
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoelectronic control of oxidation potentials of 3,7-bis(diarylamino)phenothiazines†
Ayumu Karimataa,
Shuichi Suzuki *ab,
Masatoshi Kozakia and
Keiji Okada*a
*ab,
Masatoshi Kozakia and
Keiji Okada*a
aGraduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: okadak@sci.osaka-cu.ac.jp
bGraduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: suzuki-s@chem.es.osaka-u.ac.jp
First published on 12th December 2017
Abstract
The influence of diarylamino (Ar2N–) substituents on the oxidation potential of 3,7-bis(diarylamino)phenothiazines (Ar2N)2–PTZ (1a–f, a: carbazolyl; b: dihydrodibenzoazepinyl; c: dibenzoazepinyl; d: diphenylamino; e: phenothiazinyl; and f: phenoxazinyl) is investigated, where the Ar2N-substituent sequence a→f is aligned in the increasing order of their electron-donating ability. Interestingly, a different sequence of electron-donating ability for Ar2N-substituents was observed for the oxidation potentials of (Ar2N)2–PTZ: 1a (Eox1 = +0.35 V vs. Fc/Fc+) > 1f (+0.30 V) > 1e (+0.15 V) > 1d (−0.05 V) > 1c (−0.19 V) > 1b (−0.22 V). The observed sequence can be explained by the stereoelectronic effect of the Ar2N-substituents to stabilize (Ar2N)2–PTZ˙+. Clear-cut examples are observed in the crystal structure of 1c˙+ and 1e˙+, for which coplanar conformation is observed between the PTZ˙+-plane and the planes of the sp2-hybridized nitrogen atoms in Ar2N-substituents through a large conformational change during the oxidation process of (Ar2N)2–PTZ.
Introduction
Phenothiazines (PTZs) are typical electron donors and have been used in charge transporting materials,1 molecular spin materials,2 dye-sensitized solar cells,3 π-electron systems for photo-induced electron transfer,4 organic emitting devices,5 and charge transfer complexes.6 Tuning of the oxidation potential of these PTZs is important to widen their applicability. Several 3- or 3,7-substituted PTZs have been synthesized for various purposes.7 Müller reported the oxidation potentials and optical properties of various 3,7-diamino PTZs.8 Although PTZ derivatives with considerably low oxidation potentials have been synthesized, systematic substituent-effects based on their conformations confirmed by crystal structure analyses have not been reported.In this paper, we investigated the diarylamino (Ar2N–) substituent effect on the oxidation potentials of 3,7-bis(diarylamino)-10-methyl-10H′-phenothiazines (Ar2N)2–PTZ (1a–f, a: carbazolyl, b: dihydrodibenzoazepinyl, c: dibenzoazepinyl, d: diphenylamino, e: phenothiazinyl, and f: phenoxazinyl in Scheme 1), where the Ar2N-substituent sequence a→f is arranged in the ascending order of the electron-donating ability estimated from the oxidation potentials of the corresponding N-phenyldiarylamines Ar2N-Ph (2a–f) (Scheme 1). Interestingly, a different sequence was observed for the oxidation potentials of 1a–f: 1a > 1f > 1e > 1d > 1c > 1b. The difference between 2a–f and 1a–f is based on the oxidation moieties in the first oxidation potential; the oxidation potentials are primarily controlled by Ar2N-substituents in Ar2N-Ph, whereas the PTZ moieties are oxidized in (Ar2N)2–PTZ, except for 1f (vide infra). In the latter, a large stabilization is obtained through the conjugation between Ar2N– and PTZ˙+ moieties. For instance, in compound 1d˙+, the phenyl groups in the Ar2N-substituent can rotate around the N–C(sp2) bonds to avoid steric repulsion between the PTZ˙+ and the phenyl moieties, retaining the conjugation between the p-orbital on the nitrogen atom in the Ar2N-substituent and the singly occupied molecular orbital (SOMO) on the PTZ˙+ moiety; such a conformation is not possible with the carbazolyl group in 1a˙+ because of the planar conformation. Furthermore, the Ar2N-substituents in 1b˙+ and 1c˙+ can uniquely adopt a coplanar conformation about the PTZ˙+ plane and the plane of sp2 nitrogen atom in Ar2N-substituents. Previously, we have reported a unique structure of the PTZ trimer radical cation, the 10-phenyl derivative instead of the 10-methyl derivative (1e); the PTZ trimer radical cation has a considerably deformed structure, which is stabilized by the conjugation between the inner PTZ˙+ moiety and the sp2-hybridized nitrogen atoms in the outer PTZs.9 This paper presents a more general relationship between structures and oxidation potentials using various Ar2N-substituents; the proposed stereoelectronic stabilization is directly demonstrated by the crystal structure analyses of a series of (Ar2N)2–PTZ˙+.
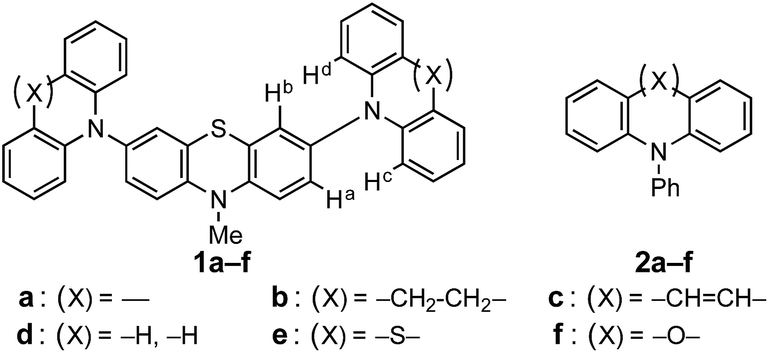 | ||
| Scheme 1 Chemical structures of (Ar2N)2–PTZ (1a–f) and Ar2N-Ph (2a–f). | ||
Results and discussion
Syntheses of 3,7-bis(diarylamino)-10-methyl-10H′-phenothiazines and their radical cationic species
Compounds (Ar2N)2–PTZ were synthesized from 3,7-dibromo-PTZ 3 (Scheme 2). Compound 3 was prepared according to the reported methods.10 Compound 3 was converted to the desired PTZ derivatives 1a–f by using Buchwald–Hartwig cross-coupling reactions with the corresponding diarylamines 4a–f.7 Single crystals of (Ar2N)2–PTZ were obtained by recrystallization under suitable conditions (see Experimental section). However, we failed to obtain single crystals of 1b. We could obtain the crystal structure of the model compound 1b′ (Scheme 3) using N-p-tolyl group instead of the N-methyl group in 1b. The reference compounds 2a–f were also synthesized from the corresponding diarylamines and bromobenzene using Buchwald–Hartwig reactions.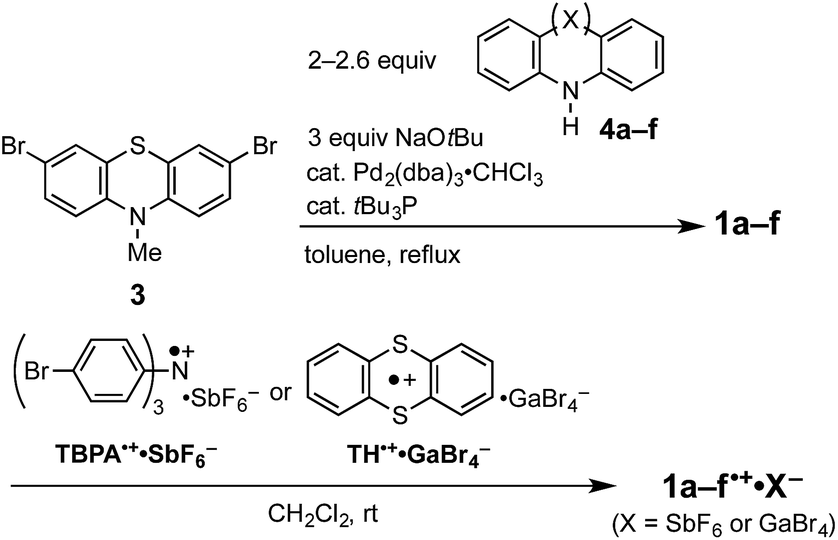 | ||
| Scheme 2 Synthetic methods of (Ar2N)2–PTZ (1a–f) and (Ar2N)2–PTZ˙+·X− (1a–f˙+·X−). | ||
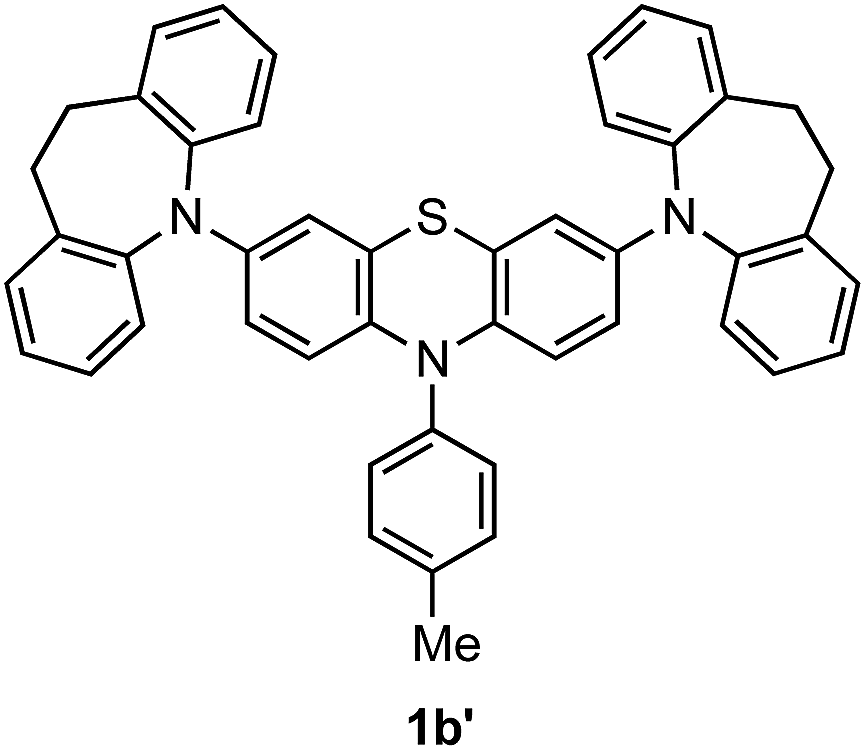 | ||
| Scheme 3 Chemical structure of 1b′. | ||
We prepared the radical cationic species of (Ar2N)2–PTZ by chemical oxidation using suitable reagents (Scheme 2) to study their detailed molecular structures by X-ray crystal structure analysis. The compounds 1a, 1b, 1d, and 1e were oxidized using tris(4-bromophenyl)aminium hexafluoroantimonate (TBPA˙+·SbF6−) to the corresponding radical cationic salts (1a˙+·SbF6−, 1b˙+·SbF6−, 1d˙+·SbF6−, 1e˙+·SbF6−). The single crystals of these radical cationic salts could be obtained by recrystallization from suitable solvents.11 Although we synthesized 1c˙+·SbF6− and 1f˙+·SbF6−, we could not obtain their single crystals for crystal structure analysis. Alternatively, we could obtain single crystals of 1c˙+·GaBr4− and 1f˙+·GaBr4−, which could be prepared from 1c and 1f by treatments with thianthrenium tetrabromogallate (TH˙+·GaBr4−).11,12 These radical cationic species are quite stable for days, even under aerated conditions in both solution state and solid state.
Oxidation potentials of 3,7-bis(diarylamino)-10-methyl-10H′-phenothiazines
The cyclic voltammograms are shown in Fig. 1 for 1a–e and Fig. S1† for 1f, and their oxidation potentials are listed in Table 1. In order to gain insight into the electron donating ability of the Ar2N-substitutents, oxidation potentials of Ar2N-Ph (2a–f) and N-methyl-10H-phenothiazine (5) were also measured (Table 1), which reflects the electron donating ability of the Ar2N-substituent: Eox1 (2a–f) is aligned in the sequence of a ≈ b > c > d ≫ e > f (Table 1).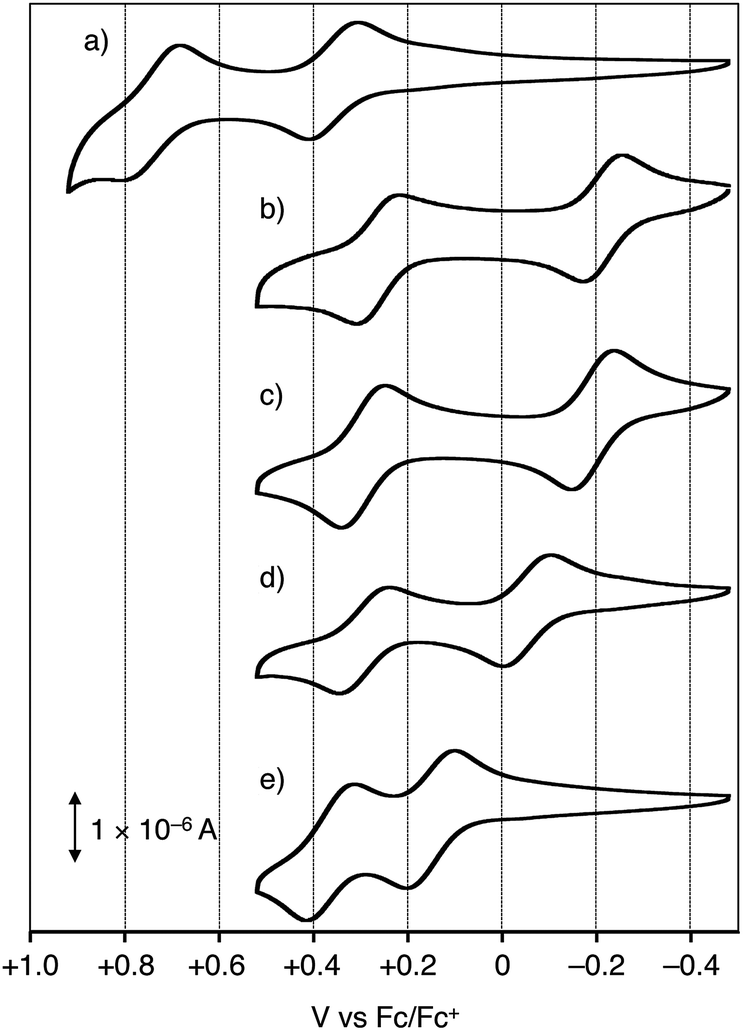 | ||
| Fig. 1 Cyclic voltammograms of (a) 1a, (b) 1b, (c) 1c, (d) 1d, and (e) 1e in dichloromethane: conditions of cyclic voltammetry: nBu4N+·ClO4− as an electrolyte; glassy carbon and Pt wire as a working and counter electrode; in CH2Cl2; Fc/Fc+ = +0.49 V vs. SCE. | ||
| Compound | Eox1 [V vs. Fc/Fc+] | Eox2 [V vs. Fc/Fc+] |
|---|---|---|
| a Conditions of cyclic voltammetry: nBu4N+·ClO4− as an electrolyte; glassy carbon and Pt wire as a working and counter electrode, respectively; in dichloromethane; Fc/Fc+ = +0.49 V vs. SCE.b Half wave potential of a reversible oxidation wave.c Two-electron oxidation processes of phenoxazine moieties.d Peak potential, respectively. Eox1, Eox2: first and second oxidation potentials. | ||
| 5 | +0.31b | — |
| 1a | +0.35b | +0.75b |
| 1b | −0.22b | +0.27b |
| 1c | −0.19b | +0.30b |
| 1d | −0.05b | +0.29b |
| 1e | +0.15b | +0.36b |
| 1f | +0.30c | — |
| 2a | +1.08d | — |
| 2b | +1.08d | — |
| 2c | +0.94d | — |
| 2d | +0.80d | — |
| 2e | +0.26b | — |
| 2f | +0.24b | — |
The oxidation potentials of (Ar2N)2–PTZ were considerably lower than those of Ar2N-Ph, except for 1f (Table 1). Compound 1a has a lower oxidation potential (+0.35 V vs. Fc/Fc+) than that (+1.08 V) of 2a, because the oxidation in 1a occurs in the central PTZ moiety rather than the outer carbazolyl-moieties (cf. Eox1 for 5 and 2a). The oxidation potentials of 1b and 1c with seven-membered ring systems (Eox1 ≈ −0.2 V) are considerably lower than those of 2b and 2c (Eox1 ≈ +1.0 V) and even lower than that of 5 (+0.31 V). The observed sequence of the oxidation potentials in 1a–f (Eox1(1a) > Eox1(1f) > Eox1(1e) > Eox1(1d) > Eox1(1c) > Eox1(1b)) is totally different from those of the Ar2N-substituents.13
X-ray diffraction studies revealed that the compounds with low oxidation potential (1b, 1c, and 1d) can adopt a coplanar conformation about the PTZ˙+ plane and the plane of sp2-hybridized nitrogen atom in the radical cation states by a structural change from the neutral states (vide infra), as observed in the PTZ trimer radical cation.9 However, 1a˙+ and 1f˙+ cannot undergo such conformational changes due to their rigid planar structures of Ar2N-substituents,14 which causes steric repulsion between Ha (Hb) and Hc (Hd) in the structure of (Ar2N)2–PTZ, as shown in Scheme 1.
It is to be noted that the oxidation wave for 1f (+0.3 V) is assigned to sequential two-electron oxidations of outer phenoxazines (Table 1, Fig. S1†). This assignment is compatible with the following three observations; (1) slightly lower oxidation potential of 2f (+0.24 V) than those of 2e (+0.26 V) and 5 (+0.31 V), (2) crystal structure of 1f˙+·GaBr4− exhibiting neutral PTZ with a butterfly structure (vide infra, Fig. 3(l) and S2†), and (3) UV-vis-NIR spectrum of 1f˙+·GaBr4− showing a strong absorption at ∼500 nm assigned to the terminal phenoxazine radical cation (Fig. S3†). These results indicate that the oxidation potential of the central PTZ moiety in 1f should be more positive than +0.30 V.
X-ray crystal structure analysis and substituent effect in the oxidation of (Ar2N)2–PTZ
To demonstrate the conformation-dependent substituent effect, we analyzed the X-ray crystal structure of both neutral and radical cationic species (Fig. 2 and 3).11 The molecular structures of neutral species 1a, 1b′ (as a model compound of 1b), 1c–f, and radical cationic species 1a–f˙+ are shown in Fig. 3. | ||
| Fig. 2 Specific names of atoms and planes for discussion of crystal structures. | ||
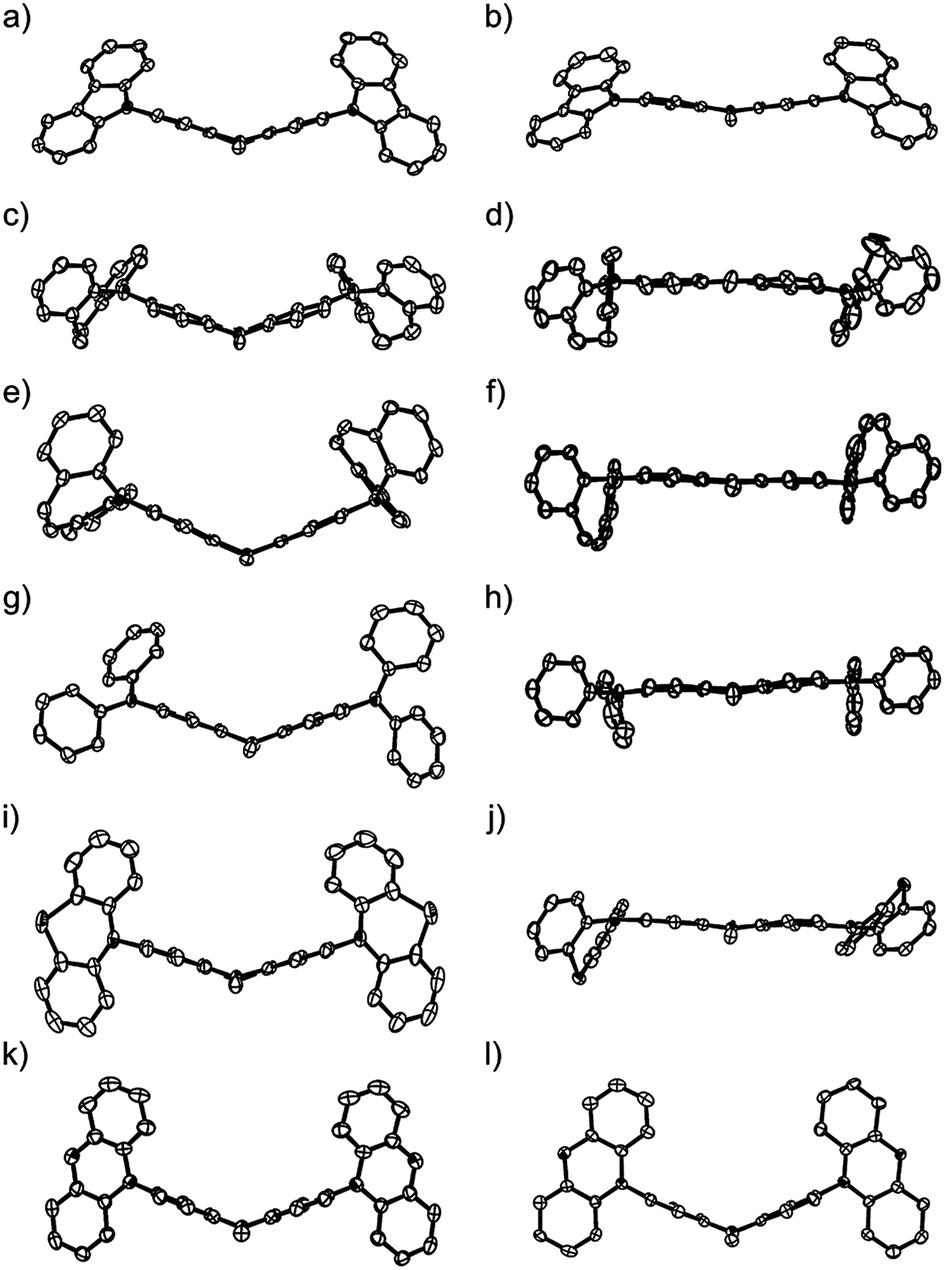 | ||
| Fig. 3 ORTEP views along the N–S axis (50% probabilities); (a) 1a, (b) 1a˙+·SbF6−, (c) 1b′ (the p-tolyl group was omitted for clarity), (d) 1b˙+·SbF6−, (e) 1c, (f) 1c˙+·GaBr4−, (g) 1d, (h) 1d˙+·SbF6−, (i) 1e, (j) 1e˙+·SbF6−, (k) 1f, (l) 1f˙+·GaBr4− hydrogen atoms, counter anions, and crystal solvents were omitted for clarity. | ||
The central PTZ moiety of (Ar2N)2–PTZ takes a butterfly structure (dihedral angles between planes A/B: 35–50°) (Table 2, Fig. 2 and 3),4a,9 whereas that of (Ar2N)2–PTZ˙+ except for 1f˙+ has an almost planar structure (A/B < 17.7°), indicating that the central PTZ moiety of 1a–e˙+ is in the radical cationic state. The structure of radical cation 1f˙+ is different from those of the other radical cations 1a–e˙+. The central PTZ moiety of 1f˙+ has a butterfly structure (A/B: 45.4°) similar to that of 1f (A/B: 45.4°), showing that the central PTZ moiety of 1f˙+ is in the neutral state. It is also interesting that one of the two outer phenoxazine moieties has a planer structure with shorter C–O bond lengths (Fig. S2†). These results indicate that the planar phenoxazine moiety is in the radical cationic state, whereas the other phenoxazine moiety is in the neutral state.
| Neutral species | Cationic species | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b′ | 1c | 1d | 1e | 1f | 1a˙+ | 1b˙+c | 1c˙+ | 1d˙+d | 1e˙+ | 1f˙+ | |
| a See Fig. 2 for naming of atoms and planes in (Ar2N)2–PTZ.b Dihedral angles between two planes.c Averaged values of two crystallographically independent species.d Averaged values of four crystallographically independent species. | ||||||||||||
| S–C1(12) [Å] | 1.76 | 1.76 | 1.77 | 1.77 | 1.76 | 1.75 | 1.73 | 1.73 | 1.73 | 1.72 | 1.74 | 1.77 |
| N1–C6(7) [Å] | 1.41 | 1.43 | 1.42 | 1.42 | 1.41 | 1.41 | 1.39 | 1.38 | 1.39 | 1.39 | 1.38 | 1.42 |
| N2(3)–C3(10) [Å] | 1.42 | 1.41 | 1.42 | 1.43 | 1.43 | 1.44 | 1.40 | 1.38 | 1.38 | 1.37 | 1.39 | 1.44 |
| Plane A/B [°]b | 33.0 | 34.5 | 48.7 | 38.3 | 34.9 | 40.5 | 14.9 | 17.7 | 3.7 | 6.0 | 7.1 | 45.4 |
| Plane A/C [°]b | 56.4 | 6.8 | 21.7 | 59.0 | 85.7 | 80.9 | 40.6 | 5.6 | 2.8 | 11.2 | 6.0 | 86.4 |
| Plane B/D [°]b | 62.0 | 3.3 | 22.4 | 41.8 | 88.1 | 80.9 | 41.4 | 4.0 | 10.0 | 4.3 | 9.1 | 88.1 |
Table 2 lists the bond lengths R(N1–C6(7)) and R(S–C1(12)) of the central PTZ moiety in the neutral and the radical cationic states. These N–C(sp2) and S–C(sp2) bond lengths in the radical cationic state are slightly shorter than those in the neutral state by reflection of the antibonding character of these bonds in SOMOs of 1a–e˙+ (Fig. S4†).
In these radical cations, the small dihedral angle between planes A and C (A/C) (or B and D (B/D)) permits the ease of conjugation between the PTZ π-orbitals and the adjacent nitrogen p-orbital in the Ar2N-substituents. Interestingly, compounds 1c˙+ and 1e˙+ experienced large conformational changes to diminish A/C and B/D. The compounds having low oxidation potentials, i.e. 1b, 1c, and 1d, have small average dihedral angle in their radical cationic state: 4.8° (1b˙+), 6.4° (1c˙+), and 7.8° (1d˙+). Compound 1a had the largest dihedral-angle in the radical cation state except for 1f˙+ and exhibited the most positive oxidation potential. Furthermore, effective conjugations about sp2 nitrogen atom and the central PTZ˙+ for the low-oxidation-potential compounds (1b, 1c, and 1d) are reflected by shorter bond lengths R(N2(3)–C3(10)) (1.37–1.38 Å) in the radical cation structures. These results clearly indicate the importance of radical cation structures for the oxidation potentials.
It is interesting to consider the mechanism of oxidation of the adsorbed neutral state (Ar2N)2–PTZ for cyclic voltammogram measurements; oxidation is accompanied by the large conformational change to stabilize the (Ar2N)2–PTZ˙+. The oxidation of neutral (Ar2N)2–PTZ on the electrode surface starts from the oxidation of the adsorbed molecular edge of the Ar2N moiety rather than the central PTZ moiety covered by the bulky groups to produce Ar2N˙+–PTZ; it then quickly changes to more stable hole-shifted Ar2N–PTZ˙+ with large conformational change. These processes occur very rapidly, so that the cyclic voltammograms exhibit usual reversible waves (Fig. 1). In order to obtain insight into such hole-shift processes, we previously investigated photo-induced electron transfer of a (PTZ trimer)–anthraquinone (PTZ3–PTZ2–PTZ1–B–AQ, B: bridge) dyad4a in which electron transfer via through-bond interaction from PTZ trimer to the excited anthraquinone (AQ) to give PTZ3–PTZ2–PTZ1˙+–B–AQ˙−. The hole shift process (PTZ3–PTZ2–PTZ1˙+–B–AQ˙− → PTZ3–PTZ2˙+–PTZ1–B–AQ˙−) with large conformational change was directly monitored by the transient absorption spectroscopy using a time constant of 6 ns. Thus, the oxidation of (Ar2N)2–PTZ is associated with both neutral and radical cation geometries.
We also carried out theoretical calculation on the stereoelectronic effects by theoretical calculation using Gaussian 09 on the basis of (U)B3LYP/6-31G** level of theory in order to obtain a general relationship between structures and oxidation potentials.15,16 The calculated HOMO energy levels of 1a–e are not correlated to the oxidation potentials (HOMO energy of 1a (−5.18 eV) > 1e (−5.00 eV) > 1d (−4.58 eV) > 1b (−4.24 eV) > 1c (−4.19 eV)) (Fig. S6†). The calculated SOMO energy levels of 1a–e˙+ are in the order of 1a˙+ (−7.92 eV) > 1e˙+ (−7.64 eV) > 1c˙+ (−7.54 eV) > 1b (−7.53 eV) > 1d˙+ (−7.46 eV) (Fig. S4†) are also not correlated to oxidation potentials; the sequence agrees with the experimentally observed oxidation potentials except for the position of 1d (Fig. 4, inset). This consideration also indicates the importance of radical cationic geometry in the oxidation potentials of 1a–e. Finally, the stabilization-energy gain from the neutral to the radical cation state, i.e. the calculated heat of formation differences between optimized structures in neutral and radical cation states (ΔHF) are considered: the plots of ΔHF (−139.23 kcal mol−1 (1a), −119.70 (1b), −118.97 (1c), −124.98 (1d), −128.85 (1e)) vs. Eox1 provides a good correlation (correlation coefficient: R2 = 0.983; Fig. 4).
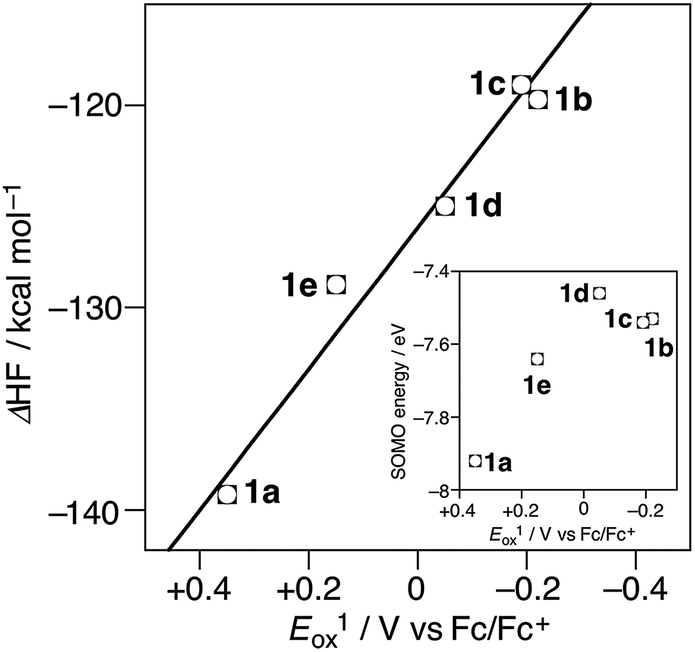 | ||
| Fig. 4 The plots of Eox1 of 1a–e vs. calculated heat of formation difference (ΔHF) between 1a–e and 1a–e˙+. (inset) The plots of Eox1 vs. SOMO energy of 1a–e˙+. | ||
As apparent from the crystal structures of 1a–e˙+, the central PTZ˙+ can be stabilized by the conjugation with the nitrogen p-orbitals in Ar2N-substituents. These π-conjugation gives rise to broad and intense intramolecular charge transfer (CT) bands in near infrared region: 1a˙+ (λmax = 1200 nm in dichloromethane), 1b˙+ (950 nm), 1c˙+ (918 nm), 1d˙+ (1118 nm), and 1e˙+ (963 nm) (Fig. 5). These differences are almost consistent with the results of theoretical calculations by TDDFT method (see, ESI†).
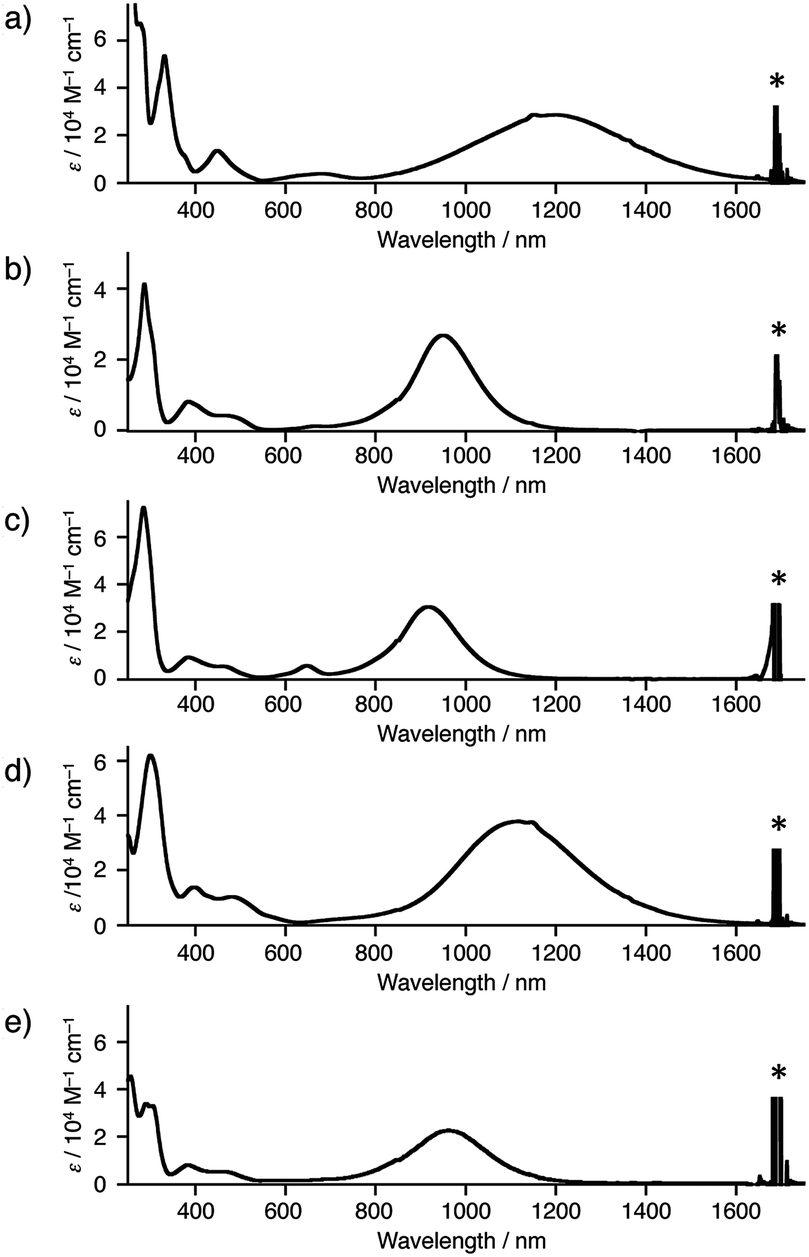 | ||
| Fig. 5 UV-vis NIR spectra of (a) 1a˙+·SbF6−, (b) 1b˙+·SbF6−, (c) 1c˙+·GaBr4−, (d) 1d˙+·SbF6−, and (e) 1e˙+·SbF6− in dichloromethane. Asterisks were denoted peaks of solvent. | ||
Conclusions
We designed and prepared a series of 3,7-bis(diarylamino)phenothiazines (Ar2N)2–PTZ (1a–f) and investigated relationship between their oxidation potentials and their molecular structures. X-ray crystal structural analyses and theoretical calculations indicated a unique stereoelectronic effect on PTZs, which strongly depends on the conformation of Ar2N-substituent.The oxidations of 1c and 1e are accompanied by the large conformational changes. In compound 1a, the freedom of Ar2N-substituent is restricted only by rotation of the carbazolyl group. Although the manner of stabilization of (Ar2N)2–PTZ˙+ (1a–e˙+) is highly substituent-dependent, these substituent-dependent oxidation potentials were shown to be proportional to the energy gain from the neutral state to the radical cation state.
The radical cationic species (Ar2N)2–PTZ˙+ were quite stable under aerobic conditions in both solution and the solid states. In addition, the radical cationic species 1a–e˙+ showed intense intermolecular CT bands in near infrared region.
Although this study was particularly investigated for the PTZ derivatives, the observed stereoelectronic substituent-effect must be uniformly applicable to electron rich aromatic or benzo-condensed π-conjugated systems.
Experimental
General information
1H and 13C NMR spectra were recorded on a Bruker NanoBay 300 spectrometer. MALDI-TOF-MS was measured on a Shimadzu-Kratos AXIMA-CFR Plus spectrometer using dithranol as a matrix reagent. Absorption spectra were measured with a JASCO V-750 UV-vis spectrometer and Hitachi U-3500L spectrometer. X-ray data were collected by a Rigaku Saturn CCD system with graphite monochromated Mo-Kα radiation. Melting points were measured with a hot-stage apparatus and the values are uncorrected. Redox potentials were measured using ALS electrochemical analyzer MODEL 610A in a conventional three-electrode cell equipped with a glassy carbon as a working electrode and a platinum wire as a counter electrode with a SCE reference electrode. The measurements were carried out with a sweep rate of 100 mV s−1 in suitable solvent containing 0.1 M tetra-n-butylammonium perchlorate (nBu4N+·ClO4−) as an electrolyte. The redox potentials were finally corrected by the ferrocene/ferrocenium couple (Fc/Fc+).Silica gel 60 (100–200 mesh) was used for column chromatography. All commercially available compounds were reagent grade and used without further purification. Dichloromethane (CH2Cl2), and acetonitrile (CH3CN) were dried and distilled over calcium hydride. Toluene, benzene, tetrahydrofuran (THF), and diethyl ether (Et2O) were dried and distilled over sodium. Ethanol (EtOH) and methanol (MeOH) were dried and distilled over magnesium.
Tris(4-bromophenyl)aminium hexafluoroantimonate (TBPA˙+·SbF6−) was prepared from tris(4-bromophenyl)amine and silver hexafluoroantimonate according to the literature with some modification.17 3,7-Dibromo-10-methyl-10H-phenothiazine (3) was prepared according to the literature.10 5,5'-(10-Tolyl-10H-phenothiazine-3,7-diyl)bis(10,11-dihydro-5H-dibenzo[b,f]azepine) (1b′) was prepared from 3,7-dibromo-10-p-tolyl-10H-phenothiazine and 10,11-dihydro-5H-dibenzo[b,f]azepine (4b) according to the synthetic method of 1b. Single crystals of 1b′ suitable for X-ray crystal structure analysis were obtained by recrystallization from toluene–EtOH.
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as eluent to give 1a as a white solid (218 mg, 80%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from CH2Cl2–MeOH. 1a: C37H25N3S; MW 543.69; mp 293–294 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.24 (d, J = 7.6 Hz, 4H), 7.51–7.26 (m, 18H), 3.54 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 147.93, 142.65, 142.06, 128.92, 128.55, 124.72, 124.15, 124.07, 123.00, 122.55, 121.98, 114.51, 34.60; HRMS (MALDI-TOF+) m/z calcd for C37H25N3S: 543.1769, found: 543.1764 [M+]; anal. calcd for C37H25N3S: C, 81.74; H, 4.64; N, 7.73. Found: C, 81.57; H, 4.72; N, 7.68.:1 v/v) as eluent to give 1b as a white solid (102 mg, 85%). 1b: C41H33N3S; MW 599.80; mp 271 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.33–7.24 (m, 16H), 6.68 (d, J = 8.9 Hz, 2H), 6.27 (dd, J = 8.9 and 2.6 Hz, 2H), 6.18 (d, J = 2.6 Hz, 2H), 3.10 (s, 3H), 2.89 (s, 8H); 13C NMR (75 MHz, THF-d8): δ (ppm) 144.82, 143.76, 138.45, 138.14, 130.69, 129.97, 126.76, 126.69, 124.08, 113.45, 111.67, 111.53, 34.12, 30.75; HRMS (MALDI-TOF+) m/z calcd for C41H33N3S: 599.2395, found: 599.2391 [M+]; anal calcd for C41H33N3S: C, 82.10; H, 5.55; N, 7.01. Found: C, 82.47; H, 5.77; N, 6.77.:1 v/v) as eluent to give 1c as a white solid (219 mg, 73%). Single crystals suitable for X-ray structure analysis were obtained by recrystallization from THF–MeOH. 1c: C41H29N3S; MW 595.76; mp 291 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57–7.41 (m, 16H), 6.87 (s, 4H), 6.52 (d, J = 8.8 Hz, 2H), 5.86 (dd, J = 8.8 and 2.8 Hz, 2H), 5.75 (d, J = 2.8 Hz, 2H), 2.99 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 144.43, 143.61, 138.36, 136.76, 130.52, 130.33, 130.29, 129.45, 126.82, 123.59, 112.93, 110.65, 110.51, 34.01; HRMS (MALDI-TOF+) m/z calcd for C41H29N3S: 595.2082, found: 595.2076 [M+].:1 v/v) as eluent to give 1d as a white solid (276 mg, 99%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from CH2Cl2–MeOH. 1d: C37H29N3S; MW 547.72; mp 219–220 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.26 (t, J = 7.7 Hz, 8H), 7.00–6.93 (m, 16H), 6.80 (s, 2H), 3.29 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 143.22, 139.30, 130.71, 127.70, 123.84, 123.81, 122.76, 121.42, 118.07, 117.75, 113.31, 107.57, 33.14; HRMS (MALDI-TOF+) m/z calcd for C37H29N3S: 547.2082, found: 547.2077 [M+]; anal. calcd for C37H29N3S: C, 81.14; H, 5.34; N, 7.67. Found: C, 80.88; H, 5.38; N, 7.52.:1 v/v) as eluent to give 1e as a white solid (161 mg, 82%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from toluene–hexane. 1e: C37H25N3S3; MW 607.81; mp 293 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.34–7.24 (m, 6H), 7.05 (dd, J = 7.5 and 1.4 Hz, 4H), 6.95 (td, J = 7.5 and 1.4 Hz, 4H), 6.85 (td, J = 7.5 and 1.4 Hz, 4H), 6.25 (dd, J = 7.5 and 1.4 Hz, 4H), 3.48 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 143.59, 142.49, 133.87, 128.41, 127.44, 124.79, 124.43, 123.35, 120.33, 118.22, 114.08, 113.95, 33.13; HRMS (MALDI-TOF+) m/z calcd for C37H25N3S3: 607.1211, found: 607.1206 [M+]; anal. calcd for C37H25N3S3: C, 73.12; H, 4.15; N, 6.91. Found: C, 73.28; H, 4.44; N, 6.81.:1) as eluent to give 1f as a white solid (103 mg, 90%). Single crystals suitable for X-ray structure analysis were obtained by recrystallization from THF–water. 1f: C37H25N3O2S; MW 575.69; mp > 300 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.31–7.23 (m, 6H), 6.74–6.65 (m, 12H), 5.96–5.93 (m, 4H), 3.46 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 145.57, 143.96, 134.38, 133.89, 129.94, 128.86, 125.58, 123.07, 121.06, 116.26, 115.03, 113.29, 34.99; HRMS (MALDI-TOF+) m/z calcd for C37H25N3O2S: 575.1667, found: 575.1663 [M+].
:1 v/v) as eluent to give 1a as a white solid (218 mg, 80%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from CH2Cl2–MeOH. 1a: C37H25N3S; MW 543.69; mp 293–294 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.24 (d, J = 7.6 Hz, 4H), 7.51–7.26 (m, 18H), 3.54 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 147.93, 142.65, 142.06, 128.92, 128.55, 124.72, 124.15, 124.07, 123.00, 122.55, 121.98, 114.51, 34.60; HRMS (MALDI-TOF+) m/z calcd for C37H25N3S: 543.1769, found: 543.1764 [M+]; anal. calcd for C37H25N3S: C, 81.74; H, 4.64; N, 7.73. Found: C, 81.57; H, 4.72; N, 7.68.:1 v/v) as eluent to give 1b as a white solid (102 mg, 85%). 1b: C41H33N3S; MW 599.80; mp 271 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.33–7.24 (m, 16H), 6.68 (d, J = 8.9 Hz, 2H), 6.27 (dd, J = 8.9 and 2.6 Hz, 2H), 6.18 (d, J = 2.6 Hz, 2H), 3.10 (s, 3H), 2.89 (s, 8H); 13C NMR (75 MHz, THF-d8): δ (ppm) 144.82, 143.76, 138.45, 138.14, 130.69, 129.97, 126.76, 126.69, 124.08, 113.45, 111.67, 111.53, 34.12, 30.75; HRMS (MALDI-TOF+) m/z calcd for C41H33N3S: 599.2395, found: 599.2391 [M+]; anal calcd for C41H33N3S: C, 82.10; H, 5.55; N, 7.01. Found: C, 82.47; H, 5.77; N, 6.77.:1 v/v) as eluent to give 1c as a white solid (219 mg, 73%). Single crystals suitable for X-ray structure analysis were obtained by recrystallization from THF–MeOH. 1c: C41H29N3S; MW 595.76; mp 291 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57–7.41 (m, 16H), 6.87 (s, 4H), 6.52 (d, J = 8.8 Hz, 2H), 5.86 (dd, J = 8.8 and 2.8 Hz, 2H), 5.75 (d, J = 2.8 Hz, 2H), 2.99 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 144.43, 143.61, 138.36, 136.76, 130.52, 130.33, 130.29, 129.45, 126.82, 123.59, 112.93, 110.65, 110.51, 34.01; HRMS (MALDI-TOF+) m/z calcd for C41H29N3S: 595.2082, found: 595.2076 [M+].:1 v/v) as eluent to give 1d as a white solid (276 mg, 99%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from CH2Cl2–MeOH. 1d: C37H29N3S; MW 547.72; mp 219–220 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.26 (t, J = 7.7 Hz, 8H), 7.00–6.93 (m, 16H), 6.80 (s, 2H), 3.29 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 143.22, 139.30, 130.71, 127.70, 123.84, 123.81, 122.76, 121.42, 118.07, 117.75, 113.31, 107.57, 33.14; HRMS (MALDI-TOF+) m/z calcd for C37H29N3S: 547.2082, found: 547.2077 [M+]; anal. calcd for C37H29N3S: C, 81.14; H, 5.34; N, 7.67. Found: C, 80.88; H, 5.38; N, 7.52.:1 v/v) as eluent to give 1e as a white solid (161 mg, 82%). Single crystals suitable for X-ray crystal structure analysis were obtained by recrystallization from toluene–hexane. 1e: C37H25N3S3; MW 607.81; mp 293 °C (decomp.); 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.34–7.24 (m, 6H), 7.05 (dd, J = 7.5 and 1.4 Hz, 4H), 6.95 (td, J = 7.5 and 1.4 Hz, 4H), 6.85 (td, J = 7.5 and 1.4 Hz, 4H), 6.25 (dd, J = 7.5 and 1.4 Hz, 4H), 3.48 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 143.59, 142.49, 133.87, 128.41, 127.44, 124.79, 124.43, 123.35, 120.33, 118.22, 114.08, 113.95, 33.13; HRMS (MALDI-TOF+) m/z calcd for C37H25N3S3: 607.1211, found: 607.1206 [M+]; anal. calcd for C37H25N3S3: C, 73.12; H, 4.15; N, 6.91. Found: C, 73.28; H, 4.44; N, 6.81.:1) as eluent to give 1f as a white solid (103 mg, 90%). Single crystals suitable for X-ray structure analysis were obtained by recrystallization from THF–water. 1f: C37H25N3O2S; MW 575.69; mp > 300 °C; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.31–7.23 (m, 6H), 6.74–6.65 (m, 12H), 5.96–5.93 (m, 4H), 3.46 (s, 3H); 13C NMR (75 MHz, THF-d8): δ (ppm) 145.57, 143.96, 134.38, 133.89, 129.94, 128.86, 125.58, 123.07, 121.06, 116.26, 115.03, 113.29, 34.99; HRMS (MALDI-TOF+) m/z calcd for C37H25N3O2S: 575.1667, found: 575.1663 [M+].Theoretical calculations
All calculations were carried out on the basis of DFT with the (U)B3LYP exchange–correlation functional and using Gaussian 09 program packages.15 The basis set used was 6-31G** for all atoms. The criterion for the SCF convergence was 10−9. Molecular orbitals for 1a–e and 1a–e˙+ were calculated by using the molecular geometries obtained from the optimized structures. The transition energies of 1a–e˙+ were estimated by TD-DFT calculation (UB3LYP/6-31G**).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by Grant-in-Aid for Scientific Research on Innovative Areas “π-System Figuration: Control of Electron and Structural Dynamism for Innovative Functions (2601)” (JSPS KAKENHI Grant Number JP26102005 for S. S.), “Stimuli-responsive Chemical Species for the Creation of Functional Molecules (2468)” (JSPS KAKENHI Grant Number JP15H00956 for K. O.), Grant-in-Aid for Scientific Research (JSPS KAKENHI Grant Number JP26288041 for K.O. and JP17K05783 for S. S.), and Iketani science and technology foundation (for S. S.). The authors thank Prof. Dr Hiroyuki Miyake and Prof. Dr Satoshi Shinoda of Graduate School of Science, Osaka City University for measuring UV-vis-NIR spectra.Notes and references
- (a) Y. Shirota, J. Mater. Chem., 2000, 10, 1 RSC; (b) M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442 CrossRef CAS; (c) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953 CrossRef CAS PubMed; (d) Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC.
- (a) P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895 RSC; (b) S. C. Blackstock and T. D. Selby, in Magnetic Properties of Organic Materials, ed. P. M. Lahti, Marcel Dekker, New York, 1999, ch. 9, p. 165 Search PubMed; (c) R. J. Bushby, in Magnetic Properties of Organic Materials, ed. P. M. Lahti, Marcel Dekker, New York, 1999, ch. 10, p. 179 Search PubMed; (d) S. Suzuki, K. Yoshida, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2013, 52, 2499 CrossRef CAS PubMed; (e) K. Yoshida, S. Suzuki, M. Kozaki and K. Okada, Chem. Lett., 2016, 45, 203 CrossRef CAS.
- (a) L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC; (b) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC; (c) Y. Hua, S. Chang, D. Huang, X. Zhou, X. Zhu, J. Zhao, T. Chen, W.-Y. Wong and W.-K. Wong, Chem. Mater., 2013, 25, 2146 CrossRef CAS; (d) G. Kim, H. R. Yeom, S. Cho, J. H. Seo, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 1847 CrossRef CAS; (e) G. Sang, Y. Zou and Y. Li, J. Phys. Chem. C, 2008, 112, 12058 CrossRef CAS.
- (a) Y. Karimata, S. Suzuki, M. Kozaki, K. Kimoto, K. Nozaki, H. Matsushita, N. Ikeda, K. Akiyama, D. Kosumi, H. Hashimoto and K. Okada, J. Phys. Chem. A, 2014, 118, 11262 CrossRef PubMed; (b) R. Sugimura, S. Suzuki, M. Kozaki, K. Keyaki, K. Nozaki, H. Matsushita, N. Ikeda and K. Okada, Res. Chem. Intermed., 2013, 39, 185 CrossRef CAS; (c) S. Suzuki, M. Kozaki, K. Nozaki and K. Okada, J. Photochem. Photobiol., C, 2011, 12, 269 CrossRef CAS.
- (a) H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985 CrossRef CAS; (b) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181 CrossRef CAS PubMed; (c) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677 RSC.
- (a) T. J. LePage and R. Breslow, J. Am. Chem. Soc., 1987, 109, 6412 CrossRef CAS; (b) S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 3683 CrossRef CAS PubMed.
- For 3-dimethylamino-substituted phenothiazines for instance: J. Bae, L. E. McNamara, M. A. Nael, F. Mahdi, R. J. Doerksen, G. L. Bidwell III, N. I. Hammer and S. Jo, Chem. Commun., 2015, 51, 12787 RSC ; For 3-alkyloxyphenothiazines: See, ref. 3c; For 3-arylphenothiazines: L. Yao, Y. Pan, X. Tang, Q. Bai, F. Shen, F. Li, P. Lu, B. Yang and Y. Ma, J. Phys. Chem. C, 2015, 119, 17800 Search PubMed.
- I. S. Pereţeanu and T. J. J. Müller, Org. Biomol. Chem., 2013, 11, 5127 Search PubMed.
- T. Okamoto, M. Kuratsu, M. Kozaki, K. Hirotsu, A. Ichimura, T. Matsushita and K. Okada, Org. Lett., 2004, 6, 3493 CrossRef CAS PubMed.
- H.-H. Lin and C.-C. Chang, Dyes Pigm., 2009, 83, 230–236 CrossRef CAS.
- CCDC 1572168 (1a), 1572169 (1a˙+·SbF6−), 1572170 (1b′), 1572171 (1b˙+·SbF6−), 1572172 (1c), 1572173 (1c˙+·GaBr4−), 1572175 (1d), 1572186 (1d˙+·SbF6−), 1572187 (1e), 1572188 (1e˙+·SbF6−), 1572500 (1f), and 1572190 (1f˙+·GaBr4−) contain the supplementary crystallographic data for this paper.†.
- M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Inorg. Chem., 2007, 46, 10153 CrossRef CAS PubMed.
- The Cyclic voltammograms of 1a–e showed second one-electron oxidation process, suggesting the strong donating abilities of the Ar2N-substituents. The detailed investigations for molecular structures and electronic properties of the two-electron oxidized species are underway.
- Neutral phenoxazine has a more planar structure because of the shorter C−O bond length (∼1.41 Å, Fig. S2†) compared to the C−S bond length (∼1.76 Å) in neutral phenothiazine.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- The geometries of 1a–e and their radical cations were optimized using Gaussian 09 with density functional theory at the (U)B3LYP/6–31G* level of theory.
- S. Suzuki and K. Okada, in Organic Redox Systems, ed. T. Nishinaga, John Wiley & Sons, New Jersey, 2016, ch. 8, p. 269 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1572168, 1572169, 1572170, 1572171, 1572172, 1572173, 1572175, 1572186, 1572187, 1572188, 1572190 and 1572500. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12600j |
| This journal is © The Royal Society of Chemistry 2017 |