P5. | |
4. Conclusions
In summary, a series of fluorene-containing functionalized polyacetylenes with different molecular structure was successfully prepared using [Rh(nbd)Cl]2-Et3N as the catalyst. The incorporation of conjugated styryl/stilbene–fluorene group into polyacetylene backbone has endowed the resultant polyacetylenes with novel near-infrared laser protection. The optical limiting properties for near infrared laser protection are influenced by their molecular structures. The functional polyacetylene with longer conjugation structure and stronger terminal acceptor NO2 group exhibits better optical limiting properties. The investigation of optical limiting mechanism shows that the optical limiting properties are mainly originated from two-photon absorption of the polymeric molecules. In addition, the thermal stabilities of the resultant polymers are significantly improved due to “jacket effect” resulting from strong intra and inter molecular electronic interaction between polyacetylenes main chain and substituted chromophoric pendants. This work provides an important foundation for design of new materials for preparing soluble functional polyacetylenes with near-infrared laser protection and high thermal properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by the National Natural Science Fund of China (Grant No. 21671037, 21471030, and 21271040).
Notes and references
- Y. L. Du, K. Zhu, Y. Fang, S. L. Zhang, X. R. Zhang and Y. N. Lu, RSC Adv., 2015, 5, 48311–48322 RSC.
- G.-K. Lim, R. G. S. Goh, Z.-L. Chen, J. Clark, W.-H. Ng and L.-L. Chua, Nat. Photonics, 2011, 5, 554–560 CrossRef CAS.
- M. Feng, H. Zhan and Y. Chen, Appl. Phys. Lett., 2010, 96, 033107 CrossRef.
- J. Wang, Y. Hernandez, M. Lotya, J. N. Coleman and W. J. Blau, Adv. Mater., 2009, 21, 2430 CrossRef CAS.
- Y. Chen, Y. Lin, Y. Liu, J. Doyle, N. He, X. Zhuang and W. J. Blau, J. Nanosci. Nanotechnol., 2007, 7, 1268–1283 CrossRef CAS PubMed.
- S. Rahman, S. Mirza, A. Sarkar and G. W. Rayfield, J. Nanosci. Nanotechnol., 2010, 10, 4805–4823 CrossRef CAS PubMed.
- Y. X. Liu, P. Zhang, C. J. He, X. F. Duan, Y. Yu and J. Q. Yao, Acta Phys. Sin., 2005, 54, 3661–3664 Search PubMed.
- J. Wang, Y. Chen, R. Li, Y. Chen, R. Li and W. J. Blau, J. Inorg. Organomet. Polym., 2011, 21, 736–746 CrossRef CAS.
- J. Wang, Y. Chen and W. J. Blau, J. Mater. Chem., 2009, 19, 7425–7443 RSC.
- R. K. Rekha and A. Ramalingam, J. Mod. Opt., 2009, 56, 1096–1102 CrossRef CAS.
- J. Gao, Q. Li, H. Zhao, L. Li, C. Liu and L. Qi, Chem. Mater., 2008, 20, 6263–6269 CrossRef CAS.
- X. Yu, C. Cao, H. Zhu, Q. S. Li, C. L. Liu and Q. H. Gong, Adv. Funct. Mater., 2007, 17, 1397–1401 CrossRef CAS.
- C. H. Li, Z. M. Huo and H. P. Zeng, Chem. Res. Chin. Univ., 2010, 31, 1554–1559 CAS.
- S. H. Chi, J. M. Hales, M. Cozzuol, C. Ochoa, M. Fitzpatrick and J. W. Perry, Opt. Express, 2009, 17, 22062–22072 CrossRef CAS PubMed.
- S. M. O'Flaherty, S. V. Hold, T. Torres, Y. Chen, M. J. Cook, M. Hanack and W. J. Blau, Adv. Mater., 2003, 15, 19–20 CrossRef.
- M. Calvete, G. Y. Yang and M. Hanack, Synth. Met., 2004, 141, 231–243 CrossRef CAS.
- M. O. Senge, M. Zawadzka, E. G. Notaras, W. J. Blau, M. Fazekas, O. B. Locos and E. M. Ni Mhuircheartaigh, Adv. Mater., 2007, 19, 2737–2774 CrossRef CAS.
- J. Perry, I.-Y. Lee, K. Mansour, P. Bedworth, X.-L. Wu and T. Wada, Science, 1996, 273, 1533–1536 CAS.
- G. J. Zhou, W. Y. Wong, S. Y. Poon, C. Ye and Z. Lin, Adv. Funct. Mater., 2009, 19, 531–544 CrossRef CAS.
- G. J. Zhou, W. Y. Wong, C. Ye and Z. Lin, Adv. Funct. Mater., 2007, 17, 963–975 CrossRef.
- P. F. Zhang, J. Nonlinear Opt. Phys. Mater., 2011, 20, 229–236 CrossRef CAS.
- J. C. Bischof and Z. Qin, Chem. Soc. Rev., 2012, 41, 1191–1217 RSC.
- D. Chateau, F. Chaput, G. Berginc, O. Maury, C. Andraud and S. Parola, J. Mater. Chem. C, 2014, 2, 5105–5110 RSC.
- Y. Xiong, L. Yan, J. Si, W. Ding, X. Liu, F. Chen and X. Hou, J. Appl. Phys., 2014, 115, 083111–083112 CrossRef.
- N. Liaros, S. Couris and E. Koudoumas, Appl. Phys. Lett., 2014, 104, 19112–19113 CrossRef.
- Z. Yang, A. D. Xia, J. S. Ma, Q. S. Li, Z. K. Wu, C. L. Liu and Q. H. Gong, Appl. Phys. Lett., 2005, 86, 061903–061904 CrossRef.
- S. Husaini, J. M. Murray, L. P. Gonzalez and R. G. Bedford, Appl. Phys. Lett., 2013, 102, 191112–191113 CrossRef.
- J. Staromlynska, T. J. McKay and P. Wilson, J. Appl. Phys., 2000, 88, 1726–1732 CrossRef CAS.
- Q. S. Li, Z. G. Liu, C. L. Liu and Q. H. Gong, Opt. Express, 2005, 13, 1833–1838 CrossRef CAS PubMed.
- J. S. Shirk, H. Heckmann, S. R. Flom and M. Hanack, J. Phys. Chem. A, 2000, 104, 1438–1449 CrossRef CAS.
- C. Li, C. L. Liu, Q. S. Li and Q. H. Gong, Chem. Phys. Lett., 2004, 400, 569–572 CrossRef CAS.
- M. Cha, A. J. Heeger, J. C. Hummelen, N. S. Sariciftci and F. Wudl, Appl. Phys. Lett., 1995, 67, 3850–3852 CrossRef CAS.
- G.-J. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- G.-J. Zhou, W.-Y. Wong, D. Cui and C. Ye, Chem. Mater., 2005, 17, 5209–5217 CrossRef CAS.
- N. Liaros, E. Koudoumas and S. Couris, Appl. Phys. Lett., 2014, 104, 19112 CrossRef.
- D. Chateau, Q. Bellier, F. Chaput, P. Feneyrou, G. Berginc, O. Maury, C. Andraud and S. Parola, J. Mater. Chem. C, 2014, 2, 5105–5110 RSC.
- S. Husaini, J. E. Slagle, J. M. Murray, S. Guha, L. P. Gonzalez and R. G. Bedford, Appl. Phys. Lett., 2013, 102, 191112 CrossRef.
- Z. Yang, Z. K. Wu, J. S. Ma, A. D. Xia, Q. S. Li, C. L. Liu and Q. H. Gong, Appl. Phys. Lett., 2005, 86, 061903 CrossRef.
- Z. G. Xiao, Y. F. Shi, R. Sun, J. F. Ge, Z. G. Li and Y. L. Song, J. Mater. Chem. C, 2016, 4, 4647–4653 RSC.
- W. S. Fann, S. Benson, J. M. Madey, S. Etemad, G. L. Baker and F. Kajar, Phys. Rev. Lett., 1989, 62, 1492–1495 CrossRef CAS PubMed.
- S. Masashi, S. Fumio and M. Toshio, Polym. Chem., 2011, 2, 1044–1058 RSC.
- J. Z. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- Z. Q. Yan, Y. F. Chen, S. Y. Guang, H. Y. Xu and L.
F. Li, Polym. Sci., Ser. B, 2011, 53, 535–539 CrossRef CAS.
- X. Wang, Y. Yan, T. Liu, X. Y. Su, L. W. Qian and H. Y. Xu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5498–5504 CrossRef CAS.
- X. Wang, J. C. Wu, P. Wang and B. Z. Tang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2072–2083 CrossRef CAS.
- X. Y. Su, N. B. Lin and Y. L. Song, Polymer, 2008, 49, 3722–3730 CrossRef CAS.
- X. Wang, S. Y. Guang, X. Y. Su and X. Y. Liu, J. Mater. Chem., 2008, 18, 4204–4209 RSC.
- X. Y. Su, J. Y. Yang and X. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4529–4541 CrossRef CAS.
- S. C. Yin, H. Y. Xu, X. Y. Su, L. Wu, Y. L. Song and B. Z. Tang, Dyes Pigm., 2007, 75, 675–680 CrossRef CAS.
- S. Y. Guang, S. C. Yin, W. J. Zhu, Y. C. Gao and Y. L. Song, Dyes Pigm., 2007, 73, 285–291 CrossRef CAS.
- S. C. Yin, Y. C. Gao, Y. L. Song and B. Z. Tang, Dyes Pigm., 2007, 72, 119–123 CrossRef.
- A. Shukla, Chem. Phys., 2004, 300, 177–188 CrossRef CAS.
- C. W. Li, K. Yang and H. Y. Xu, J. Phys. Chem. B, 2009, 113, 15730–15733 CrossRef CAS PubMed.
- G. S. He, L. S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- Q. Zheng, G. S. He and P. N. Prasad, Chem. Mater., 2005, 17, 6004–6011 CrossRef CAS.
- X. H. Ouyang, H. P. Zeng and W. Ji, J. Phys. Chem. B, 2009, 113, 14565–14573 CrossRef CAS PubMed.
- F. G. Haj, B. G. Neel and P. I. H. Bastiaens, Science, 2002, 295, 1708–1711 CrossRef CAS PubMed.
- B. Z. Tang, W. Y. Lam and H. S. Kwok, Macromolecules, 1998, 31, 2419–2432 CrossRef CAS.
- Y. Fujita, Y. Misumi and M. Tabata, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 3157–3163 CrossRef CAS.
- H. Goto, X. M. Dai, T. Ueoka and K. Akagi, Macromolecules, 2004, 37, 4783–4793 CrossRef CAS.
- J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS PubMed.
- H. Balcar, J. Sedlacek and J. Vohlidal, Polymer, 2001, 42, 6709–6721 CrossRef CAS.
- B. Z. Tang and H. Y. Xu, Macromolecules, 1999, 32, 2569–2576 CrossRef CAS.
- B. Gu, S. M. Dharmaprakash and H. T. Wang, Appl. Phys. Lett., 2008, 92, 091118–091121 CrossRef.
- M. S. Bahae, A. A. Said, T. H. Wei, D. J. Hagan and E. W. V. Stryland, IEEE J. Quantum Electron., 1990, 26, 760–769 CrossRef.
- N. Lin, X. Y. Liu and W. Ji, Adv. Funct. Mater., 2012, 22, 361–368 CrossRef CAS.
- M. Teraguchi and T. Masuda, Macromolecules, 2000, 33, 240–242 CrossRef CAS.
- Z. Yan, H. Xu and X. Y. Liu, Adv. Funct. Mater., 2012, 22, 345–352 CrossRef CAS.
- F. Y. Zhang, J. Li and H. Y. Xu, RSC Adv., 2016, 6, 448–455 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, 1H NMR spectra of M1–M3, M5 and P1–P3, P5; 13C NMR spectra of M2–M5 and P2–P5; normalized UV-vis absorption and FL emission spectra of M5 (blue solid line) and P5 (red dash dot) in THF solutions; open-aperture Z-scans of P1, P3–P5 at the different pulse energy in THF solution. See DOI: 10.1039/c7ra08499d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Naibo Lin
*a,
Naibo Lin