
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling vaginal microbial genetic diversity and abundance between Holstein and Fleckvieh cattle
Lucky T.
Nesengani†
 ,
Jun
Wang†
,
Yujiang
Yang
,
Lianyu
Yang
and
Wenfa
Lu
*
,
Jun
Wang†
,
Yujiang
Yang
,
Lianyu
Yang
and
Wenfa
Lu
*
College of Animal Science and Technology, Jilin Agricultural University, Changchun, 130118, China. E-mail: wenfa2004@163.com
First published on 12th December 2017
Abstract
Characterization of bovine vaginal microbial genetic diversity can lead to better understanding of cattle physiology, which is of economic interest in improving cattle reproduction health. However, there is limited knowledge on the factors affecting the microbial community harbored in the vaginal tract. The aim of the current study is to evaluate and compare the microbial genetic diversity and abundance between the Holstein and Fleckvieh cattle using metagenomic approach. The main bacterial phyla found were firmicutes (51.70–58.80%), proteobacteria (16.70–11.70%) and bacteroidetes (13.20–19.50%) for Holstein and Fleckvieh. Notable significant difference (p < 0.05) was observed for Fusobacteria, which was more abundant in Holstein (9.73–0.81%). At the achaea phyla, Euryarchaeota was significantly different (p < 0.05) and was more abundant in Fleckvieh than in Holstein cattle (0.86–0.22%). At the genus level, Turicibacter, Ruminococcaceae_UCG-010, Clostridium_sensu_stricto_1, Dorea, Methanobrevibacter, Acetitomaculum, Ruminococcaceae_NK4A214_group and Ruminiclostridium_5 were found to be significantly (p < 0.05) different between the two cattle breeds and all of them were more abundant in Fleckvieh than in Holstein cattle. The current results indicate that the breed can influence bovine vaginal microbial abundance and diversity. Our study characterized and provided pivotal scientific knowledge to understand the microbial community that harbors the bovine virginal flora.
Introduction
Cattle production is amongst the most important factors in global economy, contributing extensively to the improvement and sustenance of human life.1 Despite its pivotal role in human life, cattle practice has been embroiled in a vast number of known and unknown challenges since the dawn of domestication. Till date, their biological aspects and influences are not fully understood, particularly the vaginal microbial community.2 Microbial community harboured in the vaginal tract have been mostly evaluated using traditional methods3–5 with some recent studies using advanced molecular techniques such as metagenomics.6,7 Since the dawn of molecular-based techniques, several studies conducted in human and other species have revealed a great deal of vaginal microbial community structure.8,9Conversely, in the current era of next-generation sequencing, metagenomics provide access to the total microbial genetic material from any environment without prior culture.10,11 Numerous studies have evaluated the bovine gastrointestinal microbial community12–14 and that of other species, such as pigs,15,16 through metagenomics approach. The human vaginal and gastrointestinal microflora have also been explored using metagenomics approach.17,18 Previous studies have shown that vaginal bacterial abundance and diversity in humans is shaped by different factors, such as pregnancy.6 Moreover, factors such as biogeographical and ethnic differences have been observed to influence vaginal bacterial abundance during pregnancy and postpartum in humans.19 However, knowledge of factors that influence vaginal bacterial genetic diversity and abundance in cattle is limited, particularly on a breed effect level.
Few studies have evaluated the cow vaginal microbial community. Till date, a study has evaluated the composition of vaginal microbial genetic diversity in the Nellore cattle using the metagenomic approach without taking into account the breed effect.20 Hence, the current study aims at evaluating the breed effect on the vaginal microbial diversity and abundance by describing and comparing the vaginal microbiomes structure of the Holstein and Fleckvieh cattle.
Materials and methods
Animals and sample collections
An elite herd (Guangyuan farm, Jilin Province, People's Republic of China) was selected, which is composed of Hereford and Fleckvieh cattle breed. Within the herd, a total of 12 cattle were randomly chosen from Holstein (n = 6) and Fleckvieh (n = 6) cattle. The cattle considered in the current study were all reared under the same conditions. All animals were healthy and did not receive any antibiotic treatment in a month prior to sampling. Vaginal samples were collected from all 12 cows using swabs and stored at −80 °C in a refrigerator until further use. This study was performed at the College of Animal Science and Technology, Jilin Agricultural University. All animal studies were conducted following the guidelines and regulations for the care and use of experimental animals established by the Ministry of Agriculture, China. Ethical approval for the present study was obtained from the Ethical Committee of Jilin Agricultural University, China.Total genomic DNA extraction and 16S rDNA gene amplification
Total microbial genomic DNA was extracted from the swab of each sample using a QIAamp DNA mini kit (Qiagen, Valencia, CA, USA) following the manufacturer's recommendation. DNA concentration was evaluated by optimal density using Nano-Drop 2000 (Thermo Electron Corporation, USA) at wavelengths of 260 and 280 nm. The integrity of the DNA was assessed by electrophoresis on 1.0% agarose gels and visualized with E-Gel Imager (Tanon-2500, Shanghai, China). V4 hypervariable regions of 16S rDNA were PCR amplified from microbial genomic DNA harvested from the vaginal samples. The barcoded fusion forward primer was 515F 5′-GTGCCAGCMGCGGTAA-3′ and the reverse primer was 806R 5′-GGACTACHVGGGTATCTAAT-3′. The PCR reactions were carried out in 30 μL reactions with 15 μL of Phusion® High-Fidelity PCR MasterMix (New England Biolabs), 0.2 μM of forward and reverse primers, and 10 ng template DNA. The PCR conditions were as follows: initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 30 s and post elongation at 72 °C for 5 min. The PCR amplicon products were separated on 2% agarose gels and extracted from the gels. Samples with bright main strip between 200–450 bp were chosen for further experiment. PCR products were purified with GeneJET Gel Extraction Kit (Thermo Scientific, Waltham, USA).Library preparation and sequencing
Sequencing libraries were generated using Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, USA) following manufacturer's recommendations and index codes were added. The library quality was assessed on the Qubit 2.0 Fluorometer (Thermo Scientific, Waltham, USA) and Agilent Bioanalyzer 2100 system. The library was sequenced on an Illumina HiSeq platform, and 250 bp paired-end reads were generated.Data analysis
The paired-end reads from the DNA fragments were merged using FLASH,21 which is an accurate analysis tool designed to merge paired-end reads when there are overlaps between the reads. Reads were first assigned to each sample according to the unique barcodes. Sequences were analyzed using QIIME22 software package (Quantitative Insights into Microbial Ecology). In-house perl scripts were used to analyze alpha-(within samples) and beta-(among samples), and reads were filtered by QIIME quality filters. Pick_de_novo_otus.py was used to pick operational taxonomic units (OTUs). Sequences with ≥97% similarity were assigned to the same OTUs. A representative of sequences for each OTU was classified using the RDP classifier23 to annotate taxonomic information for each representative sequence. All other statistical analyses were carried out using R, version 3.2.0.24 To compute alpha diversity, the OTU was rarified and metrics were calculated; chao1, Simpson index, ACE, goods coverage and Shannon's diversity index were generated based on these metrics. Both the weighted and unweighted unifrac that are phylogenetic measures of beta diversity were calculated. We used weighted unifrac for principal coordinate analysis (PCA) with arithmetic mean (UPGMA) clustering. The significant differences were tested at p < 0.05 and p < 0.01. The difference between the two groups was accessed by analysis of similarity (ANOSIM) in R Package “vegan”. Heatmaps were generated using gplot in R with taxonomic data. A Venn diagram was generated to compare unique and common OTUs between the two cattle breeds using R package “VennDiagram”.Results
Sequencing overview
Twelve cattle (n = 6 for Holstein and n = 6 for Fleckvieh) were sampled to evaluate and compare the vaginal microbial genetic diversity and abundance. Samples were used to generate V4 16S rDNA gene profiles. A total of 906![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 914 reads were obtained in all samples with an average number of 75576 reads per sample.
914 reads were obtained in all samples with an average number of 75576 reads per sample.
Significant difference between Holstein and Fleckvieh cattle
Analysis of the phylogenetic composition of vaginal bacterial community between Holstein and Fleckvieh cattle was characterized by 16S rDNA gene sequencing and the alpha- and beta-diversity of microbiota across the two cattle breeds were compared. The data indicated a diverse structure, which is summarized in Table 1, indicating the diversity and abundance indexes (chao1, Shannon diversity index, and ACE and Simpson index).| Sample name | Observed species | Shannon | Simpson | Chao1 | ACE | Goods coverage |
|---|---|---|---|---|---|---|
| a H1 to H6 refers to the samples per individual from the Holstein cattle and F1 to F6 refers to the samples from the Fleckvieh cattle group. | ||||||
| H1 | 1573 | 7.924 | 0.983 | 1716.5 | 1738.151 | 0.996 |
| H2 | 1402 | 6.319 | 0.931 | 1552.785 | 1560.483 | 0.996 |
| H3 | 1473 | 8.126 | 0.987 | 1581.247 | 1592.57 | 0.997 |
| H4 | 1338 | 7.482 | 0.979 | 1428.017 | 1438.394 | 0.997 |
| H5 | 1610 | 7.97 | 0.982 | 1739.814 | 1754.923 | 0.996 |
| H6 | 1373 | 6.963 | 0.907 | 1447.072 | 1443.439 | 0.998 |
| F1 | 1553 | 8.415 | 0.992 | 1658.174 | 1663.541 | 0.997 |
| F2 | 1602 | 8.547 | 0.992 | 1711.48 | 1710.816 | 0.997 |
| F3 | 1342 | 8.62 | 0.994 | 1395.382 | 1404.364 | 0.998 |
| F4 | 1481 | 8.573 | 0.993 | 1598.56 | 1580.378 | 0.997 |
| F5 | 1462 | 6.595 | 0.877 | 1554.557 | 1561.573 | 0.997 |
| F6 | 1785 | 8.26 | 0.981 | 2412.338 | 2122.975 | 0.993 |
The Fleckvieh cattle had a significantly higher Shannon index compared to the Holstein cattle (p < 0.05, Fig. 1A). There was a clear variation in data distribution between the two groups. The significant difference between the two groups was observed using ANOSIM analysis (p < 0.05, Fig. 1B). The variation of phylogenetic diversity of the tested samples was evaluated. There was a slight mixture in clustering, in which sample F2 and F5 from the Fleckvieh were clustered into the samples from the Holstein group and sample H6 was clustered into the samples from Fleckvieh group in principal component analysis (PCA) (Fig. 1C). However, significant difference between the two clustered groups was observed (p < 0.05, Fig. 1C). Most of the samples indicated higher similarity in phylogenetic composition between the samples from the two groups when analysing the alpha-diversity. However, few of the samples indicated a slight distinct phylogenetic composition between the Holstein and Fleckvieh samples (Fig. 1D). Greater variation between the two breeds was observed while analysing the beta-diversity, the t-test bars for the significant difference between the two groups at the phylum level are reported in Fig. 2A. Fusobacteria and Euryarchaeota were the notable significant differences between the two groups at the phylum level. Fusobacteria was more abundant in the Holstein cattle, while the Euryarchaeota was more abundant in the Fleckvieh cattle. The significant difference at the genus level is reported in Fig. 3B; the Ruminococcaceae_UCG_010 amount showed significant difference between the two groups (p < 0.05, Fig. 2B). It was more abundant in the Fleckvieh than in the Holstein cattle. The most significant differences (p < 0.01, Fig. 2B) at the genus level were in Clostridium_sensu_stricto_1, Methanobrevibacter, Turicibacter, Dorea, Ruminiclostridium_5 and Ruminococcaceae_NK4A214_group. All the significant differences at the genus level were more abundant in the Fleckvieh cattle than in the Holstein cattle.
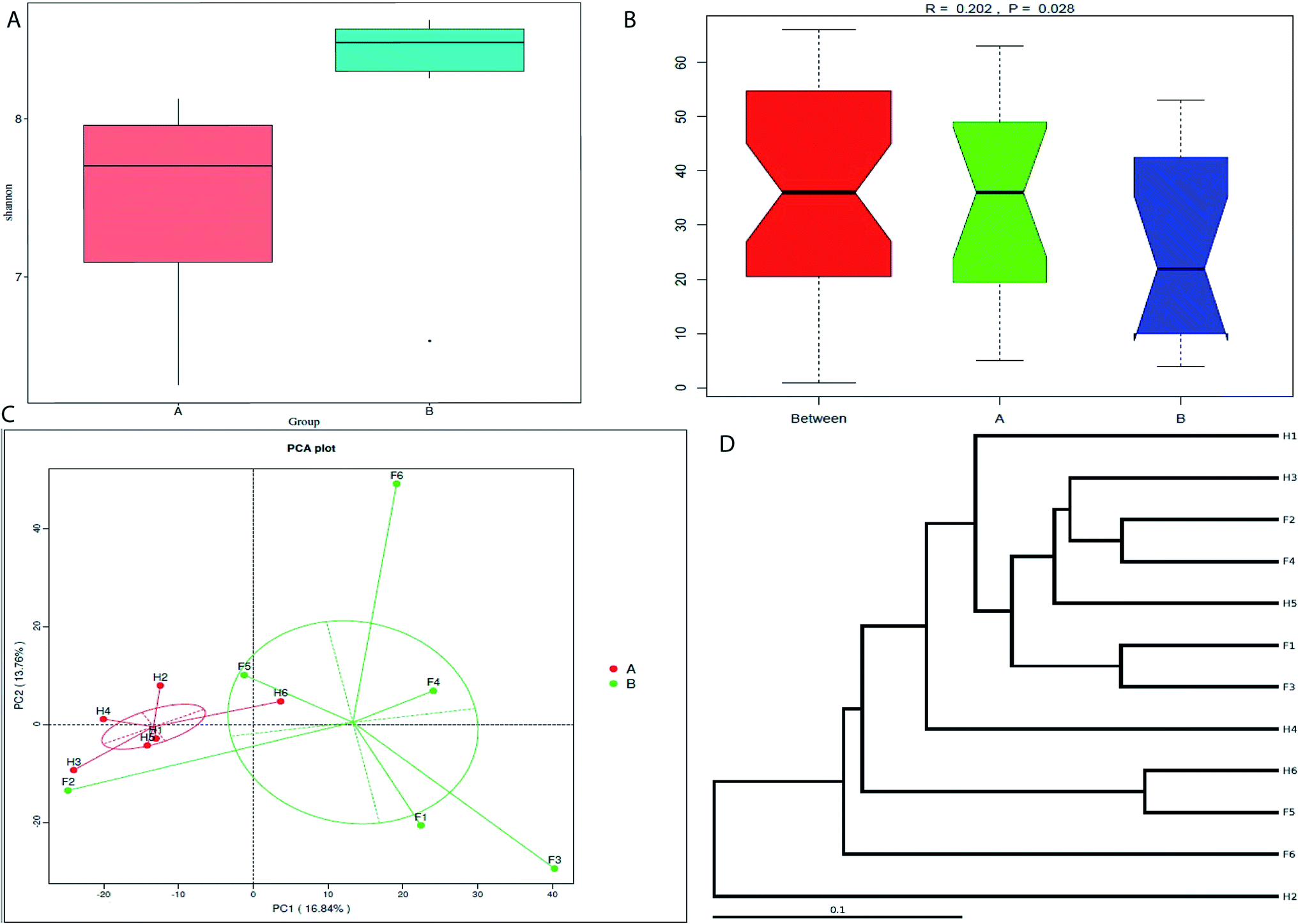 | ||
| Fig. 1 Alpha- and beta-diversity comparison of the vaginal microbiomes of the Holstein and Fleckvieh cattle breeds. The analyses were performed on 16S rRNA V4 region data. The coding names are as follows: group A refers to the Holstein cattle group and group B refers to the Fleckvieh cattle group. H1 to H6 refers to the samples per individual from the Holstein cattle and F1 to F6 refers to the samples from the Fleckvieh cattle group. (A) Alpha-diversity comparison based on Shannon's diversity index, grouped by sampling cattle breed (mean ± SEM). Compared to the Holstein, the Fleckvieh had a higher microbial richness. (B) Analysis of similarities between the two groups (Holstein and Fleckvieh). (C) Principal component analysis of vaginal bacterial microflora from two groups. (D) Weighted_unifrac cluster tree. Samples indicated a mixture of cluster with some from Holstein tending to cluster with Fleckvieh. | ||
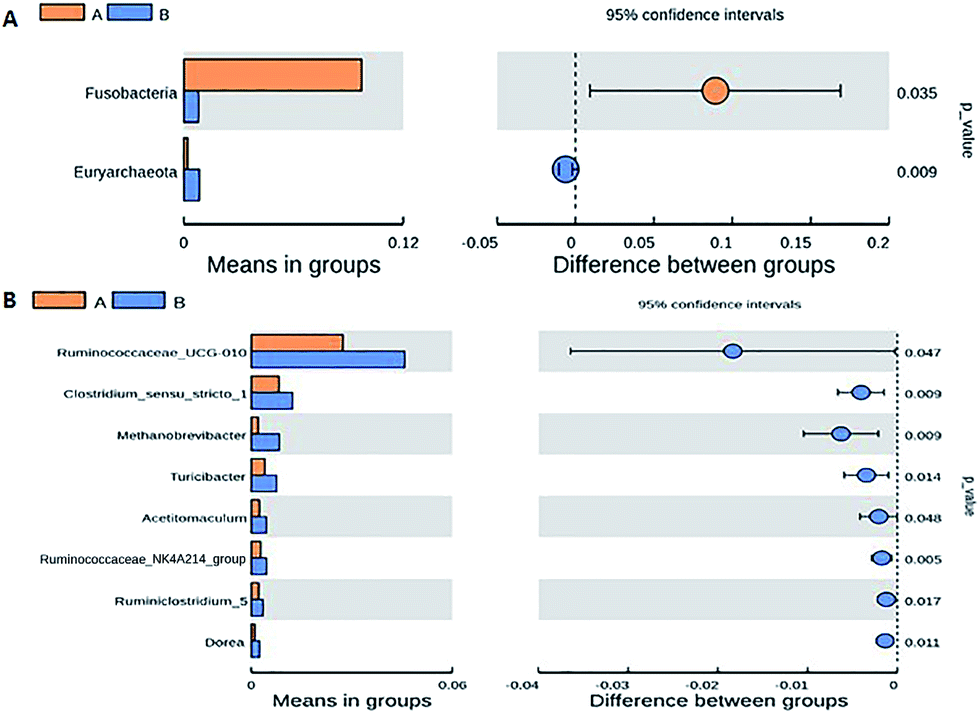 | ||
| Fig. 2 Beta-diversity (among samples) comparison of the vaginal microbiomes of the Holstein and Fleckvieh cattle breeds. The t-test bars plot indicate significant difference between the two groups. (A) Beta-diversity comparison based on t-test significance results at the phylum level. (B) Beta-diversity comparison based on t-test significance results at the genus level. | ||
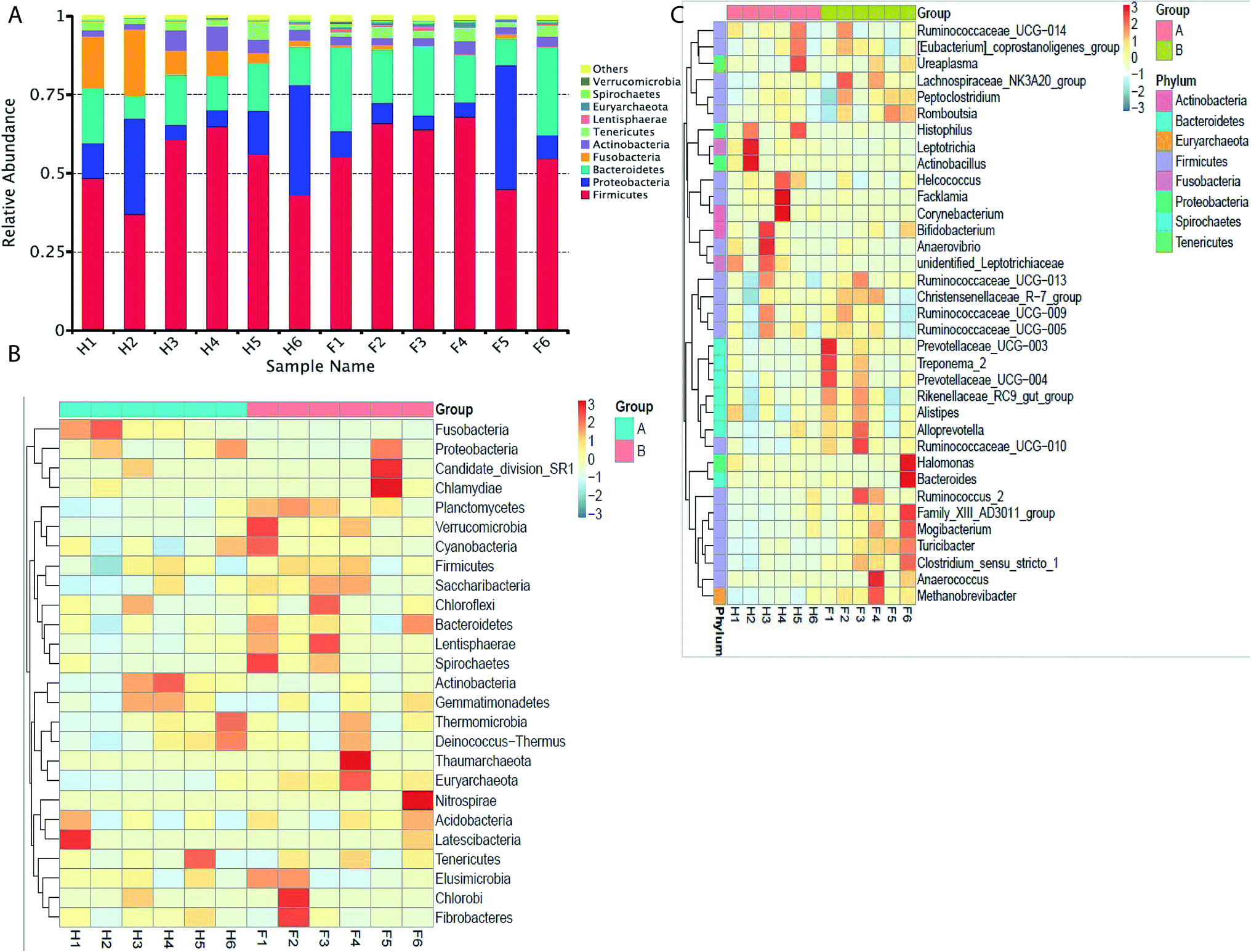 | ||
| Fig. 3 Taxonomic profiles of the microbial communities of the Holstein and Fleckvieh cattle derived from 16S rRNA gene sequencing of V4 region data. (A) Microbial composition at the phylum level. The samples names are as previously explained. Samples are presented along with the horizontal axis and relative abundance at the vertical axis. (B) Heatmap showing the 26 phyla with significant differences in relative abundances among the two breeds. Heatmap is color-coded based on the scale of −3 to 3. (C) Heatmap showing the 35 genera with significant differences of relative abundances among the two breeds. Heatmap is color-coded based on the scale of −3 to 3. | ||
Core bacterial abundance between the cattle breeds
The taxonomic distribution of the numerically abundant bacteria derived from the 16S rRNA gene sequences was further evaluated. At the phylum level, firmicutes was the highest abundant phenotype in the Holstein and Fleckvieh group, which accounted for 51.70% and 58.80% of relative abundance respectively, followed by proteobacteria that accounted for 16.70% and 11.70% of relative abundance and bacteroidetes that accounted for 13.20% and 19.50% of relative abundance (Fig. 3A). Conversely, bacteria abundance was further evaluated using a heat map analysis for the top 26 bacteria at phylum level (Fig. 3B), which indicated similar results as previously explained with Fusobacteria indicating to be more abundant in the Holstein group and less abundant in the Fleckvieh group, while others indicated slight variation between the two groups at a similar magnitude. At the genus level, Actinobacillus had 4.70% abundance in the Holstein group, but only 0.06% abundance in the Fleckvieh group; however, there was no significant difference (p > 0.05). Moreover, it should be noted that the Facklamia and Histophilus accounted for 2.3% and 3.8% of abundance in the Holstein, while they accounted for 0.03% and 0.07% of abundance in the Fleckvieh group respectively (p > 0.05, Fig. 3C).Common and unique operational taxonomic units (OTU)
The common and unique operational taxonomic units (OTUs) between the two groups were evaluated. A total of 2574 OTUs were observed from all the samples. About 348 OTUs were identified to be unique in Fleckvieh cattle, while a total of 151 OTUs were identified to be unique in Holstein cattle; in addition, 2075 of OTUs exist in both groups and are defined as the core (Fig. 4A). Furthermore, the OTUs were evaluated based on each sample. Samples from the Fleckvieh cattle were found to have 22 OTUs and above, while the samples from Holstein were found to contain 18 OTUs and below with the only exception of H6 sample that contained 24 OTUs (Fig. 4B). | ||
| Fig. 4 Comparison of the OTUs in Holstein and Fleckvieh cattle. The number of observed OTUs sharing ≥97% nucleotide sequence identity. (A) A Venn diagram was generated to describe the common and unique OTUs among the two cattle breeds. (B) A flower diagram was used to identify the common and unique OTUs within each individual from the two breeds. | ||
Discussion
Bovine microbial composition has been fairly studied primarily in the gut with few studies in the vaginal microbial composition.14,25,26 To the best of our knowledge, there is no report based on bovine metagenomic studies that evaluated the vaginal microbial genetic diversity and abundance between cattle breeds. Cattle vaginal microbiota has been evaluated primarily based on the effects of diseases through culture-depended technique.4,9,27 In this study, we compared vaginal microbial community structure between the Holstein and the Fleckvieh cattle breeds. The two breeds were chosen since Holstein is the most common and leading breed in dairy breeds across the world and in China, while the Fleckvieh is the common breed used for beef in Northern China.Bacterial composition and diversity are indicated to be breed type-dependent between the two groups (Holstein and Fleckvieh cattle). The results of the bacterial composition are comparable to the study conducted by Laguardia-Nascimento et al.20 The group evaluated vaginal microbiota composition in Nellore cattle and found that firmicutes (40–50%) was predominant in the vaginal microbiota, followed by bacteroidetes (15–25%) and proteobacteria (5–25%). Swarts et al.7 also reported that the vaginal microbiota of ewes and dairy cows is predominated by bacteroidetes, followed by Fusobacteria and proteobacteria. The studies conducted to evaluate bovine vaginal microbiota are in general agreement with the current study. Almost all the studies reported that the bovine vaginal microbiota is predominated by similar groups of bacteria (firmicutes, bacteroidetes and proteobacteria) at different magnitudes, which could be due to environmental factors and host genotype. However, it is also worthy to note that similar microbiota composition has also been observed in other sites, such as in the gut. Jami et al.28 found that bovine microbial composition in the gut is predominated by bacteroidetes, which is followed by firmicutes and proteobacteria at 51%, 41% and 5.46% respectively.
In the current study, it was found that different breeds dictate different bacterial genetic composition, in which Holstein cattle have less abundant and diverse bacteria than the Fleckvieh cattle. Greater bacterial genetic diversity has been associated with less risk of infections in dairy cattle.29 It was also noted that Fusobacteria was more abundant in the Holstein than Fleckvieh. Higher abundance of Fusobacteria has been reported to be associated with uterine diseases and related infections.30 It was further reported that low Fusobacteria abundance in vaginal microbiota is associated with healthy cows.31 From the current observation, the Holstein cattle can be more susceptible to uterine infections than the Fleckvieh cattle.
The microbial differences were further evaluated at the genus level to gain more insight into vaginal microbial differences between the two cattle breeds. It was found that most of the significantly (p < 0.05) different genus species are more abundant and diverse in the Fleckvieh than in the Holstein. A greater difference was also observed at the family level (results not shown) as well as the class level (results not shown) at a similar magnitude, in which Fleckvieh was more in abundance.
The other difference between the two breeds was observed at the OTUs, in which the Fleckvieh cattle had greater unique OTUs than the Holstein cattle. This phenomenon was also observed in individual sequences. The Fleckvieh cattle had more OTUs per individual, while the Holstein had less OTUs per individual sample. The current results indicate that the Holstein cattle have less microbial abundance and OTUs as compared to the Fleckvieh cattle.
Conclusions
In our study, we have demonstrated, for the first time, the influence of breed type on the vaginal microbial genetic abundance and diversity. The current results are of critical importance for better understanding of vaginal microbial genetic diversity and abundance in different breeds. Such information can be used as a scientific tool to help in reducing and better understanding the physiological challenges in livestock practice to advance reproduction health methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (31672420), Jilin Province key scientific and technical project (20140203016NY; 20140203013NY) and general project of the Department of Science and Technology of Jilin province (20150204074NY; 20150519018JH; 20160209001NY).Notes and references
- A. d. B. des Roches, I. Veissier, X. Boivin, E. Gilot-Fromont and L. Mounier, J. Dairy Sci., 2016, 99, 5573–5585 CrossRef PubMed.
- J. H. H. M. van de Wijgert, H. Borgdorff, R. Verhelst, T. Crucitti, S. Francis, H. Verstraelen and V. Jespers, PLoS One, 2014, 9, e105998 CrossRef PubMed.
- B. Chaban, M. G. Links, T. P. Jayaprakash, E. C. Wagner, D. K. Bourque, Z. Lohn, A. Y. K. Albert, J. van Schalkwyk, G. Reid, S. M. Hemmingsen, J. E. Hill and D. M. Money, Microbiome, 2014, 2, 23 CrossRef PubMed.
- J. Wang, C. Sun, C. Liu, Y. Yang and W. Lu, Anaerobe, 2016, 38, 1–6 CrossRef PubMed.
- C. Gonzalez Moreno, C. Fontana, P. S. Cocconcelli, M. L. Callegari and M. C. Otero, J. Appl. Microbiol., 2016, 121, 1232–1241 CrossRef CAS PubMed.
- K. Aagaard, K. Riehle, J. Ma, N. Segata, T.-A. Mistretta, C. Coarfa, S. Raza, S. Rosenbaum, I. Van den Veyver, A. Milosavljevic, D. Gevers, C. Huttenhower, J. Petrosino and J. Versalovic, PLoS One, 2012, 7, e36466 CrossRef CAS PubMed.
- J. D. Swartz, M. Lachman, K. Westveer, T. O'Neill, T. Geary, R. W. Kott, J. G. Berardinelli, P. G. Hatfield, J. M. Thomson, A. Roberts and C. J. Yeoman, Front. Vet. Sci., 2014, 1, 19 CrossRef PubMed.
- R. F. Lamont, J. D. Sobel, R. A. Akins, S. S. Hassan, T. Chaiworapongsa, J. P. Kusanovic and R. Romero, BJOG, 2011, 118, 533–549 CrossRef CAS PubMed.
- D. N. Fredricks, Anaerobe, 2011, 17, 191–195 CrossRef CAS PubMed.
- J. Handelsman, Microbiol. Mol. Biol. Rev., 2004, 68, 669–685 CrossRef CAS PubMed.
- H. Teeling and F. O. Glöckner, Briefings Bioinf., 2012, 13, 728–742 CrossRef PubMed.
- S. Jiao, H. Cao, Y. Dai, J. Wu, J. Lv, R. Du and B. Han, J. Sci. Food Agric., 2017 DOI:10.1002/jsfa.8380.
- S. Mao, M. Zhang, J. Liu and W. Zhu, Sci. Rep., 2015, 5, 16116 CrossRef CAS PubMed.
- R. Roehe, R. J. Dewhurst, C.-A. Duthie, J. A. Rooke, N. McKain and D. W. Ross, et al. , PLoS Genet., 2016, 12(2), e1005846 CrossRef PubMed.
- W. Zhao, Y. Wang, S. Liu, J. Huang, Z. Zhai and C. He, et al. , PLoS One, 2015, 10(2), e0117441 CrossRef PubMed.
- R. Lamendella, J. W. Santo Domingo, S. Ghosh, J. Martinson and D. B. Oerther, BMC Microbiol., 2011, 11, 103 CrossRef CAS PubMed.
- A. S. Zevin, I. Y. Xie, K. Birse, K. Arnold, L. Romas, G. Westmacott, R. M. Novak, S. McCorrister, L. R. McKinnon, C. R. Cohen, R. Mackelprang, J. Lingappa, D. A. Lauffenburger, N. R. Klatt and A. D. Burgener, PLoS Pathog., 2016, 12, e1005889 CrossRef PubMed.
- W.-L. Wang, S.-Y. Xu, Z.-G. Ren, L. Tao, J.-W. Jiang and S.-S. Zheng, World J. Gastroenterol., 2015, 21, 803–814 CrossRef PubMed.
- D. A. MacIntyre, M. Chandiramani, Y. S. Lee, L. Kindinger, A. Smith, N. Angelopoulos, B. Lehne, S. Arulkumaran, R. Brown, T. G. Teoh, E. Holmes, J. K. Nicoholson, J. R. Marchesi and P. R. Bennett, Sci. Rep., 2015, 5, 8988 CrossRef CAS PubMed.
- M. Laguardia-Nascimento, K. M. G. R. Branco, M. R. Gasparini, S. Giannattasio-Ferraz, L. R. Leite and F. M. G. Araujo, et al. , PLoS One, 2015, 10(11), e0143294 CrossRef PubMed.
- T. Magoc and S. L. Salzberg, Bioinformatics, 2011, 27, 2957–2963 CrossRef CAS PubMed.
- J. G. Caporaso, J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Pena, J. K. Goodrich, J. I. Gordon, G. A. Huttley, S. T. Kelley, D. Knights, J. E. Koenig, R. E. Ley, C. A. Lozupone, D. McDonald, B. D. Muegge, M. Pirrung, J. Reeder, J. R. Sevinsky, P. J. Turnbaugh, W. A. Walters, J. Widmann, T. Yatsunenko, J. Zaneveld and R. Knight, Nat. Methods, 2010, 7, 335–336 CrossRef CAS PubMed.
- Q. Wang, G. M. Garrity, J. M. Tiedje and J. R. Cole, Appl. Environ. Microbiol., 2007, 73, 5261–5267 CrossRef CAS PubMed.
- R. C. Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2014, http://www.R-project.org/ Search PubMed.
- R. J. Wallace, J. A. Rooke, N. McKain, C.-A. Duthie, J. J. Hyslop, D. W. Ross, A. Waterhouse, M. Watson and R. Roehe, BMC Genomics, 2015, 16, 839 CrossRef PubMed.
- A. Kumar and D. Kumar, Anaerobe, 2015, 33, 117–123 CrossRef CAS PubMed.
- V. S. Machado, G. Oikonomou, M. L. S. Bicalho, W. A. Knauer, R. Gilbert and R. C. Bicalho, Vet. Microbiol., 2012, 159, 460–469 CrossRef CAS PubMed.
- E. Jami and I. Mizrahi, PLoS One, 2012, 7, e33306 CrossRef CAS PubMed.
- S. F. Lima, A. G. V. Teixeira, F. S. Lima, E. K. Ganda, C. H. Higgins, G. Oikonomou and R. C. Bicalho, J. Dairy Sci., 2017, 100, 3031–3042 CrossRef CAS PubMed.
- M. L. S. Bicalho, T. Santin, M. X. Rodrigues, C. E. Marques, S. F. Lima and R. C. Bicalho, J. Dairy Sci., 2017, 100, 3043–3058 CrossRef CAS PubMed.
- M. L. S. Bicalho, S. Lima, C. H. Higgins, V. S. Machado, F. S. Lima and R. C. Bicalho, J. Dairy Sci., 2017, 100, 3863–3874 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2017 |