
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic graphene solid-phase extraction in the determination of polycyclic aromatic hydrocarbons in water†
Liling Panga,
Wanfeng Zhangb,
Weiya Zhangc,
Pin Chena,
Jing Yua,
Gang-Tian Zhu a and
Shukui Zhu*a
a and
Shukui Zhu*a
aState Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan, 430074, PR China. E-mail: shukuizhu@126.com
bState Key Laboratory of Isotope Geochemistry, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, P. O. Box 1131, Wushan, Guangzhou 510640, PR China
cTesting & Technology Centre for Industrial Products, Shenzhen Entry-exit Inspection and Quarantine Bureau, 518067, PR China
First published on 22nd November 2017
Abstract
In this work, magnetic graphene nanocomposite Fe3O4/rGO was fabricated by using a solvothermal method and applied to the magnetic solid-phase extraction (MSPE) of polycyclic aromatic hydrocarbons (PAHs) in environmental water samples, followed by gas chromatography-mass spectrometry (GC-MS). Several parameters affecting the extraction efficiency, such as amount of magnetic graphene, extraction time, desorption conditions, were studied in detail. The results showed that Fe3O4/rGO has an extremely high adsorption capacity for PAHs, only 10 mg of the adsorbent was needed for 100 mL of water sample, and the PAHs could be completely desorbed with 100 μL of desorption solvent. Under the optimized conditions, a good linearity was obtained in the concentration range of 0.005–5 μg L−1 for 10 low ring PAHs from naphthalene to chrysene, 0.1–5 μg L−1 for benzo[b]fluoranthene, benzo[k]fluoranthene and benzo(a)pyrene. The limits of detection (LOD, S/N = 3) of the method were in the range between 0.02–14.3 ng L−1. The recoveries for PAHs tested in spiked water samples were in the range 75.6–112.4% with relative standard deviations (RSD) ranging from 0.1–9.5%. Finally, the method was successfully applied for the analysis of real water samples in the search for PAHs.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a class of semivolatile organic compounds with two or more fused aromatic rings. As we know, PAHs are formed by the incomplete combustion of organic matter and generated whenever fossil fuels or vegetation are burned. These environmental pollutants are ubiquitous under the influence of atmospheric circulation and the water cycle, even in the Arctic and Antarctica in which there is almost no human activity.1 The NHANES have shown that humans, starting from fetuses, are under the toxic effects of PAHs, which can increase the risk of adverse health effects.2 Moreover, DNA adducts in all cell types and carcinogenic PAHs and/or B[a]P in the inhaled air showed positive significant correlations (r = 0.38–0.45, P < 0.001).3 Hence, the toxicity of PAHs has justified their inclusion in environmental legislation worldwide. For instance, the U.S. Environmental Protection Agency (EPA) developed ambient water quality criteria and gave priority to 16 PAHs in control in 1985.The relevance of water in environmental research is related to the hydrophobic properties of organic pollutants, referring to the octanol–water partition coefficient, and their exchange between water and particulate phase in water. Particularly, PAHs barely have an affinity for water, resulting in a low pollution concentration, and so sample enrichment is essential before instrument analysis. Meanwhile, more sensitive analytical methods aimed at improving the current ones are required to meet environment quality and eco-toxicological considerations. Traditional methods such as liquid–liquid extraction (LLE) are laborious, time-consuming, uneconomic and solvent-wasting.4,5 In our group, gas purge microsyringe extraction (GP-MSE) and micro-solid phase extraction based on a glass pipette have been developed recently and shows some outstanding advantages, but needs to couple other sample pretreatment steps when analyzing water samples.6–9
Magnetic solid phase extraction (MSPE), developed from SPE, has drawn great attention for its advantages of extraction efficiency and material reuse.10–14 Magnetic or magnetizable adsorbents can be dispersed in sample matrix for preconcentration of target analytes, and isolated from matrix by using an external magnet. Besides, the magnetic substrates, such as nano-iron oxide, can enhance the extraction efficiency for their large uniform nanometer-scale pore structure, high specific surface area and large pore volume.15–17 However, an unavoidable problem associated with particles in this size range is their intrinsic instability, which tends to lead to formation of agglomerates. Moreover, naked metallic nanoparticles are easily oxidized in air, which result in loss of magnetism and dispersibility.18 Fortunately, the magnetic substrates can be assembled specialized with different functional groups for various analytes and shown positive effect.18–21 Furthermore, not only the specific surface area, but the high π-electron density of adsorptions is important for phenyl adsorbates.22–24 For example, although the surface area is only 53.8 m2 g−1, the adsorption capacity of STY/DVB/GMA polyHIPE monolith is 16.21 mg Ant per g polyHIPE polymers.24 As a single layer sp2 bonded carbon materials with two dimensional lattices, graphene has excellent physico-chemical properties including a remarkably stable structure and properties, large surface area and delocalized π-electron system. Many studies have shown that graphene can stabilize the dispersion of metals and metal oxides, in which graphene coupled with Fe3O4 has been widely used in battery, medical imaging and sensors etc.13,25–30 There are also applications in analysis of PAHs,30 but they were limited to PAHs with low molecular weight.31–33
In this study, a novel nanocomposite of reduced graphene oxide (Fe3O4/rGO) was synthesized as an adsorbent for MSPE, and coupled with GC-MS for the simultaneous determination of PAHs from low to high molecular weight (2–5 rings). Several parameters affecting the extraction efficiency, such as amount of magnetic graphene, extraction time, the type and volume of desorption solvent, desorption time and organic modifier, were studied in detail. The method showed high sensitivity for analysis of PAHs in real water samples.
2. Experimental section
2.1 Chemicals and materials
PAHs standards dissolved in binary mixed solvents benzene/dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) were purchased from ANPEL Laboratory Technologies Inc. (Shanghai, China), including naphthalene (Nap), acenaphthylene (Acp), acenaphthene (Ace), fluorene (Flu), phenanthrene (Phe), anthracene (Ant), fluoranthene (Flt), pyrene (Pyr), benz[a]anthracene (BaA), chrysene (Cry), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF) and benzo[a]pyrene (BaP). The concentration of each compound in the mixture was 2000 mg L−1. HPLC-grade toluene and n-hexane were purchased from Tedia Company (Fairfield, OH, USA), methanol was purchased from Merck Kgaa (Darmstadt, Germany) and acetone was purchased from Mreda Technology Inc. (USA). Graphite was purchased from Aladdin (Shanghai, China), analytical-grade 2-propanol, ethylene glycol, concentrated sulfuric acid (H2SO4), hydrochloric acid (HCl), potassium thiosulfate (K2S2O4), phosphorus pentoxide (P2O5), potassium permanganate (KMnO4), ferric trichloride hexahydrate (FeCl3·6H2O), sodium acetate (NaAc) and ascorbic acid (VC) were purchased from Sinopharm Chemical Reagents Co., Ltd (Shanghai, China). The ultrapure water was obtained from a Smart-N (Heal Force NW, China) water purification system.
:1 v/v) were purchased from ANPEL Laboratory Technologies Inc. (Shanghai, China), including naphthalene (Nap), acenaphthylene (Acp), acenaphthene (Ace), fluorene (Flu), phenanthrene (Phe), anthracene (Ant), fluoranthene (Flt), pyrene (Pyr), benz[a]anthracene (BaA), chrysene (Cry), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF) and benzo[a]pyrene (BaP). The concentration of each compound in the mixture was 2000 mg L−1. HPLC-grade toluene and n-hexane were purchased from Tedia Company (Fairfield, OH, USA), methanol was purchased from Merck Kgaa (Darmstadt, Germany) and acetone was purchased from Mreda Technology Inc. (USA). Graphite was purchased from Aladdin (Shanghai, China), analytical-grade 2-propanol, ethylene glycol, concentrated sulfuric acid (H2SO4), hydrochloric acid (HCl), potassium thiosulfate (K2S2O4), phosphorus pentoxide (P2O5), potassium permanganate (KMnO4), ferric trichloride hexahydrate (FeCl3·6H2O), sodium acetate (NaAc) and ascorbic acid (VC) were purchased from Sinopharm Chemical Reagents Co., Ltd (Shanghai, China). The ultrapure water was obtained from a Smart-N (Heal Force NW, China) water purification system.
2.2 Sample preparation
A stock solution (containing 5 mg L−1 of each analyte) was prepared by diluting the standard PAHs solution with methanol and was stored at 4 °C in the refrigerator. Working solutions were prepared daily by appropriate dilution of the stock solution with ultrapure water before use.Real water samples, including drinking water and river water, were collected from commercially mineral water and Hanjiang River, which is the branch of Yangtze River. The river water sample was filtered through a 0.45 μm membrane to eliminate particulate matters and stored in brown glass bottles before analysis.
2.3 Preparation of Fe3O4/rGO and MSPE procedure
The synthetic routes for Fe3O4/rGO and MSPE procedure are illustrated in Scheme 1. GO was synthesized from natural graphite based on the modified Hummers method, which is widely used to prepared GO.25,31 The GO colloid was prepared through two stages. The first stage was a modest and slow pro-oxide process as follows: 3.0 g of natural graphite, 2.5 g of K2S2O4 and 2.5 g of P2O5 were added into 15 mL of concentrated H2SO4. With vigorous stirring, the temperature of the mixture was maintained at 80 °C for 6 h.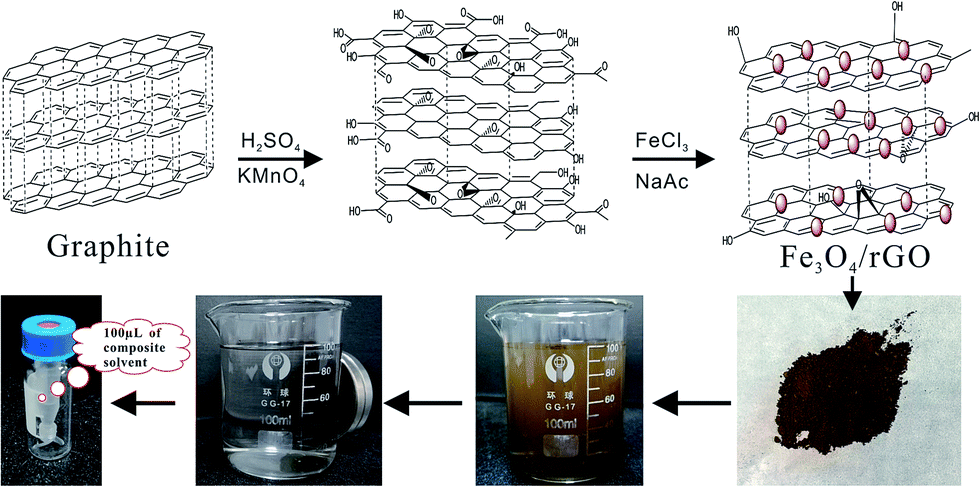 | ||
| Scheme 1 Schematic of the synthesis of Fe3O4/rGO and MSPE procedure for analysis of PAHs in water. | ||
The resulting suspension could not be filtered until it was cooled and thoroughly precipitated. The solid mixture was washed with ultrapure water and dried at 60 °C under vacuum as pre-oxidation product. The second stage was a further oxide process. Briefly, 0.6 g of pre-oxidation product was added into 21.8 mL of concentrated H2SO4. With vigorous stirring, 3 g of KMnO4 was gradually added, and the temperature of the mixture was controlled under 5 °C for 1 h. The ice bath was then removed, and the temperature of the mixture was maintained at 35 °C for 2 h. Next, 45 mL of ultrapure water was slowly added into the mixture in ice bath to prevent the temperature rising violently. The temperature of the mixture was maintained at 35 °C for 2 h once again. Then vast water was used to terminate reaction, and shortly, 4 mL of 30% H2O2 aqueous solution were added with stirring. The resulting suspension was filtered and washed with dilute HCl aqueous solution under the filter as well as with ultrapure water under centrifuge. Finally, the resulting solution was dried naturally and the khaki GO product was obtained.
Fe3O4/rGO was synthesized by the in-site solvothermal method in the presence of graphene.34 Firstly, a stably dispersed graphite oxide ethylene glycol solution was prepared by exfoliation of 75 mg of graphite oxide via ultrasonication for 2 h. Secondly, 10 mg of VC was added in the solution, and kept it under ultrasonic field for 1 h to reduce the GO initially. Thirdly, 1.35 g of FeCl3·6H2O and 3.6 g of NaAc were added and a uniform solution was formed. Then transmit the solution to Teflon-lined stainless steel autoclave and heat at 180 °C for 8 h. Finally, the precipitate was alternately washed with water and ethanol for final cleaning. The product Fe3O4/rGO was obtained after drying at 50 °C under vacuum for 24 h.
10 mg of Fe3O4/rGO was dispersed into 100 mL of aqueous sample under ultrasound for 8 min for extraction. Then, the NdFeB magnet was held at the bottom of the flask and the Fe3O4/rGO adsorbent was isolated from the suspension. After 10 min, the suspension became clear and then was decanted. The residual adsorbent was transmitted into a 5 mL vial, washed with ultrapure water and dried under a mild stream of nitrogen. Then it was eluted with 100 μL of binary mixed solvents of toluene and acetone (3:1 v/v) under water bath at 50 °C for 5 min. After cooling to room temperature, 1.0 μL of the final extract is injected in the GC/MS system for analysis.
2.4 Instrumentation and methods
Powder X-ray diffraction (XRD) patterns of materials were obtained with a diffractometer (Rigaku D/max-β B) using Cu Kα radiation source. The microcrystalline structure and surface characteristics of the samples were characterized by using Hitachi S-4800 scanning electron microscopy (SEM). Fourier transform infrared (FTIR) spectra were obtained with a Bruker Equinox 55 FT-IR instrument.GC-MS analysis was performed on a 7890A GC interfaced to 5975C mass spectrometer system (Agilent Technologies, USA). The GC separation was equipped with a DB-5MS column (30 m × 0.25 mm × 0.25 μm, Agilent Technologies, USA). Helium (purity 99.999%) was employed as the carrier gas at a flow rate of 1.0 mL min−1. Samples (1 μL) were injected in splitless mode. The injector temperature was set at 300 °C. The GC oven was initially held at 40 °C for 1 min and then programmed to 300 °C at 7 °C min−1 and held for 5 min. The solvent delay time was 10 min. PAHs standards and samples were analyzed in selective ion monitoring (SIM) mode for quantitative determination of the analytes. The masses monitored by the detector were set as follows: 10–15 min, m/z 128, 129, 127, 102; 15–20 min, m/z 152, 153, 151, 154; 20–22 min, m/z 166, 165, 167, 139; 22–26 min, m/z 178, 176, 179, 152; 26–30 min, m/z 202, 203, 200, 101; 30–35 min, m/z 228, 226, 229, 227, 252; 35–43 min, m/z 253, 252, 250, 126. PAHs standards and samples were analyzed in SIM mode for quantitative determination of the analytes: Nap, m/z 128, 129, 127, 102; Acp, m/z 152, 153, 151; Ace, 153, 154, 152; Flu, m/z 166, 165, 167; Phe, m/z 178, 176, 179; Ant, m/z 178, 179, 176; Flt, m/z 202, 203, 200, 101; Pyr, m/z 202, 200, 203, 101; Cry, m/z 228, 226, 229; BaA, m/z 228, 226, 227, 229, 252; BbF, m/z 252, 253, 250; BkF, m/z 252, 250, 126; BaP, m/z 252, 253, 250, 126.
3. Results and discussion
3.1 Characterization of Fe3O4/rGO nanoparticles
The structures of Fe3O4/rGO nanoparticles were analysed by FTIR, Raman, XRD and SEM. Fig. 1A is FTIR image of GO, Fe3O4 and Fe3O4/rGO, which showed the changes of group functions accompanying with the synthesis of Fe3O4/rGO. It can be seen from the FTIR image of pristine GO that the signals of four groups of obvious transmission peak at 3430 cm−1 (O–H stretching), 1627 cm−1 (O–H bending vibration), 1220 cm−1 and 1052 cm−1 (C–O stretching), 1700 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching). Based on the proofs that the O–H stretching was much stronger than C–O stretching and CO stretching, even the CO stretching was too weak to discern, the modified Hummers method was mild enough to prepare GO with fewer unrecoverable defects to aromatic rings. The functional groups of Fe3O4 nanoparticles were Fe–O (584 cm−1) and O–H (3390 cm−1 and 1630 cm−1), which was turned out by FTIR image. The spectrum of the Fe3O4/rGO was assumed integrated characteristics similar to that of Fe3O4 and GO. Thereinto, the intensity of O–H stretching vibration (3390 cm−1) increased. These spectral characteristics proved that the combination of Fe3O4 and GO was successful. It's worth noting that the bands attributed to the C–O stretching (1220 cm−1 and 1052 cm−1) strengthened, and CC stretching (1660 cm−1), C–C stretching in benzene rings (1600 cm−1, 1580 cm−1 and 1440 cm−1) appeared. The FTIR results thus demonstrated that the GO was successfully reduced with VC and functionalized with Fe3O4.
O stretching). Based on the proofs that the O–H stretching was much stronger than C–O stretching and CO stretching, even the CO stretching was too weak to discern, the modified Hummers method was mild enough to prepare GO with fewer unrecoverable defects to aromatic rings. The functional groups of Fe3O4 nanoparticles were Fe–O (584 cm−1) and O–H (3390 cm−1 and 1630 cm−1), which was turned out by FTIR image. The spectrum of the Fe3O4/rGO was assumed integrated characteristics similar to that of Fe3O4 and GO. Thereinto, the intensity of O–H stretching vibration (3390 cm−1) increased. These spectral characteristics proved that the combination of Fe3O4 and GO was successful. It's worth noting that the bands attributed to the C–O stretching (1220 cm−1 and 1052 cm−1) strengthened, and CC stretching (1660 cm−1), C–C stretching in benzene rings (1600 cm−1, 1580 cm−1 and 1440 cm−1) appeared. The FTIR results thus demonstrated that the GO was successfully reduced with VC and functionalized with Fe3O4.
 | ||
| Fig. 1 (A) FTIR spectra of GO, Fe3O4 and Fe3O4/rGO; (B) Raman spectra of Fe3O4/rGO and GO; (C) X-ray diffraction patterns of GO, Fe3O4 and Fe3O4/rGO; SEM images of (D) GO, (E) Fe3O4 and (F) Fe3O4/rGO. | ||
The Raman spectra of graphite is very sensitive to the structural changes during the chemical reactions.35 It is stated that the spectra of graphite with high crystallinity has two sharp band at 1580 cm−1 (G band) and 1350 cm−1 (D band), respectively.36 The intensity ratio of the D and G band (ID/IG) is a useful indicator to evaluate the ordered and disordered crystal structures of carbon, and the larger the ID/IG ratio, the higher the defect density.36,37 The value of ID/IG is 0.34 for graphite, and it increases to 1.99 for GO (Fig. 1B), indicating the introduction of many defect sites to GO by the oxidative treatment of graphite. As a comparison, The ID/IG for Fe3O4/rGO reduces to 1.7, which could be attributed to reduction with ascorbic acid and EDG for solvothermal reduction, and still be larger than graphite, suggesting that D band is dominated by graphitic carbon.
XRD image indicated that GO, Fe3O4 and Fe3O4/rGO had integrated structure (Fig. 1C). A sharpen reflection peak at ca. 9.4° corresponding to the (002) reflection was examined in GO. The intraplanar lattice spacing was 0.94 nm calculated by the Debye–Scherrer formula, much higher than graphite (0.34 nm). A number of prominent Bragg reflections at ca. 30.3°, 35.6°, 43.2°, 57.3°, 62.9° matched with the standards indices of crystal face of magnetic Fe3O4, inverse spinel, corresponding to (220), (311), (400), (511) and (440) planes. However, the only special part was that the reflection peak of GO disappeared. It resulted from the low concentration of rGO and the inserting of Fe3O4 between rGO layers dispersion of magnetite between layers of graphene, which has been proved by the SEM image (Fig. 1D–F). It can be seen that sheet-like structure was smooth surfaces and crumpled edges in GO, globular clusters had a uniform size in Fe3O4 and the magnetite dispersed in interlamination and surface of graphene.
3.2 Optimization of the extraction parameters
Hydrophilic–lipophilic balance and binding energy between PAHs and adsorbent are the key factors to affect the ability of extraction and desorption. In order to obtain high extraction efficiency, some parameters including the amount of sorbent material, the extraction time and extraction temperature, the desorption solvent and desorption time, the effect of organic modifier, were evaluated. All the experiments were performed using 5 μg L−1 of PAHs aqueous samples by mixing an appropriate amount of the stock solution with ultrapure water. The detailed extraction conditions in the following experiments were shown in (ESI†).In order to choose the optimum dosage of the adsorbent (Fe3O4/rGO) for the adsorption of the PAHs, different amounts of the Fe3O4/rGO were investigated in the range from 3.0 to 15.0 mg. Fig. 2A indicated that the peak areas increased for 2–3 rings PAHs with increasing sorbent material, while high ring PAHs decreased. When the amount of adsorbent materials was more than 10 mg, the test signal of PAHs was not improved. On the contrary, the peak area of most PAHs decreased, especially 4–5 rings PAHs. It is considered that more rings for PAHs could achieve stronger π-π conjugation with rGO, to the extent, were more difficult to elute with more sorbents.16,38 On all account, 10 mg of Fe3O4/rGO was chosen for optimal dosage in optimization experiments.
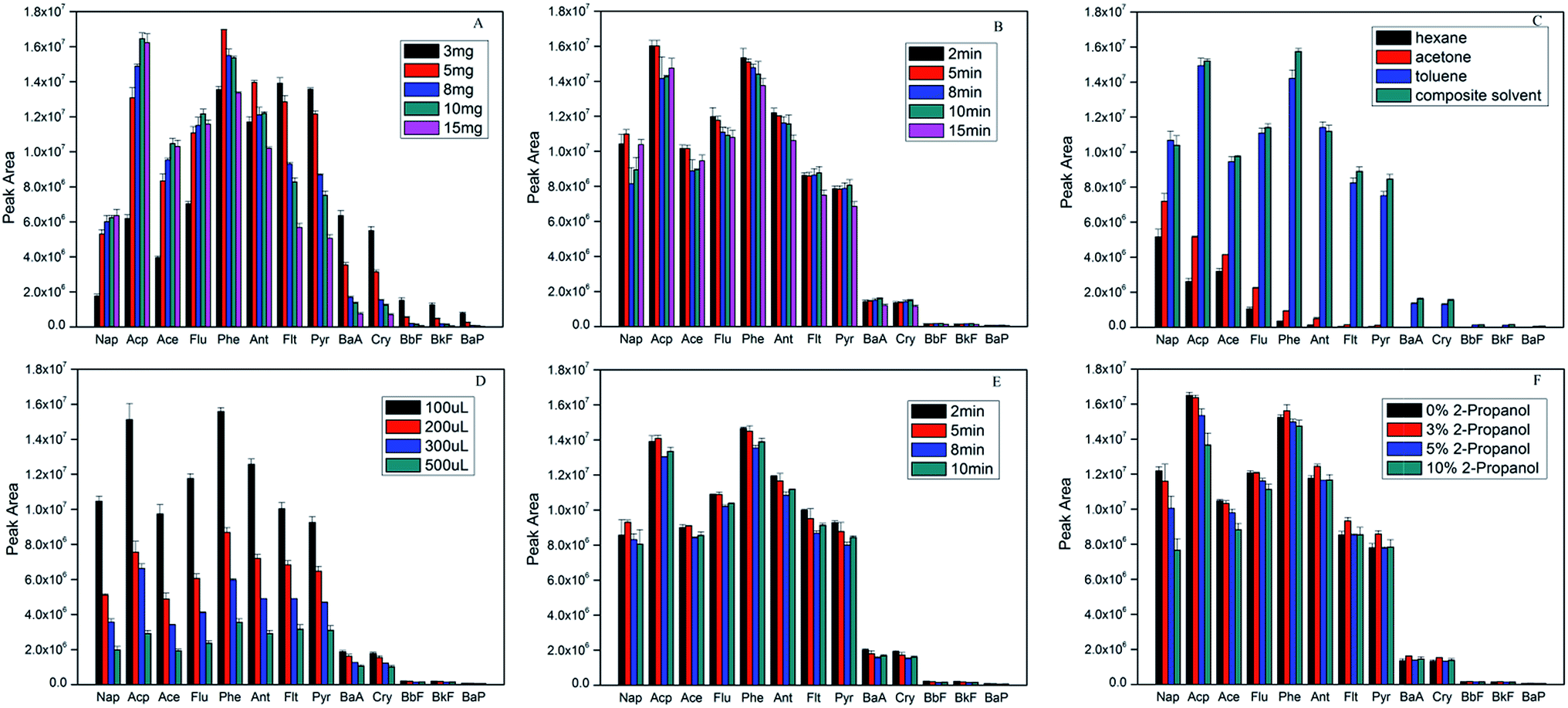 | ||
| Fig. 2 Effect of (A) sorbent amount, (B) extraction time, (C) desorption solvent type, (D) desorption solvent amount, (E) desorption time and (F) organic modifier on extraction efficiency. | ||
Generally, adsorption and desorption happen simultaneously during the process of extraction. The extracting efficiency will increase until adsorption and desorption come to dynamic balance. In this study, the effect of the extraction time was investigated in the range of 2 to 15 min. Fig. 2B showed the extraction time profile. Once the materials were dispersed evenly, the signals were almost of no differences even with more extraction time than 5 min. However, the peak areas decreased little by little after 8 min. It is believed that ultrasonic may strengthen the back-diffusion of analytes, which frequently occurred in many microextraction procedures.33
Desorption capabilities of solvents were assessed with four solvents including n-hexane, toluene, acetone and a binary mixed solvents of toluene and acetone (3:1 v/v). Fig. 2C showed the results of the comparative studies. It was observed that composite solvent gave the highest chromatographic response, followed by toluene, acetone and n-hexane. Taken into consideration that hexane and acetone have the strongest hydrophobicity and polarity respectively, the strong van der Waals interaction between the analytes and benzene rings might be responsible for this observation. In addition, the interaction in a model complex of HCHO-benzene is much stronger (1.87 kcal mol−1) than a CH4–benzene interaction (0.53 kcal mol−1).39 Obviously, composite solvent as desorption solvent for PAHs analysis in this current method was borne out by the experimental observations.
The volume of desorption solvent and desorption time are vital for the desorption efficiency. Fig. 2D showed the influence of the volume of desorption solvent evaluated ranging from 100 μL to 500 μL. It could be seen that the smaller the volume of binary mixed solvents was used, the higher concentration of PAHs can be obtained in the desorption solution. Therefore, 100 μL of composite solvent was adopted in this study.
Moreover, desorption time was also studied from 2 to 10 min (shown in Fig. 2E). The peak areas of analytes did not significantly increase for most PAHs with more desorption time. At the same time, all signals slightly decreased after 5 min for desorption. All things considered, 5 min is a reasonable compromise.
Organic modifier was studied to evaluate whether PAHs can adsorb onto the surfaces of containers or not.1,40,41 Fig. 2F showed the results of adding different percentages of 2-propanol to the sample solution. The results demonstrated that the peak areas of 2–3 rings of PAHs hardly changed while those of high ring PAHs significantly increased with 3% 2-propanol. On the other hand, the peak areas for all PAHs decreased when adding 2-propanol more than 3%. It can be explained that high ring PAHs was more hydrophobic so that they were easier to be adsorbed onto the surfaces of containers.33 However, the excessive 2-propanol could probably lead up to the increase of PAHs solubility in the solvent that ironically reduced their availability to the extraction device.1,40
On the basis of the foregoing discussion, the optimal extraction conditions were as follows: 10 mg of Fe3O4/rGO per 100 mL solution, sample solution containing 3% 2-propanol (v/v), 5 min extraction time, 100 μL of binary mixed solvents (toluene:acetone = 3:1, v/v) as the desorption solution, 5 min desorption time. All the following experiments were carried out under these optimal conditions.
3.3 Method validation
Under the optimal extraction conditions mentioned above, proposal method was estimated and validated. Using spiked ultrapure water samples, the factors that affect the analytical performance, such as linearity (R2), linear range, limits of detection (LODs) were investigated. The analytical results were listed in Table 1 and the GC/MS-SIM chromatogram of PAHs spiked at 5 μg L−1 was shown in Fig. 3.| Analytes | Linear range (μg L−1) | Correlation coefficient (R2) | LOD (ng L−1) | RSD (%) n = 5 |
|---|---|---|---|---|
| a Results obtained from the analysis of samples spiked at a concentration of 5 μg L−1. | ||||
| Nap | 0.005–5 | 0.9977 | 0.25 | 3 |
| Acp | 0.005–5 | 0.9998 | 0.15 | 2.6 |
| Ace | 0.005–5 | 0.9993 | 0.1 | 2.5 |
| Flu | 0.005–5 | 1 | 0.02 | 2.9 |
| Phe | 0.005–5 | 1 | 0.02 | 2.7 |
| Ant | 0.005–5 | 0.9973 | 0.03 | 3.9 |
| Flt | 0.005–5 | 0.9964 | 0.15 | 5.4 |
| Pyr | 0.005–5 | 0.9963 | 0.12 | 8.4 |
| Cry | 0.005–5 | 0.982 | 0.42 | 7.2 |
| BaA | 0.005–5 | 0.984 | 0.38 | 6.8 |
| BbF | 0.1–5 | 0.978 | 5 | 5.9 |
| BkF | 0.1–5 | 0.977 | 5.1 | 5.7 |
| BaP | 0.1–5 | 0.978 | 14.3 | 6.4 |
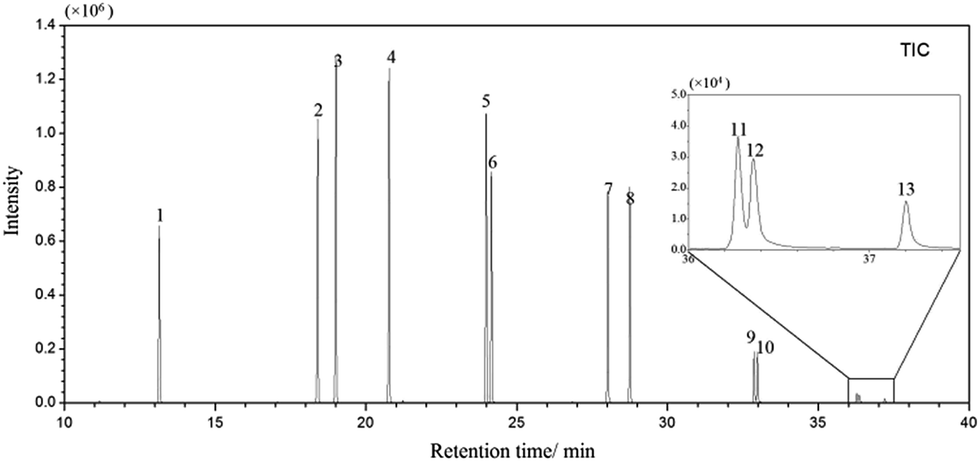 | ||
| Fig. 3 GC/MS-SIM chromatogram of PAHs in river water sample spiked with standards at 5 μg L−1 after MSPE. Sample volume, 100 mL; peak identities: (1) Nap, (2) Acp, (3) Ace, (4) Flu, (5) Phe, (6) Ant, (7) Flt, (8) Pyr, (9) Cry, (10) BaA, (11) BbF, (12) BkF, (13) BaP. | ||
The linearity was studied using ultrapure water samples spiked with PAHs at a series of concentration. The curves of different ring PAHs were linear in the concentration range of 0.005–5 μg L−1, 0.1–5 μg L−1, respectively. The linearity was assessed by correlation coefficients (R2), and were higher than 0.99 for most PAHs. The LODs calculated based on S/N of 3, ranged from 0.02 to 14.3 ng L−1. The results of RSD were calculated by adopting five replicate runs in the range of 2.5–8.4%.
Moreover, five kinds of different methods used for the study of polycyclic aromatic hydrocarbons, containing SPE, μ-SPE, SPME, IT-SPME, MSPE, were summarized.24,31,33,42–44 Table 2 summarized the method of specific information, such as sorbents, material dosage, desorption solvents, solvent consumption/sample volume, test number of PAHs, LODs and instrumentation. Compared with SPE and SPME, MSPE showed a merit in material dosage, volume of desorption solvent and stability. Previous studies based on MSPE focused on lower ring PAHs, but failed in the analysis of high PAHs. The present method shows an extremely good analytical performance, especially for high ring PAHs.
| Extraction method | Sorbents | Determination | LOD (ng L−1) | Desorption solvents | Material dose (mg) | Solvent consumption/sample volume (mL mL−1) | Number of PAHs | Ref. |
|---|---|---|---|---|---|---|---|---|
| Monolithic column | STY/DVB/GMA polyHIPE monoliths | HPLC-FLD | 4–228 | Ethanol/methylenechloride (1:1, v/v) |
61 | 3/500 | 13 | 36 |
| MSPE | Fe3O4@SiO2-G | HPLC-FLD | 0.5–5.0 | Acetone | 15 | 0.3/250 | 5 | 28 |
| μ-SPE | Magnetic CS functionalized GO | GC-MS | 0.2–1.8 | n-Hexane | 100 | 0.15/10 | 7 | 26 |
| IT-SPME | TRB-5 | HPLC-FLD | 2.3–28 | Acetonitrile | Not reported | 2/5 | 15 | 37 |
| SPE | Cyclodextrin-silica microporous composite | HPLC-FLD | 50–800 | Hexane + methanol | 200 | 8/100 | 6 | 38 |
| SPE | Sulfur microparticles | HPLC-UV | 7–48 | Acetonitrile | 1500 | 2/100 | 10 | 39 |
| MSPE | Fe3O4/rGO | GC-MS | 0.02–14.3 | Toluene/acetone (3:1, v/v) |
10 | 0.1/100 | 13 | This work |
3.4 Analysis of real water samples
In order to further evaluate the application of method, two real water samples were analyzed. All samples obtained were analyzed in three replicates. The results were illustrated in Table 3. There were no PAHs found in drinking water, and trace amount of low ring PAHs were detected in river water, which was probably due to rain leaching into the Hanjiang River. To evaluate the accuracy of the method, the relevant recoveries were analyzed by spiking with PAHs into real water samples at a series of concentrations from 50 ng L−1 to 2.5 μg L−1. It can be observed that satisfactory recoveries were obtained ranging from 75.6% to 112.4%. It suggested that the developed method was suitable for the analysis of environmental water samples in the search for PAHs.| Analytes | Drinking water | Hanjing river | ||||||
|---|---|---|---|---|---|---|---|---|
| Concentration (ng L−1)(RSD, n = 3) | Recovery (RSD)a | Recovery (RSD)b | Recovery (RSD)c | Concentration (ng L−1) (RSD, n = 3) | Recovery (RSD)a | Recovery (RSD)b | Recovery (RSD)c | |
| a Spiked at a concentration of 50 ng L−1.b Spiked at a concentration of 250 ng L−1.c Spiked at a concentration of 2.5 μg L−1; – non-detected or below LOD. | ||||||||
| Nap | — | 101.6 (5.5) | 104.4 (5.6) | 84.4 (1.7) | 15 | 98.3 (7.9) | 105.8 (3.5) | 88.1 (1.2) |
| Acp | — | 99.8 (6.8) | 95.2 (6.0) | 95.0 (6.1) | — | 98.0 (4.1) | 84.4 (2.1) | 92.2 (0.1) |
| Ace | — | 91.0 (7.5) | 93.7 (7.1) | 108.9 (6.3) | — | 92.0 (4.1) | 108.0 (2.3) | 103.2 (0.5) |
| Flu | — | 100.0 (6.5) | 100.5 (5.7) | 106.8 (4.4) | — | 103.3 (2.5) | 93.9 (2.4) | 99.7 (0.5) |
| Phe | — | 105.0 (5.4) | 99.9 (3.9) | 99.5 (3.9) | — | 93.1 (0.6) | 98.3 (3.5) | 109.4 (0.8) |
| Ant | — | 106.9 (0.3) | 89.3 (5.6) | 98.4 (3.5) | — | 110.6 (1.8) | 81.6 (4.6) | 95.4 (1.9) |
| Flt | — | 98.3 (1.1) | 83.7 (1.3) | 92.9 (6.1) | — | 90.4 (6.6) | 88.2 (8.6) | 93.7 (3.1) |
| Pyr | — | 84.4 (2.6) | 87.7 (2.7) | 93.8 (6.7) | — | 84.1 (6.6) | 86.4 (8.8) | 86.9 (3.1) |
| Cry | — | 96.9 (9.5) | 108.2 (7.0) | 86.9 (5.1) | — | 76.4 (2.1) | 104.8 (9.2) | 75.6 (1.4) |
| BaA | — | 89.1 (5.3) | 98.7 (7.8) | 82.2 (6.6) | — | 78.0 (1.6) | 103.1 (7.9) | 77.3 (0.5) |
| BbF | — | — | 112.4 (3.2) | 91.7 (3.1) | — | — | 96.9 (7.5) | 92.5 (2.3) |
| BkF | — | — | 109.4 (7.5) | 101.7 (1.9) | — | — | 102.7 (7.8) | 110.9 (1.2) |
| BaP | — | — | 83.0 (9.2) | 88.2 (7.1) | — | — | 83.0 (3.4) | 97.1 (3.7) |
4. Conclusions
In the present study, an extraction method suitable for the simultaneous determination of PAHs from low to high molecular weight (2–5 rings) in water was developed based on a magnetic nanocomposite of reduced graphene oxide (Fe3O4/rGO). By comparing elution of PAHs using various solvents with different polarity, hydrophobic property and characteristic functional groups, a conclusion had been drawn that effect of elution was mainly associated with the polarity and functional groups of the solvents. Phenyl in toluene and carbonyl in acetone increase the extraction efficiency due to π–π interactions and p–π interactions, respectively. In addition, only 100 μL of binary mixed solvents could effectively desorb PAHs on the surface of magnetic materials. The decrease of organic solvent is influential, not only for researcher's health and economy, but also reducing the damage to the environment and conforming to trend of environment-friendly technologies. Conceivably, the procedure provided a simple, sensitive and efficient method for the simultaneous determination of diverse PAHs in environmental water samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21077039, 41603045) and the Programme of Introducing Talents of Discipline to Universities (No. B14031).References
- L. Guo and H. K. Lee, J. Chromatogr. A, 2011, 1218, 9321–9327 CrossRef CAS PubMed.
- T. J. Woodruff, A. R. Zota and J. M. Schwartz, Environ. Health Perspect., 2011, 119, 878–885 CrossRef PubMed.
- B. Binková, J. Topinka, G. Mracková, D. Gajdosová, P. N. Vidová, Z. Stávková, V. T. Peterka, T. Pilcík, V. R. Rimár, L. R. Dobiás, P. B. Farmer and R. J. Rám, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 1998, 416, 67–84 CrossRef.
- Z. Li, L. C. Romanoff, D. A. Trinidad, N. Hussain, R. S. Jones, E. N. Porter, D. G. Patterson and A. Sjodin, Anal. Chem., 2006, 78, 5744–5751 CrossRef CAS PubMed.
- R. Fan, R. Ramage, D. Wang, J. Zhou and J. She, Talanta, 2012, 93, 383–391 CrossRef CAS PubMed.
- X. Gao, S. Zhu, W. Zhang, D. Li, W. Dai and S. He, Fuel, 2016, 182, 788–797 CrossRef CAS.
- X. Gao, L. Pang, S. Zhu, W. Zhang, W. Dai, D. Li and S. He, Org. Geochem., 2017, 106, 30–47 CrossRef CAS.
- W. Zhang, S. Zhu, L. Pang, X. Gao, G. Zhu and D. Li, J. Chromatogr. A, 2016, 1478, 75–83 CrossRef CAS PubMed.
- G. Zhu, S. He, X. He, S. Zhu and Y. Feng, RSC Adv., 2017, 7, 40608–40614 RSC.
- S. Zhang, H. Niu, Y. Cai and Y. Shi, Anal. Chim. Acta, 2010, 665, 167–175 CrossRef CAS PubMed.
- Q. Zhao, F. Wei, Y. Luo, J. Ding, N. Xiao and Y. Feng, J. Agric. Food Chem., 2011, 59, 12794–12800 CrossRef CAS PubMed.
- S. Huo and X. Yan, Analyst, 2012, 137, 3445–3451 RSC.
- G. Giakisikli and A. N. Anthemidis, Anal. Chim. Acta, 2013, 789, 1–16 CrossRef CAS PubMed.
- M. Wierucka and M. Biziuk, TrAC, Trends Anal. Chem., 2014, 59, 50–58 CrossRef CAS.
- S. Sadeghi, H. Azhdari, H. Arabi and A. Z. Moghaddam, J. Hazard. Mater., 2012, 215–216, 208–216 CrossRef CAS PubMed.
- Q. Zhao, F. Wei, N. Xiao, Q. Yu, B. Yuan and Y. Feng, J. Chromatogr. A, 2012, 1240, 45–51 CrossRef CAS PubMed.
- M. Ghaedi, S. Hajjati, Z. Mahmudi, I. Tyagi, S. Agarwal, A. Maity and V. K. Gupta, Chem. Eng. J., 2015, 268, 28–37 CrossRef CAS.
- L. Xie, R. Jiang, F. Zhu, H. Liu and G. Ouyang, Anal. Bioanal. Chem., 2014, 406, 377–399 CrossRef CAS PubMed.
- C. Chen, Y. Yu, W. Li, C. Cao, P. Li, Z. Dou and W. Song, J. Mater. Chem., 2011, 21, 12836–12841 RSC.
- W. Li, D. Chen, F. Xia, J. Tan, P. Huang, W. Song, N. Nursam and R. Caruso, Environ. Sci.: Nano, 2016, 3, 94–106 RSC.
- L. Chen, T. Wang and J. Tong, TrAC, Trends Anal. Chem., 2011, 30, 1095–1108 CrossRef CAS.
- S. Lamichhane, K. Bal Krishna and R. Sarukkalige, Chemosphere, 2016, 148, 336–353 CrossRef CAS PubMed.
- K. Yuan, J. Wang, H. Zhai, Z. Chen, L. Huang and Z. Su, Anal. Methods, 2015, 7, 8297–8303 RSC.
- R. Su, G. Ruan, H. Nie, T. Xie, Y. Zheng, F. Du and J. Li, J. Chromatogr. A, 2015, 1405, 23–31 CrossRef CAS PubMed.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- Q. Wu, G. Zhao, C. Feng, C. Wang and Z. Wang, J. Chromatogr. A, 2011, 1218, 7936–7942 CrossRef CAS PubMed.
- X. Cao, J. Chen, X. Ye, F. Zhang, L. Shen and W. Mo, J. Sep. Sci., 2013, 36, 3579–3585 CrossRef CAS PubMed.
- B. Zawisza, R. Sitko, E. Malicka and E. Talik, Anal. Methods, 2013, 5, 6425–6430 RSC.
- Y. Zhao, X. Li, X. Zhou and Y. Zhang, Sens. Actuators, B, 2013, 231, 324–340 CrossRef.
- R. Sitko, B. Zawisza and E. Malicka, TrAC, Trends Anal. Chem., 2013, 51, 33–43 CrossRef CAS.
- W. Wang, R. Ma, Q. Wu, C. Wang and Z. Wang, J. Chromatogr. A, 2013, 1293, 20–27 CrossRef CAS PubMed.
- J. Wang, Z. Chen and B. Chen, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed.
- N. N. Naing, S. F. Yau Li and H. K. Lee, J. Chromatogr. A, 2016, 1440, 23–30 CrossRef CAS PubMed.
- H. Bagheri, A. Afkhami, P. Hashemi and M. Ghanei, RSC Adv., 2015, 5, 21659 RSC.
- L. Zhang, W. Li, Z. Cui and W. Song, J. Phys. Chem. C, 2009, 113, 20594–20598 CAS.
- H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CAS.
- W. Li, L. Zhang, Q. Wang, Y. Yu, Z. Chen, C. Cao and W. Song, J. Mater. Chem., 2012, 22, 15342–15347 RSC.
- J. Ma, R. Xiao, J. Li, J. Yu, Y. Zhang and L. Chen, J. Chromatogr. A, 2010, 1217, 5462–5469 CrossRef CAS PubMed.
- A. K. Mallik, S. Guragain, H. Hachisako, M. M. Rahman, M. Takafuji and H. Ihara, J. Chromatogr. A, 2014, 1324, 149–154 CrossRef CAS PubMed.
- L. Wolska, M. Rawa-Adkonis and J. Namieśnik, Anal. Bioanal. Chem., 2005, 382, 1389–1397 CrossRef CAS PubMed.
- W. Li, F. Xia, J. Qiu, P. Li, D. Chen, Z. Chen, Y. Yu, Y. Lu, R. Caruso and W. Song, Nano Res., 2014, 7, 903–916 CrossRef CAS.
- V. Khalili-Fard, K. Ghanemi, Y. Nikpour and M. Fallah-Mehrjardi, Anal. Chim. Acta, 2012, 714, 89–97 CrossRef CAS PubMed.
- M. Fernández-Amado, M. C. Prieto-Blanco, P. López-Mahía, S. Muniategui-Lorenzo and D. Prada-Rodríguez, Talanta, 2016, 155, 175–184 CrossRef PubMed.
- A. Mauri-Aucejo, P. Amorós, A. Moragues, C. Guillem and C. Belenguer-Sapiña, Talanta, 2016, 156–157, 95–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10551g |
| This journal is © The Royal Society of Chemistry 2017 |