
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical study on the photophysical properties of boron-fused double helicenes†
Yanling Si *a,
Yan Chengb,
Nan Qua,
Xinyu Zhaoa and
Guochun Yang*c
*a,
Yan Chengb,
Nan Qua,
Xinyu Zhaoa and
Guochun Yang*c
aCollege of Resource and Environmental Science, Jilin Agricultural University, Changchun 130118, China
bDepartment of Ophthalmology, Second Hospital of Jilin University, Changchun, 132400, China
cCentre for Advanced Optoelectronic Functional Materials Research, Key Laboratory for UV Light-Emitting Materials and Technology of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: yanggc468@nenu.edu.cn
First published on 15th December 2017
Abstract
Heterohelicenes have attracted much attention because the involvement of heteroatom into helicene skeleton effectively modulates physical properties and greatly widens the application of helicenes. Recently, boron-fused double helicenes (compounds 1 and 2), exhibiting unique photophysical properties, were reported [J. Am. Chem. Soc., 2016, 138, 5210]. Fully understanding their microelectronic structures is rather important for further performance optimization or improvement. Here, we employed density functional theory to investigate the electronic transition properties, circular dichroism (CD), charge transport, and second-order nonlinear optical (NLO) response of the seven boron-fused double helicenes. Our simulated UV-vis/CD spectra of compound 2 are in good agreement with experimental ones. Different electron-donor or electron-acceptor substituents have considerable effect on frontier molecular orbital energy level, absorption wavelength, electron transition property, and second-order NLO response values. The designed compound 7 with two electron donors (TTF) and two electron acceptors (TCNQ) is expected to be excellent second-order NLO material in view of large first hyperpolarizability value and inherent asymmetric structure. The study of charge transport indicates that incorporation boron and oxygen atoms into intermolecular π–π packing units is an effective way to realize ambipolar charge transport.
1. Introduction
The design and synthesis of organic materials with desirable properties has become a rather active research area because of the advantages of these materials such as low cost, light weight, large area, and flexibility.1–5 Up to now, some organic materials have exhibited broad applications, including organic light emitting diodes,6 organic field effect transistors,7 organic photovoltaic cells,8 and nonlinear optics.9 The continuous performance improvement strongly depends on the discovery of new materials with desirable properties and the full understanding of the structure and property relationships at the quantum mechanical level.Compared with inorganic and semiconductor nonlinear optical (NLO) materials, organic NLO materials exhibit a variety of merits, for example, larger NLO response coefficients, higher laser damage thresholds, lower dielectric constants, and faster response times.10,11 Among the various NLO response processes, second-order NLO closely and immediately correlates with the practical application. Based on the extensive studies, it is widely recognized that the two following strategies are the effective ways to enhance the NLO response of organic materials. One is the introduction of asymmetry substitutions (i.e. donor and acceptor) into the π-conjugated skeleton. The other is to use the chiral compounds, which can satisfy not only the requirement of non-centrosymmetric electron structure but also keep the merits of organic materials. More information about the role of chiral NLO materials has been overviewed.12–14
Helicenes have received extensive attention in the past few years, due to their inherent chirality and extended conjugation structure, which leads to broad applications in organic optoelectronic field.15–19 In particular, the involvement of heteroatoms into helicene and synthesis of helicene with multihelicity have been proved to be an effective way to further improve their performance and modulate the photophysical properties. The long-desirable target, the introduction of boron atom into multihelical π-conjugated skeleton, has been recently synthesized, which exhibits unique three-dimensional brickwork stacking.20 Intriguingly, these compounds possess the bipolar carrier transport characters, which can greatly simplify device manufacturing process. Notably, their photophysical properties and relationship behind the structural characters, and the charge transport property remain elusive especially at the microscopic level, however it is very important to further elevate the performance.
Now, density functional theory (DFT)/time-dependent DFT calculations become a powerful tool not only to gain insight into the observed properties but also to design new materials with outstanding properties.21–25 In this paper, we mainly focus on determining the geometry structures, electron transition properties, chiroptical properties, and electron and hole transport of boron-fused double helicenes, named as compounds 1 and 2, and establishing their structure–property relationship. On the other hand, these compounds might provide some new opportunities to the second-order NLO materials in view of the inherent chirality (asymmetry), extended π-conjugated structure, and heteroatom boron.26–28 As a consequence, the other five compounds 3–7, including different electron donor/acceptor units (i.e. NH2, NO2, tetrathiafulvalene (TTF) and 7,7,8,8-tetracyanoquinodimethane (TCNQ)) and their combinations were designed to probe the charge transfer cooperativity and find excellent second-order NLO materials.
2. Computational details
The ground state geometries of the studied compounds were fully optimized at the B3LYP/6-31G(d) level of theory as performed in the Gaussian 09 program package.29 During the geometry optimization, no any symmetry or internal coordination constraints was done. The B3LYP functional is a combination of Becke's three-parameter hybrid exchange functional30 and the Lee–Yang–Parr31 correlation functional. All the positive vibrational frequencies indicate that our optimization structures are the local minima.To ensure the accuracy of our calculation method, four well-accepted functionals (PBE0,32 B3LYP, CAM-B3LYP33 and BHandHLYP34) combined with the polarizable continuum model (PCM) in CH2Cl2 solvent were performed to study the UV-visible absorption spectra of compound 2. The results indicated that PBE0 is the most suitable in reproducing the experimental data (Table S1, ESI†). Therefore, TDDFT-PBE0/6-31+G(d) was selected in the calculation of the electron excitation energies, oscillator strengths, and rotational strengths of the studied compounds. Both length and velocity representations were used to obtain the rotational strengths. It is noted that the velocity-gauge representation of the dipole operator is gauge origin independent. Gaussian bandshapes35 with a bandwidth of 0.20 eV were used to compare the calculated UV-vis/CD spectra with experimental ones. To test the effect of solvent on the UV-vis/CD spectra, the polarizable continuum model36,37 was utilized as implemented in Gaussian 09. According to the experimental condition, dichloromethane (CH2Cl2) was treated as the continuous dielectric environment.
Each tensor of second-order NLO response was calculated as implemented in the Gaussian 09 program package, which was used to calculate second-order NLO response coefficients. Here, hyper-Rayleigh scattering (HRS) was used to measure the NLO response. In the case of plane-polarized incident light and observations made perpendicular to the propagation plane without polarization analysis of the scattered beam, the second-order NLO response that can be extracted from HRS data can be described as:38,39
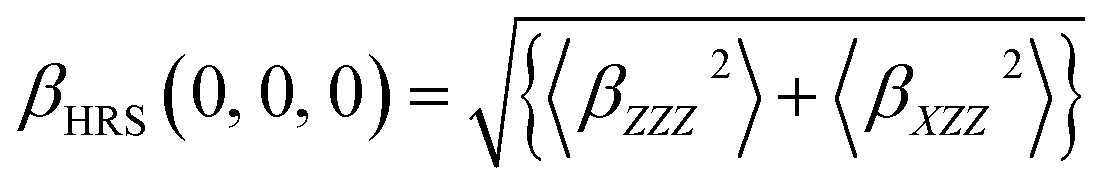 | (1) |
Here, electron band structure and Projected Density of States (PDOS) calculations were carried out as implemented in the Vienna Ab initio simulation package (VASP).41 Band model was used to describe charge transportation property.42 Perdew–Burke–Emzerhof functional and a plane-wave basis set with an energy cut-off of 400 eV were adopted.
3. Results and discussion
3.1 Geometrical and electronic structures
Here, seven boron-fused double helicenes were investigated, as shown in Fig. 1. Compounds 1 and 2 were synthesized and characterized by X-ray crystallography.20 To keep both the nonsymmetric structure and find the effective way to enhance the second-order NLO response, compounds 3–7, containing different electron-donors or acceptors and their combinations, were designed based on compound 1. The geometric structures of the studied compounds were fully relaxed without any symmetry constraints at the B3LYP/6-31G(d) and PBE0/6-31G(d) level of theory (Table S2†). Here, compound 1 was taken as an example to probe the suitability of our adopted method. Obviously, the resultant structural parameters at B3LYP/6-31G(d) level are in good agreement with experimental ones, indicating that functional B3LYP can reliably determine the geometry structures of the studied compounds. This can be further confirmed by the same average deviations of the bond lengths and the dihedral angles, obtained by functionals B3LYP and PBE0. On the other hand, the calculated vibrational frequencies of the considered compounds are all real, implying that the optimized structures are local minimum.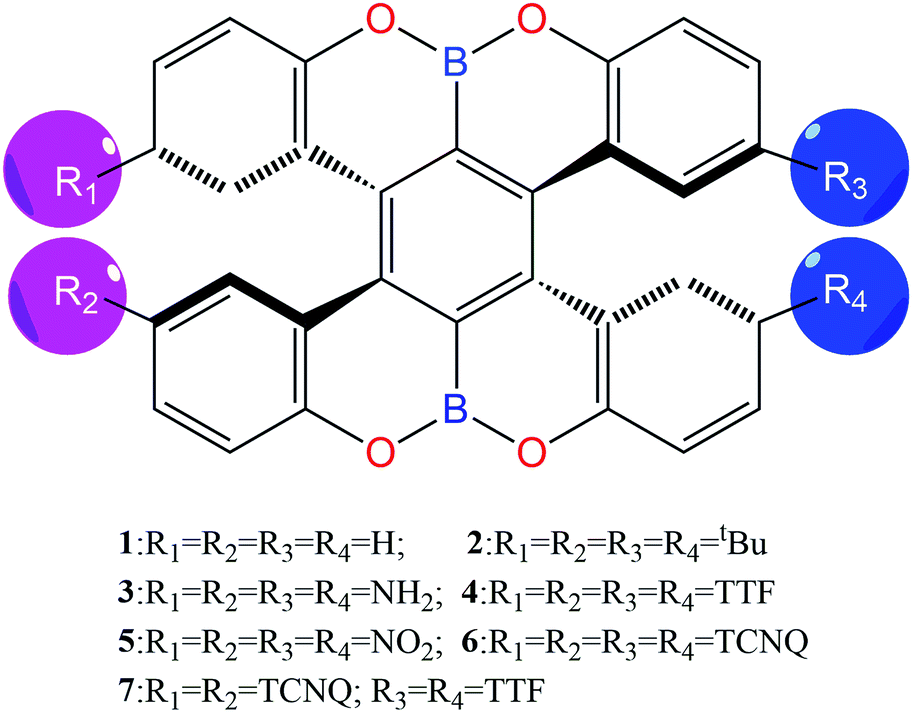 | ||
| Fig. 1 Chemical structures of the studied compounds (compounds 1 and 2 from ref. 20). | ||
It is well-known that the distribution of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), and the energy gap between HOMO and LUMO play an important role in determining the electronic properties. As a consequence, we firstly examined the character of HOMO and LUMO and the variations of energy gap of the studied compounds. Their energy level contour plots and energy gaps were shown in Fig. 2.
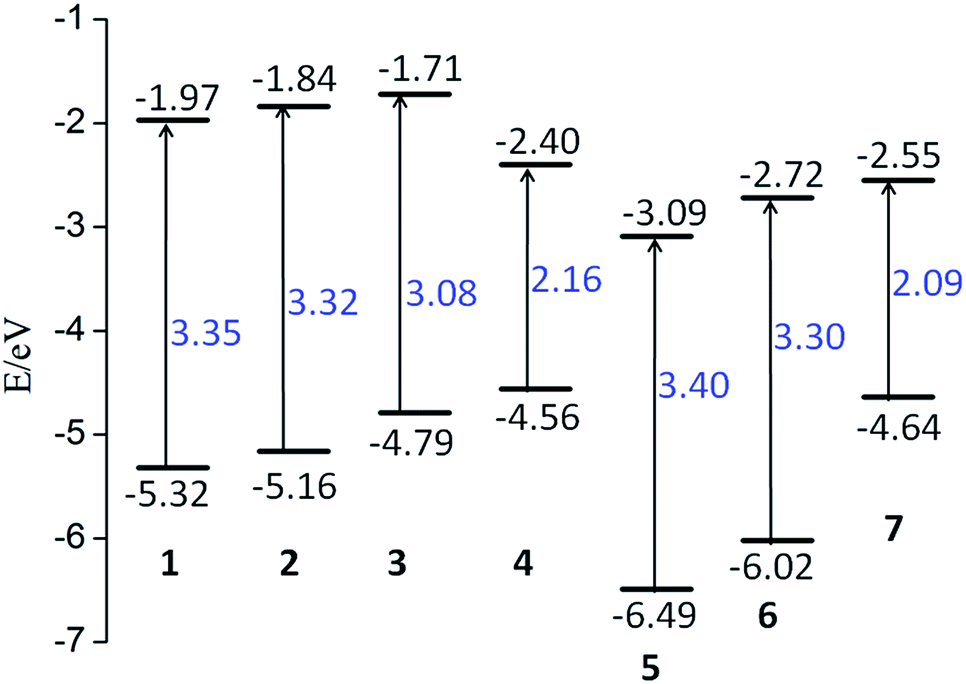 | ||
| Fig. 2 Schematic energy levels of the studied compounds. The lower short line represents the HOMO level, while the upper one is the LUMO level. | ||
The studied compounds exhibit a wide range of energy gaps from 2.09 to 3.40 eV. This indicates that different substituents of electron-donor or electron-acceptor and the combination have great effects on modulating energy gap. To be specific, compared to compound 1, tert-butyl (tBu) and NH2 groups, acting as the electron donors in compounds 2 and 3, raise the energy levels of both HOMO and LUMO. Notably, both the HOMO and LUMO energy levels of compound 2 are elevated with a similar extent, which results in slightly change of energy gap. However, for compound 3, the increased magnitude of the HOMO energy level is much larger than that of the LUMO. As a consequence, the band gap of compound 3 is significantly smaller than that of compounds 1 and 2. Intriguingly, compound 4 with TTF substitution, usually acting as electron-donor, has completely different variable trends in comparison with compounds 2 and 3. Specifically, its HOMO energy level is raised, while the LUMO energy level is greatly reduced, leading to the obvious decrease of energy gap. On the other hand, for compounds 5 and 6, in which H groups are substituted with electron-acceptors of NO2 and TCNQ groups, both the HOMO and LUMO energy levels are decreased simultaneously. Moreover, the rate of decline is almost the same. As a consequence, the band gaps of compounds 5 and 6 are unchanged compared to that of compound 1. As illustrated in Fig. 2, the band gap of compound 7 is the smallest, in which H groups are substituted with two electron donors (TTF) and two electron acceptors (TCNQ), respectively. Based on the above analysis, the effect of the substitutions of electron-donor or electron-acceptor or their combinations on frontier molecular levels are obvious. Therefore, it is expected that the studied compounds might exhibit different electronic properties.
Subsequently, we studied their HOMO and LUMO distributions. For compound 1, both HOMO and LUMO delocalizes over π-system of the benzene rings, whereas the contribution of the boron and oxygen atoms are vanishing small (Fig. 3). For the compounds 2, 3, 5 and 6, the distributions of the HOMO and LUMO are almost unchanged compared to that of compound 1, which can be found in Fig. S1.† Interestingly, for the compound 7, the LUMO is mainly localized at the benzene rings, whereas the HOMO is obviously different, which mainly resides on the one of the TTF groups (Fig. 3). This character is favor of intramolecular charge transfer, as shown in the electron transition analysis. And the HOMO and LUMO distribution of the compound 4 (Fig. S1†) are similar to those of the compound 7.
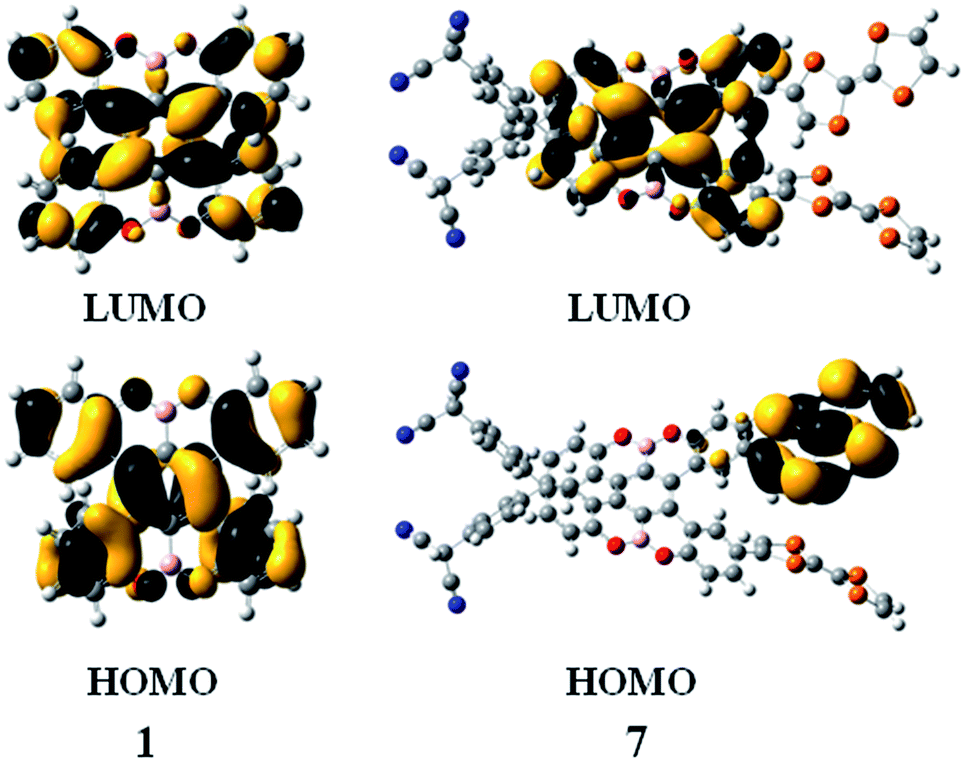 | ||
| Fig. 3 HOMO and LUMO of compounds 1 and 7. | ||
3.2 UV-vis/CD spectra
To further reveal the effect of different electron donors/acceptors on the photophysical properties and assign the electron transition properties, we calculated UV-vis/CD spectra of the studied compounds by employing the TDDFT method, which has become a widely accepted tool to investigate the electronic transition and chiroptical properties.43–47 In general, different basis sets and DFT functionals might influence the electronic excitation energies and transition properties. Based on the previous studied experiences, the 6-31+G(d) basis set has proved to be enough for organic compounds.48,49 However, the different functionals usually have great effect on electronic excitation energy and the electronic transition properties.50 Therefore, the 6-31+G(d) basis set combined with four popular DFT functionals were adopted to calculate the electron absorption wavelength of compound 2, which has been fully characterized by experiment.20 As mentioned in the computational details, TDDFT/PBE0 exhibits the best performance in reproducing the experimental absorption wavelength. In other words, there is just 4 nm difference between calculated and measured values (Table S1†). Thus, PBE0/6-31+G(d) was used in the following calculations.According to the calculated data, we simulated the UV-vis/CD spectra of compound 2 by using a Gaussian band shape with a bandwidth of 0.20 eV along with the experimental spectra20 (Fig. 4). As illustrated in Fig. 4, the experimental UV-vis/CD absorption spectra were also reproduced, which can be used to reliably assign the electron transition properties and chiroptical origin. To probe the solvent effect on the UV-vis/CD spectra,51,52 we also simulated the spectra obtained in CH2Cl2 solution. The resultant absorption spectra in the gas phase are rather similar to those of in solution, which indicates that the influence of the solvent could be negligible for this kind of compounds. It is noted that there is also small difference between simulated spectra and experimental ones. For example, there is a double peak at roughly 300 nm in experimental spectra, while it is one peak in our simulation. For compounds 1 and 2, the electron transition properties of the low-energy absorption peaks can be best described as π/π* transition (Fig. 5 and S2†). It should be noted that the tBu groups have almost no contribution to these transition orbitals. The electron transition properties of compounds 3, 5 and 6 are similar to those of compounds 1 and 2 as represented in Fig. S3,† indicating that the introduction of NH2, NO2 and TCNQ groups has little effect on the electronic transition properties, whereas has certain influence on the electron absorption wavelength. For example, the calculated absorption band of compound 3 is obviously red-shifted compared to those of compounds 1 and 2 (Table 1). Again, there are some differences of compound 4 with TTF groups. The absorption band of compound 4 arises from HOMO−4 → LUMO and HOMO−3 → LUMO+2 excitations, involving certain contribution from TTF groups. Finally, the combination of the donors and acceptors greatly alters the electron transition characters. As illustrated in Fig. 5, obvious intramolecular charge transfer has been observed in compound 7, which might lead to a large nonlinear optical response.
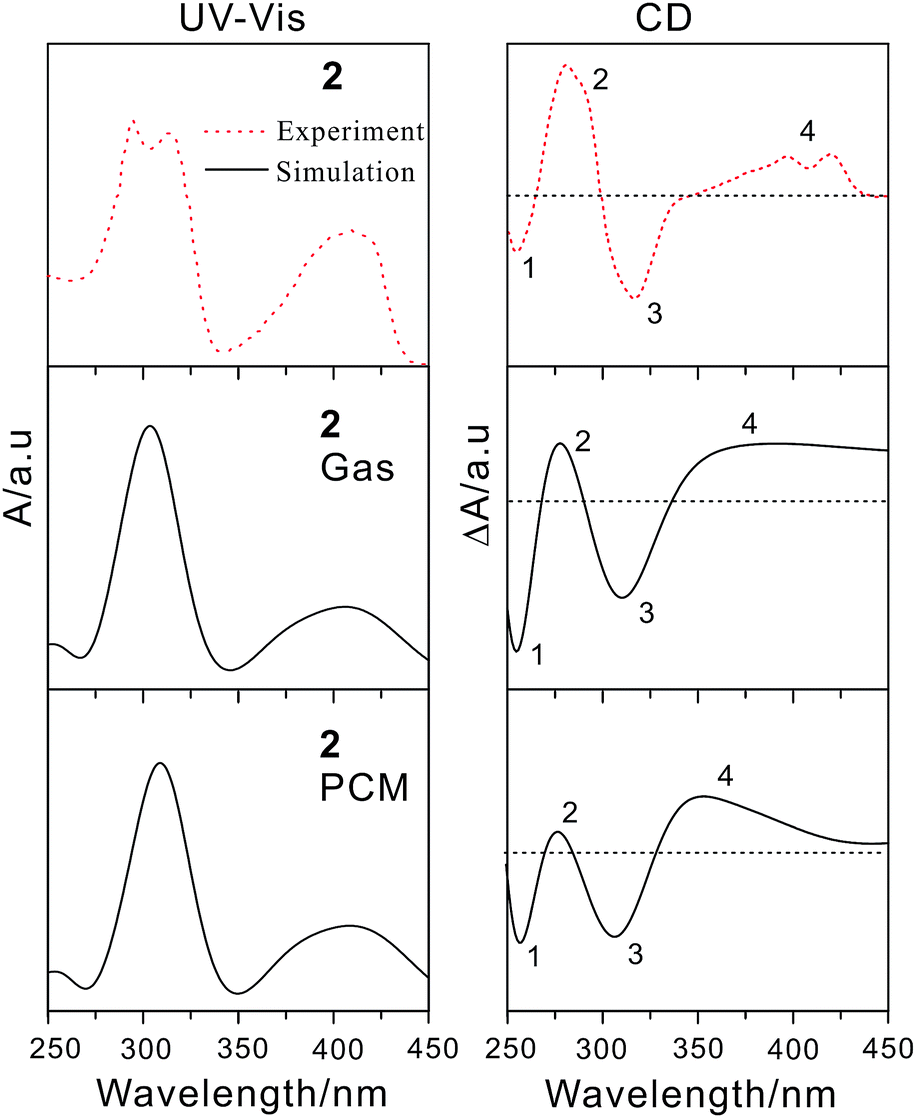 | ||
| Fig. 4 Calculated UV-vis (left) and ECD (right) spectra in both gas and solution phases of compound 2 along with experimental ones. | ||
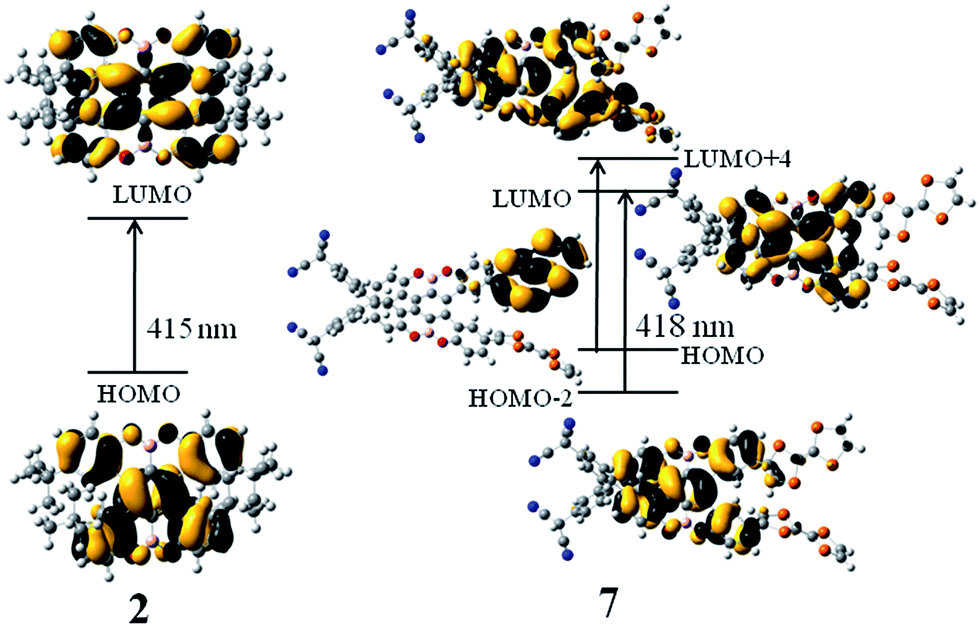 | ||
| Fig. 5 Molecular orbital isosurfaces involved in the main electron transitions of compounds 2 and 7. | ||
| Systems | λ | f | Major contribution |
|---|---|---|---|
| 1 | 408(405) | 0.3137 | HOMO → LUMO (99%) |
| 2 | 415(411) | 0.2513 | HOMO → LUMO (99%) |
| 3 | 448 | 0.2686 | HOMO → LUMO (99%) |
| 4 | 416 | 0.1330 | HOMO−4 → LUMO (35%) |
| HOMO−3 → LUMO+2 (18%) | |||
| 5 | 409 | 0.1790 | HOMO → LUMO (95%) |
| 6 | 422 | 0.1685 | HOMO → LUMO (70%) |
| 7 | 418 | 0.1154 | HOMO−2 → LUMO (41%) |
| HOMO → LUMO+4 (23%) |
In the simulated CD spectra of compound 2, there are four main bands. Among them, two negative bands are located at about 260 and 320 nm. And two positive bands are observed around 280 and 400 nm. Comparison of the experimental20 and simulated spectra shows that the experimental spectra were well reproduced by our calculated CD ones in the solution. This means that the CD spectra are more sensitive to the solvents, which has been shown in the previous studies.53–55 Besides, the differences between the rotational strengths calculated using the length- and velocity-gauge representation of the electric dipole operator are very small (Table S2†), which confirms again the suitability of the 6-31+G(d) basis set for CD calculations. To further understand the chiral origin of compound 2, the molecular orbitals involved in the main transitions were shown in Fig. S4.† The observed CD bands mainly result from exciton-coupling of aromatic benzene rings, while there is not any contribution from tBu groups. This is also in consistent with previous frontier molecular orbital analysis.
3.3 Charge transportation property
The studied compound 1, in its crystal form, has a unique packing pattern20 (i.e. an offset face to face stacking arrangement), which is a benefit for the application in organic semiconducting transportation materials. More intriguingly, hole mobility of compound 1 is only slightly larger than electron mobility. Thus, it exhibits ambipolar carrier transportation character. Subsequently, we investigated its charge transportation property from the standpoint of band model. According to band model, the mobility increases with the size of the band width.56,57 The valence band width corresponds to the hole transportation, while the conduction band width is the electron transportation. The calculated electron energy band indicates that crystal 1 is an indirect gap semiconductor with its maximum of valence band at Z point and minimum of conduction band at G point (Fig. 6). The PDOS analysis shows that the valence band mainly comes from the contribution of O-p, C-p, and B-p orbitals, whereas to conduction band, C-p orbital does. The calculated width of valence band is comparable to that of conduction band, clearly indicating the ambipolar transportation properties. This observation mainly results from the contribution of Y → A direction, corresponding to intermolecular interaction (i.e. between a pure C6 ring and another C6 ring containing the boron and oxygen atoms), as illustrated in Fig. 6. In general, pure intermolecular π–π interaction is mainly responsible for hole transport (e.g. pentacene and rubrene).58 In this case, the involvement of oxygen and born atoms into C6 ring obviously alters charge transportation property. This might be attributed to strong accepting electronic capabilities of oxygen. Similar observation has been found in phenyl–perfluorophenyl (π–πF) interaction.59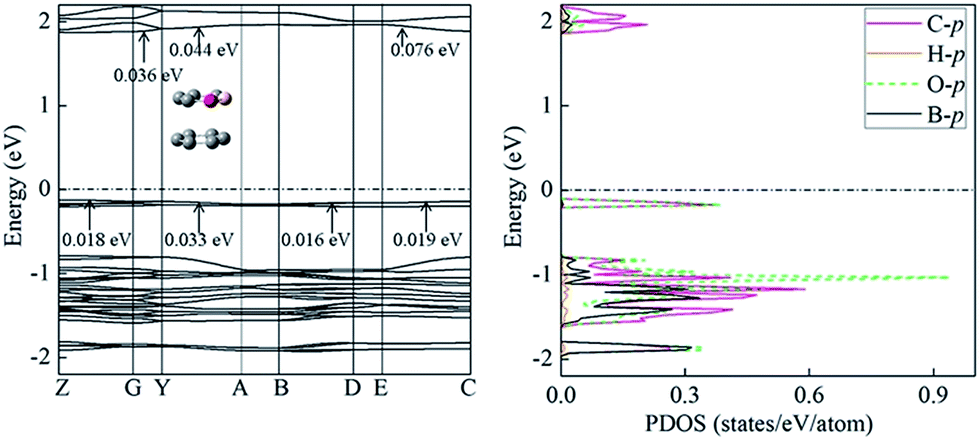 | ||
| Fig. 6 The band structures and projected density of states (PDOS) of crystal 1. High symmetry k-points in the first Brillouin zone are Z = (0, 0, 1/2), G = (0, 0, 0), Y = (0, 1/2, 0), A = (−1/2, 1/2, 0), B = (−1/2, 0, 0), D = (−1/2, 0, 1/2), E = (−1/2, 1/2, 1/2), C = (0, 1/2, 1/2). | ||
3.4 Second-order NLO property
From the standpoint of the inherent asymmetric structures and large intramolecular charge transfer character, the studied compounds are expected to exhibit excellent second-order NLO response under the external electronic field. To reliably determine the second-order NLO response, four widely used functionals (i.e. B3LYP, BHandHLYP, CAM-B3LYP and PBE0) were considered. The resultant βHRS values were shown in Fig. S5.† Although there are some differences between second-order NLO response values, the overall trend is consistent. This means that our adopted method can reliably describe the effects on βHRS values of different substituents and design the potential NLO materials. It is noted that following discussion was based on the results obtained from B3LYP/6-31+G(d) level. As shown in Table 2, their βHRS values of the studied compounds ranges from 0.34 to 25.48 × 10−33 esu, indicating that appropriate structural modifications can effectively enhance the second-order NLO response. In detail, among the studied compounds 1–4 with various electron-donor substitutions, compound 4 with TTF groups show the largest βHRS values. This means that TTF is the better donor for our studied compounds. For the electron-donor substitutions, TCNQ groups show the better performance. General speaking, the involvement of both donors and acceptors into one compound, at the opposite positions, is an effective way to enhance the second-order NLO response.60,61 As we expected, compound 7, containing TTF and TCNQ groups, shows the largest βHRS values of 25.48 × 10−33 esu. Notably, its βHRS values is about 190 times larger than that of the organic urea molecule62 and 5 times larger than that measured for the highly π-delocalized phenyliminomethylferrocene complex,63 indicating that this kind of compounds are the potential second-order NLO materials. The large NLO response of compound 7 originates from large intramolecular charge transfer, as discussed in electron structure analysis part.| Compounds | βHRS |
|---|---|
| 1 | 0.34 |
| 2 | 0.85 |
| 3 | 0.59 |
| 4 | 2.40 |
| 5 | 2.43 |
| 6 | 6.51 |
| 7 | 25.48 |
4. Conclusions
In summary, we have investigated the electronic structure, chiroptical and linear optical properties, carrier transport, and second-order NLO response of seven boron-fused double helicenes and elucidated structure–property relationships, for the first time. The experimental electron absorption wavelengths are well reproduced by our calculations. The simulated CD spectra are in good agreement with the experimental CD spectra, not only wavelength position but also the relative rotational strength. Its chiral origin mainly results from exciton-coupling between the ortho-fused aromatic rings. The first hyperpolarizability value of compound 7 is greatly larger than that of phenyliminomethylferrocene complex. The charge transfer from TCNQ to aromatic rings is responsible for the NLO response. The ambipolar carrier transportation character mainly comes from intermolecular interaction (i.e. between a pure C6 ring and another C6 ring containing the boron and oxygen atoms). Our work is also important for further designing and optimizing heterohelicenes with better performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21503091 and 21573037) and the Natural Science Foundation of Jilin Province (20170101089JC).Notes and references
- J. E. Coughlin, Z. B. Henson, G. C. Welch and G. C. Bazan, Acc. Chem. Res., 2014, 47, 257–270 CrossRef CAS PubMed.
- C. Zhang and X. Z. Zhu, Acc. Chem. Res., 2017, 50, 1342–1350 CrossRef CAS PubMed.
- R. Shintani, N. Misawa, T. Tsuda, R. Iino, M. Fujii, K. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2017, 139, 3861–3867 CrossRef CAS PubMed.
- J. L. Wang, K. K. Liu, J. Yan, Z. Wu, F. Liu, F. Xiao, Z. F. Chang, H. B. Wu, Y. Cao and T. P. Russell, J. Am. Chem. Soc., 2016, 138, 7687–7697 CrossRef CAS PubMed.
- Y. Z. Lin and X. W. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- R. P. Xu, Y. Q. Li and J. X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- M. Muccini, Nat. Mater., 2006, 5, 605–613 CrossRef CAS PubMed.
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158–162 CrossRef CAS PubMed.
- C. Wang, T. Zhang and W. B. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
- J. L. Bredas, C. Adant, P. Tackx, A. Persoons and B. M. Persoons, Chem. Rev., 1994, 94, 243–278 CrossRef CAS.
- D. E. Katsoulis, Chem. Rev., 1998, 98, 359–387 CrossRef CAS PubMed.
- A. Persoons, T. Verbiest, S. V. Elshocht and M. Kauranen, Application of Chiral Symmetries in Even-Order Nonlinear Optics, American Chemical Society, Washington, DC, 2002, ch. 11, pp. 145–156 Search PubMed.
- L. M. Haupert and G. J. Simpson, Annu. Rev. Phys. Chem., 2009, 60, 345–365 CrossRef CAS PubMed.
- T. Verbiest and A. Persoons, in Materials-Chirality, ed. M. M. Green, R. J. M. Nolte and E. W. Meijer, Wiley, Hoboken, NJ, 2004, vol. 24 Search PubMed.
- E. Louis, E. San-Fabian, M. A. Diaz-Garcia, G. Chiappe, J. A. Verges, C. F. Chen and Y. Shen, Helicene Chemistry: From Synthesis to Applications, Springer, Berlin, 2017 Search PubMed.
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed.
- E. Louis, E. San-Fabian, M. A. Diaz-Garcia, G. Chiappe and J. A. Verges, J. Phys. Chem. Lett., 2017, 8, 2445–2449 CrossRef CAS PubMed.
- D. Hait, T. Zhu, D. P. McMahon and T. Van Voorhis, J. Chem. Theory Comput., 2016, 123353–123359 Search PubMed.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CAS.
- A. R. G. Smith, M. J. Riley, P. L. Burn, I. R. Gentle, S. C. Lo and B. J. Powell, Inorg. Chem., 2012, 51, 2821–2831 CrossRef CAS PubMed.
- C. Azarias, M. Pawelek and D. Jacquemin, J. Phys. Chem. A, 2017, 121, 4306–4317 CrossRef CAS PubMed.
- G. C. Yang, Y. Liao, Z. M. Su, H. Y. Zhang and Y. Wang, J. Phys. Chem. A, 2006, 110, 8758–8762 CrossRef CAS PubMed.
- W. Y. Wang, N. N. Ma, C. H. Wang, M. Y. Zhang, S. L. Sun and Y. Q. Qiu, J. Mol. Graphics Modell., 2014, 48, 28–35 CrossRef CAS PubMed.
- A. H. Pandith and N. Islam, PLoS One, 2014, 9(12), e114125 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09W (Revision B.01), Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS; (c) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- P. A. Brooksby and W. R. Fawcett, Spectrochim. Acta, Part A, 2001, 57, 1207–1221 CrossRef CAS.
- C. Bernini, L. Zani, M. Calamante, G. Reginato, A. Mordini, M. Taddei, R. Basosi and A. Sinicropi, J. Chem. Theory Comput., 2014, 10, 3925–3933 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Chem. Rev., 1999, 99, 2161–2200 CrossRef CAS PubMed.
- F. Mançois, L. Sanguinet, J. L. Pozzo, M. Guillaume, B. Champagne, V. Rodriguez, F. Adamietz, L. Ducasse and F. Castet, J. Phys. Chem. B, 2007, 111, 9795–9802 CrossRef PubMed.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J. L. Pozzo and V. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- R. Bersohn, J. Chem. Phys., 1966, 45, 3184 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- M. Q. Long, L. Tang, D. Wang, L. Wang and Z. Shuai, J. Am. Chem. Soc., 2009, 131, 17728–17729 CrossRef CAS PubMed.
- A. Wang, Y. Wang, J. Jia, L. Feng, C. Zhang and L. Liu, J. Phys. Chem. A, 2013, 117, 5061–5072 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS PubMed.
- Y. L. Si and G. C. Yang, RSC Adv., 2013, 3, 2241–2247 RSC.
- M. Dierksen and S. Grimme, J. Chem. Phys., 2006, 124, 174301 CrossRef PubMed.
- Y. M. Sang, L. K. Yan, N. N. Ma, J. P. Wang and Z. M. Su, J. Phys. Chem. A, 2013, 117, 2492–2498 CrossRef CAS PubMed.
- L. Wang, W. Y. Wang, Y. Q. Qiu and H. Z. Lu, J. Phys. Chem. C, 2015, 119, 24965–24975 CAS.
- Y. L. Si and G. C. Yang, J. Mater. Chem. C, 2013, 1, 2354–2361 RSC.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- R. Cammi, B. Mennucci and J. Tomasi, J. Am. Chem. Soc., 1998, 120, 8834–8847 CrossRef CAS.
- D. Cavagnat, T. Buffeteau and T. Brotin, J. Org. Chem., 2008, 73, 66–75 CrossRef CAS PubMed.
- J. H. Bredehöft, N. C. Jones, C. Meinert, A. C. Evans, S. V. Hoffmann and U. J. Meierhenrich, Chirality, 2014, 26, 373–378 CrossRef PubMed.
- V. P. Nicu, E. J. Baerends and P. L. Polavarapu, J. Phys. Chem. A, 2012, 116, 8366–8373 CrossRef CAS PubMed.
- M. Q. Long, L. Tang, D. Wang, L. J. Wang and Z. G. Shuai, J. Am. Chem. Soc., 2009, 131, 17728–17729 CrossRef CAS PubMed.
- Y. X. He, Y. Y. Huang, J. Y. Li, X. Pang and G. C. Yang, Org. Electron., 2017, 50, 220–227 CrossRef CAS.
- E. G. Kim, V. Coropceanu, N. E. Gruhn, R. S. Carrera, R. Snoeberger, A. J. Matzger and J. L. Brédas, J. Am. Chem. Soc., 2007, 129, 13072–13081 CrossRef CAS PubMed.
- L. Liu, G. C. Yang, Y. Geng, Y. Wu and Z. M. Su, RSC Adv., 2014, 4, 50188–50194 RSC.
- C. Y. Liu, G. C. Yang, Y. L. Si, Y. J. Liu and X. M. Pan, J. Mater. Chem. C, 2017, 5, 3495–3502 RSC.
- G. C. Yang, Z. M. Su and C. S. Qin, J. Phys. Chem. A, 2006, 110, 4817–4821 CrossRef CAS PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- S. K. Pal, A. Krishnan, P. K. Das and A. G. Samuelson, J. Organomet. Chem., 2000, 604, 248–259 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: HOMO and LUMO of compounds 2–6. Fig. S2: molecular orbital isosurfaces involved in the main electron transitions of compounds 1, 3, 5 and 6. Fig. S3: molecular orbital isosurfaces involved in the main electron transitions of compound 4. Fig. S4: molecular orbitals involved into the main ECD transitions of compound 2. Fig. S5: the calculated second-order NLO response values obtained from the four functionals (i.e. B3LYP, BHandHLYP, CAM-B3LYP and PBE0). Table S1: calculated absorption wavelength (nm) for compound 2 with the TDDFT method at the PBE0, B3LYP, Cam-B3LYP and BHandHLYP level, respectively, together with the experimental data. Table S2: the main concerned bond length for compound 1 between experiment and calculation. Table S3: the calculated excitation energies, oscillator strengths and rotational strengths for compound 1 in the gas phase at the TD-B3LYP/6-31+G(d) level. See DOI: 10.1039/c7ra11476a |
| This journal is © The Royal Society of Chemistry 2017 |