
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface plasmon-driven catalytic reactions on a patterned Co3O4/Au inverse catalyst†
Si Woo Lee ab,
Changhwan Leeab,
Kalyan C. Goddetiab,
Sun Mi Kimb and
Jeong Young Park*ab
ab,
Changhwan Leeab,
Kalyan C. Goddetiab,
Sun Mi Kimb and
Jeong Young Park*ab
aGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 305-701, Republic of Korea. E-mail: jeongypark@kaist.ac.kr
bCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science, Daejeon, 305-701, Republic of Korea
First published on 12th December 2017
Abstract
We report hot electron- and surface plasmon-driven catalytic reactions on an inverse catalyst that is composed of a reactive cobalt oxide (Co3O4) for the catalyst portion and a metal as the source of localized surface plasmon resonance (LSPR). To achieve this scheme, a patterned Co3O4/Au nanostructure was fabricated using the nanosphere lithography technique. The enhancement of catalytic activity for CO oxidation on cobalt oxide by the transfer of plasmonic hot carriers generated from the LSPR of Au under light irradiation was observed. We found that the Co3O4/Au inverse catalyst exhibited 25–50% higher catalytic activity when under light irradiation. We attribute this enhancement of catalytic activity to the flow of surface plasmon-driven hot carriers that were created by the absorption of photons on the Au metal, followed by injection into the active Co3O4 layers.
1 Introduction
Plasmonic metal nanoparticles have been widely used for many applications (e.g. catalytic activity, water splitting reactions, biosensors, solar-cell design, and cancer diagnostics).1–6 Surface plasmon resonance (SPR) is a strong electromagnetic field near the surface of noble metal nanostructures that can occur in plasmonic metal nanostructures by resonant oscillation of free electrons with the same wavelength as the incident photons.7–9 Recently, many studies have shown the generation of hot electrons with high kinetic energy (1–3 eV) by the electromagnetic decay of surface plasmons at a femtosecond timescale in plasmonic nanostructures under light irradiation and SPR excitation.10–12 Mukherjee et al. revealed that H2 molecules on Au nanoparticles can dissociate at room temperature because of hot electrons created from surface plasmon decay under visible light irradiation.13 Lee et al. found a steady-state flow of hot electrons generated by localized surface plasmon resonance (LSPR) at a metal–semiconductor junction.14 Wang et al. reported that a high concentration of hot electrons can tunnel through the insulating layer and generate photocurrent in a metal–insulator–metal device.15To fabricate new plasmonic nanostructures with homogeneous size, shape, and interparticle distance of the structures, several nanolithography techniques have been developed (e.g. electron beam lithography, focused ion beam lithography, and nanoimprint lithography).16–18 Furthermore, the nanosphere lithography (NSL) technique is a low-cost fabrication technique that uses 2-dimensional colloidal crystals (e.g. 2D arrays of polystyrene and silica nanospheres) as a mask. The advantage of the NSL technique for plasmonic nanostructures is that the LSPR of noble metal nanostructures can be easily controlled in the visible to near-IR region because the aspect ratio that determines the extinction maximum wavelength can be accurately adjusted by varying the size of the nanospheres used as the mask and the thickness of the deposited noble metal.19
Conventional catalysts consist of a metal as the catalyst and an oxide or semiconductor as the support for the metal catalyst. It has been demonstrated that the interaction between the metal and the oxide support at their interface (i.e. strong metal–support interactions (SMSI)) can affect the catalytic activity with a dependence on the oxidation state of the metal catalyst and the doping state or reducibility of the oxide support.20–23 In addition, there are some studies that show the influence of hot electrons from the oxide or semiconductor support on the catalytic activity of metal catalysts when under light irradiation.24–26
To understand the intrinsic relation between catalytic activity and hot electron flows generated across metal–oxide interfaces, the design of a fresh catalyst model is important.27 In previous studies, the active site was the metal, which is influenced by hot electrons from an oxide or a semiconductor. The major shortcoming of this conventional catalyst is the short range of the electron mean free path for the metal, which can de-energize the hot electron before its arrival at the metal surface. Kim et al. reported that the change in catalytic activity for CO oxidation on Au/CeO2 or Pt/GaN catalysts under light irradiation decreased as the size of the Au or Pt nanoparticles increased because of the length of the mean free path of the metal.24,26 Lee et al. found that smaller Pt nanoparticles lead to higher chemicurrent yield (i.e. the number of hot electrons detected per molecule of reaction product formed on the Au/TiO2 nanodiode surface) during hydrogen oxidation, which is associated with the shorter travel length for the hot electrons, compared with their inelastic mean free path.28 Therefore, the role of the electron mean free path is critical for studying the relationship between catalytic activity and hot electron flow.
Some oxide materials (e.g. cobalt oxide (Co3O4), cerium oxide (CeO2) and cuprous oxide (Cu2O)) are catalytically reactive because these oxide materials can be easily and reversibly oxidized and reduced.29–31 Among these reactive oxide materials, Co3O4 is specifically known as the most catalytically active oxide material for CO oxidation. Haruta and co-workers demonstrated that some base metal oxides (e.g. Co3O4, MnO2, and NiO) are intrinsically reactive for CO oxidation without a metal catalyst and that the catalytic activity of base metal oxides decreases in the following order: Co3O4 > MnO > NiO > CuO > Fe2O3 > Mn2O3 > Fe3O4 > CeO2 > TiO2.32 Xie et al. found that Co3O4 nanorods with predominantly exposed [110] planes are very reactive and stable for CO oxidation at temperatures as low as −77 °C, giving 100% CO conversion because of the presence of active Co3+ species at the surface.33 Song et al. reported that mesoporous cobalt oxide nanoparticle aggregates can achieve 100% CO conversion to CO2 at −60 °C under normal conditions.34 It was found that a small Co3O4–SiO2 nanocomposite catalyst prepared using an activated carbon template exhibits very high activity for CO oxidation at −76 °C.35
To overcome the short electron mean free path problem in the metal, we used an inverse catalyst in this study that is composed of a reactive oxide for the catalytic portion and a noble metal for the LSPR source. Triangular noble metal nanostructures show much higher surface-enhanced Raman scattering (SERS) activities than flat noble metals because of the large number of confined hot spots between adjacent triangles.36 Subsequently, we grew Co3O4 on a patterned triangular Au nanostructure to make the patterned Co3O4/Au inverse catalyst using NSL (Fig. 1a). We studied the catalytic activity for CO oxidation on the patterned Co3O4/Au inverse catalyst with and without light irradiation to identify the role of hot electrons from LSPR on the catalytic reaction. The energy band diagram of the Co3O4/Au junction (Fig. 1b) shows the transfer of hot electrons and their effect on catalytic activity. The Schottky barrier height for Au/Co3O4 is 1.6 eV, which was determined by the difference between the work function of the Au (5 eV) and the electron affinity for Co3O4 (3.4 eV).37 The main merit of our inverse catalyst is the long mean free path in the oxide created by using a reactive oxide material on a plasmonic noble metal. Energetic hot electrons having high kinetic energy (2.0–2.1 eV) formed via LSPR upon the absorption of photons on the plasmonic gold. The hot electrons then travelled over the Schottky barrier and arrived at the Co3O4 surface because of their long mean free path. Thus, these plasmonic hot carriers can affect the catalytic activity of CO oxidation on Co3O4.
 | ||
| Fig. 1 (a) Schematic drawing of the patterned Co3O4–Au nanostructure and the effect of hot electrons on catalytic activity under light irradiation. (b) Energy band diagram for the Au–Co3O4 inverse catalyst representing the flow of hot electrons generated from the LSPR of the patterned Au nanostructures and their effect on the CO oxidation reaction when under light irradiation. | ||
2 Experimental
2.1 Fabrication of the two-dimensional patterned Co3O4/Au nanostructures by nanosphere lithography
2.2 Characterization, chemical modification, and structures of the patterned Co3O4/Au nanostructures
The crystalline phases of the cobalt oxide and the Au nanostructure before and after heat treatment were revealed using X-ray diffraction patterns (XRD, Rigaku D/MAX-2500) taken at a 2θ scan range of 10–70°, scan speed of 4° min−1, and step size of 0.01 Å using Cu Kα radiation. The oxidation state of the cobalt oxide (Co 2p) before and after CO oxidation was identified using X-ray photoelectron spectroscopy (XPS, Thermo VG Scientific Sigma Probe system with an Al Kα X-ray source (1486.3 eV)) with an energy resolution of 0.47 eV full width at half maximum (FWHM) under ultrahigh vacuum (10−10 Torr). The thickness and morphology of the patterned Co3O4/Au nanostructures were characterized using cross-section transmission electron microscopy (TEM, FEI Titan G2 Cubed 60-300), scanning transmission electron microscopy and high-angle annular dark field (STEM-HAADF, FEI Titan G2 Cubed 60-300), corresponding energy-dispersive X-ray spectroscopy (EDS, FEI Super X (4 SDD EDS)) elemental maps for 300 s, energy-dispersive X-ray spectroscopy (EDS) line profiles, and scanning electron microscopy (SEM, Magellan 400). The TEM specimen was prepared using the focused ion beam (FIB, Helios Nanolab 450 F1) preparation technique for cutting cross sections of the Co3O4/Au nanostructures. Two sizes of triangular Au nanostructures were created by changing the size of the SiO2 nanosphere mask. The optical properties of these Au nanostructures were characterized using a UV-vis spectrophotometer (Lambda 1050). Triangular Au nanostructures (30 nm high) were prepared on a quartz window (1 mm thick) using the same method described previously. The spectral range was 400–800 nm, and a clean quartz window was used as a reference.2.3 Measurement of the catalytic activity of CO oxidation on patterned Co3O4/Au inverse catalysts with and without light irradiation
Reaction studies were performed in an ultrahigh vacuum chamber (1 L) with a base pressure of 1 × 10−8 Torr, evacuated using rotary pumps and turbo molecular pumps. After isolating with a gate valve, the CO oxidation reaction was carried out in the batch reactor system under 40 Torr CO and 100 Torr O2 as the reactant gases and 620 Torr He as the balancing gas. All the gases were circulated continuously through the reaction line by a Metal Bellows recirculation pump at a rate of 2 L min−1. After 30 min of recirculation at room temperature for gas equilibration, a DS 6200 gas chromatograph (GC) equipped with a thermal conductivity detector and a 6 in. × 1/8 in. SS molecular sieve 5A was used to separate the products for analysis. The CO conversion was monitored at various temperatures (523–553 K) and the measured reaction rates were reported as turnover frequencies (TOF), which were measured in units of product molecules of CO2 produced per active Co3O4 surface site per second of reaction time. All the reaction data were obtained by measuring the rate of CO oxidation at low conversion (i.e., conversion below ∼15%) to check the initial reaction rate within a kinetically controlled regime, assuming different reaction conditions. The number of active sites was calculated from the geometry in the SEM measurements of the surface area of the nanoparticle arrays. A halogen lamp (Dolhan-Janner D150) was used as the light source to measure the enhancement of catalytic activity while under light irradiation. The distance between the sample and halogen lamp was about 8 cm through a sapphire window and the light intensity was measured to be 49.4 mW cm−2 at this distance by an optical power meter (ADCMT 8230).2.4 Finite-difference time domain (FDTD) simulations
The distributions of the electric field by localized surface plasmon resonance (LSPR) on the patterned Co3O4–Au nanostructure and the patterned Au nanostructure were calculated using a two-dimensional FDTD simulation with a commercial FDTD simulation package (FDTD-Lumerical™). The optical properties of the gold were obtained from the materials database in the software using the Drude model. The refractive index of Co3O4 was obtained experimentally using a spectroscopic ellipsometer (Woollam M2000D). Variable angle spectroscopic ellipsometric (VASE) data (i.e. 65.00, 70.00, 75.00°) were acquired from a Co3O4 thin film on a SiO2 wafer (173 nm cobalt oxide/1000 nm SiO2) and modelled in B-spline mode to calculate the refractive index for cobalt oxide (2.8 at 622 nm). The electric field distribution was acquired with a mesh size of 0.5 nm at an incident light of 622 nm, which is the extinction maximum wavelength (λex) of the nanostructures derived from the UV-vis absorbance spectra. The geometric model used in these calculations for the patterned Co3O4/Au nanostructure and the patterned Au nanostructure was built from the SEM and TEM images.3 Results and discussion
3.1 Characterization of the patterned Co3O4/Au inverse catalysts
We fabricated reactive oxide/plasmonic noble metal hybrid structures using the NSL technique. The Co3O4 on triangular Au nanostructures were thus fabricated: first, 300 nm SiO2 nanospheres were synthesized using the well-known Stöber method, and self-assembled monolayer arrays of silica nanospheres were prepared on a clean SiO2/Si substrate using the LB technique. Next, 30 nm of gold as the LSPR source and 40 nm of cobalt as the catalyst were deposited sequentially by electron beam evaporation on the previously fabricated closely packed SiO2 nanospheres on SiO2/Si substrate. The closely packed SiO2 nanosphere arrays act as a mask so that all the metal was deposited using the SiO2 nanospheres for patterning. Finally, the cobalt was oxidized to Co3O4 by heat treatment at 553 K for 2 h 30 min and the 300 nm SiO2 nanospheres were removed by sonication for 1 minute with deionized water, thus obtaining the patterned Co3O4–Au islands. All the fabrication procedures and SEM images are shown in Fig. 2a–d and e–h, respectively. | ||
| Fig. 2 Fabrication procedure for the patterned Co3O4–Au nanostructures and corresponding SEM images. (a and e) Deposition of 300 nm SiO2 nanospheres using the LB technique. (b and f) E-Beam deposition of 30 nm of Au. (c and g) E-Beam deposition of 40 nm of Co. (d and h) Heat treatment for oxidation of the cobalt to cobalt oxide and removal of the SiO2 nanospheres. | ||
Fig. 3a shows the crystalline phase of the cobalt oxide according to annealing temperature, as revealed by XRD patterns. After annealing at 553 K, diffraction peaks corresponding to a cubic spinel structure were confirmed, indicating that the cobalt was fully oxidized to Co3O4. Co3O4 has a cubic spinel structure and the Au nanostructure was polycrystalline after annealing at 553 K, as shown in Fig. 3a and S1 in ESI.† Fig. 3b illustrates the UV-vis extinction spectra of the 30 nm triangular Au nanostructures fabricated by NSL using 200 and 300 nm SiO2 nanospheres as masks. All the Au cones remained and there were no SiO2 nanospheres after sonication, as shown in Fig. S2 in ESI.† It is well known that the optical properties (e.g. the intensity and extinction maximum wavelength) of the LSPR for noble metal nanoparticles are influenced by shape, size, and dielectric environment. Previous studies show that the extinction maximum wavelength of triangular noble metal nanostructures fabricated by NSL can be tuned by changing the aspect ratio (i.e. the ratio of the length and the height) of the triangular nanostructures.38,39 In this work, the aspect ratio was controlled by changing the size of the SiO2 nanospheres used as the mask. The extinction maximum wavelength peak of the triangular Au nanostructures from the 300 nm SiO2 nanospheres was red-shifted from the wavelength peak of the triangular Au nanostructures from the 200 nm SiO2 nanospheres because of the increased aspect ratio from the large SiO2 nanospheres, which reveals that the extinction spectrum represents absorption by the LSPR of the triangular Au nanostructures. In addition, there was an ultraweak absorption band about 750 nm away from the extinction maximum wavelength peak. This broad LSPR band came from some defects in the triangular Au nanostructures that were caused by misalignment of the SiO2 nanospheres or by the size distribution of the SiO2 nanospheres, as shown in Fig. S2 in ESI.†39
 | ||
| Fig. 3 (a) XRD patterns of the cobalt oxide at different annealing temperatures. (b) UV-visible absorbance spectra of the patterned Au nanostructures fabricated using 200 or 300 nm SiO2 spheres. | ||
To identify the thickness and morphology of the patterned Co3O4/Au nanostructures, a cross-sectional TEM image was taken (Fig. 4a). During fabrication of the Co3O4/Au nanostructures, vacancies between three closely packed SiO2 nanosphere arrays created cobalt oxide/Au nanostructures with nanopyramidic shape. Fig. 4d–h shows a STEM-HAADF image and EDS elemental maps for 300 s of the patterned Co3O4/Au nanostructure. It clearly shows that the top layer is cobalt oxide (i.e. the catalyst portion) and that there is a Au triangle under the cobalt oxide (i.e. the LSPR source). An EDS line profile of the patterned Co3O4/Au nanostructure is shown in Fig. 4b and c. The heights of the Au triangle and cobalt oxide are 30 and 40 nm, respectively.
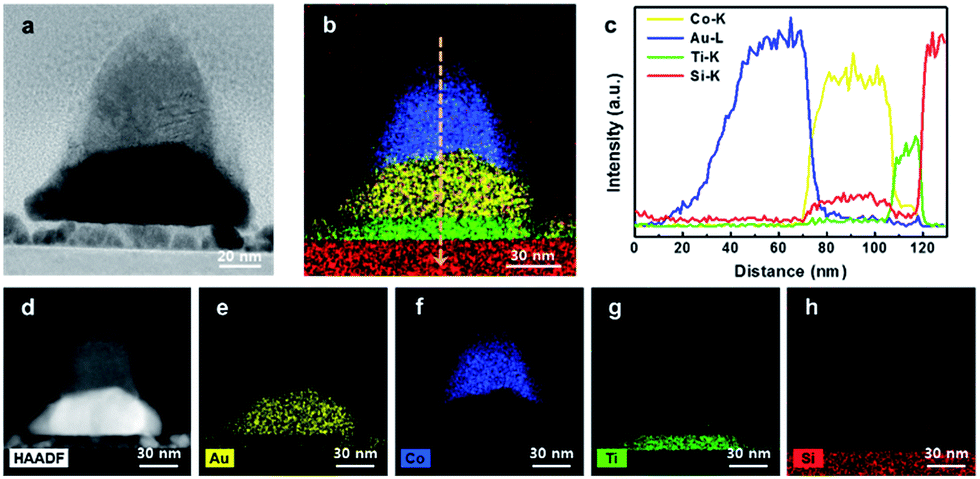 | ||
| Fig. 4 (a) TEM image of the Co3O4–Au nanostructure. (b and c) EDS line profile of the Co3O4–Au nanostructure. (d) STEM-HADDF image of the Co3O4–Au nanostructure and (e–h) EDS elemental maps of each component. | ||
3.2 Surface-plasmon driven CO oxidation on patterned Co3O4/Au inverse catalysts
Here, we carried out CO oxidation on the patterned Co3O4/Au inverse catalyst with and without light to show the intrinsic relation between catalytic activity and hot electron flow from the LSPR on the triangular Au nanostructure during light irradiation. Reaction studies were performed in an ultrahigh vacuum chamber (1 L) with a base pressure of 1 × 10−8 Torr, evacuated using rotary and turbo molecular pumps. After isolation using a gate valve, the CO oxidation reaction was carried out in the batch reactor system under 40 Torr CO and 100 Torr O2 as the reactant gases and 620 Torr He as the balancing gas. CO conversion was monitored at various temperatures (523–553 K) and the measured reaction rates were reported as TOF, which are measured in units of product molecules of CO2 produced per active Co3O4 surface site per second of reaction time.Fig. 5a and b shows the catalytic activity for CO oxidation measured on the patterned Co3O4/Au inverse catalyst at four different temperatures (523–553 K) with and without light. The patterned Co3O4/Au inverse catalyst shows a higher catalytic activity under light irradiation than in the dark. Previous studies show that the enhancement of the activity of the Pt NPs/p-doped GaN wafer under light irradiation was 11–33% which was measured using the same batch reactor system and halogen lamp as used in this study;26 thus, the enhancement under light irradiation (25–50%) observed on the Co3O4/Au inverse catalyst is significant. It is associated with hot electrons generated from LSPR excitation of the triangular Au nanostructure by light absorption that can affect the catalytic activity for CO oxidation on the reactive Co3O4. However, the thermal effect of light irradiation can also result in the enhancement of catalytic activity. We measured sample temperature data with a temperature controller with and without light at 523 K under 40 Torr CO, 100 Torr O2, and 620 Torr He (Fig. S3 in ESI†). There was no change in the sample temperature when light was irradiated on the sample, demonstrating that any heating effect by light irradiation can be ignored in our experiments. Also some previous studies show a decrease in catalytic activity for CO oxidation on Pt/n-Si and Pt nanoparticle/n-type GaN resulting from a reduced net charge on the Pt nanoparticles when under light irradiation.25,26,40 CO oxidation was also carried out on the patterned Co3O4 nanostructure with and without light under the same conditions to compare the catalytic activity of the patterned Co3O4/Au inverse catalyst (Fig. 5c). Under light irradiation, the catalytic activity of the Co3O4 exhibited an insignificant difference (i.e. within the error of measurement). Even if a hot electron is generated on the Co3O4 from electron–hole pair excitation when under light irradiation, it will eventually turn into a low-energy electron through inelastic scattering within a time scale of femtoseconds. Hot electrons from LSPR excitation on Au can be transferred when a metal–oxide interface is formed, revealing that a role for Au nanostructures is as an LSPR excitation source, leading to a change in catalytic activity for CO oxidation. The TOF values of the patterned Co3O4/Au inverse catalyst were similar to the TOF values of the patterned Co3O4 nanostructure in the same reaction temperature range, demonstrating that there was no synergetic effect between the Au and the Co3O4. We also carried out CO oxidation on the patterned Au nanostructure at 573 K without Co3O4, but there was no CO2 product although the triangular Au nanostructure was polycrystalline. Thus, the CO oxidation reaction was occurring on the Co3O4, and not on the Au nanostructure. While the triangular Au nanostructure is not reactive in this reaction temperature range, it does perform a role as the LSPR source when under light irradiation. Hence, we can confirm that the enhancement of catalytic activity only originated from hot carriers generated by LSPR excitation of the Au that were then transferred to the reactive Co3O4.
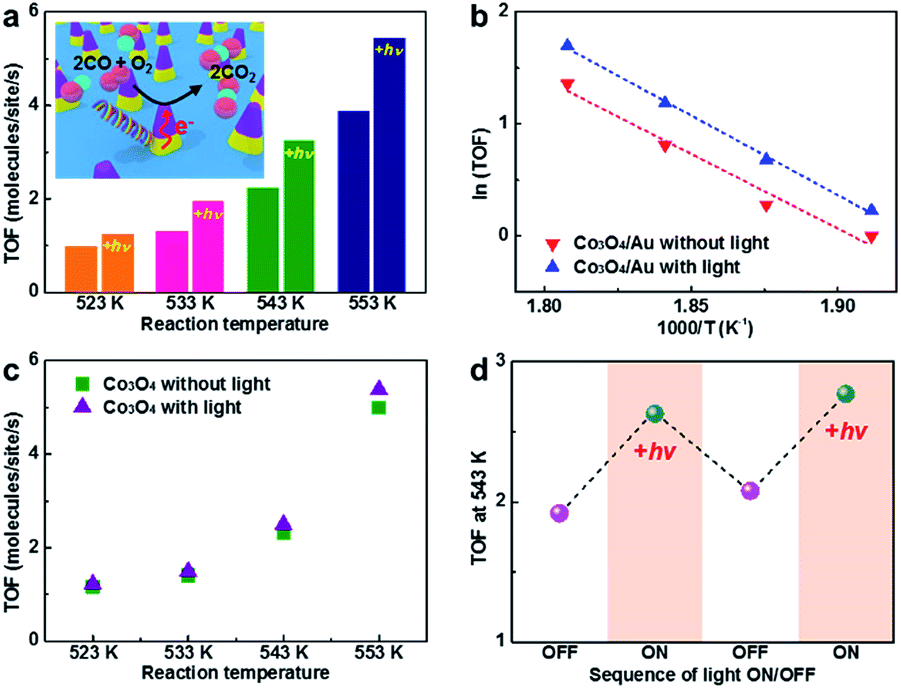 | ||
| Fig. 5 (a) Catalytic activity for CO oxidation on the patterned Co3O4–Au inverse catalyst with and without light at four different temperatures. (b) Arrhenius plots of the patterned Co3O4–Au inverse catalyst with and without light at four different temperatures. (c) Catalytic activity for CO oxidation on the patterned Co3O4 nanostructure with and without light at four different temperatures. (d) TOF for CO oxidation with sequential light ON/OFF on the patterned Co3O4/Au at 543 K. The TOF value was calculated from the slope of the turnover number with time. | ||
To confirm whether the plasmonic hot carrier effect on catalytic activity was reversible, CO oxidation was carried out continuously on the patterned Co3O4/Au inverse catalyst as the light was turned on and off at 543 K (Fig. 5d). The TOF value was similar to prior experiments (Fig. 5a): the catalytic activity was enhanced and then returned to the original catalytic activity as the halogen lamp was turned on and off, respectively, showing that the plasmonic hot carrier effect from the triangular Au nanostructure to the Co3O4 was reversible. The small increase in the TOF value in the second cycle was the result of calcination of the sample during the reaction. After the reaction, the morphology of the patterned Co3O4/Au nanostructure had minor changes, but there were no significant morphology changes, as verified by SEM images taken before and after the CO oxidation reaction (Fig. S4 in ESI†). XPS measurements showed that the oxidation state of the cobalt oxide was a mixture of Co2+ and Co3+, representing Co3O4. After the reaction, a higher percentage of Co3+ was observed (Fig. S5 in ESI†).
To understand the electric field distribution from the LSPR around the patterned Co3O4–Au nanostructure, we carried out two-dimensional FDTD simulation analysis. The electric field distribution was obtained with a mesh size of 0.5 nm and incident light of 622 nm, which corresponds to the extinction maximum wavelength of the nanostructures from the UV-vis absorbance spectra. We set the refractive index of the Co3O4 at 622 nm to 2.8, which was acquired experimentally, and the geometric model for the patterned Co3O4/Au nanostructure was built on experimentally observed structural parameters from SEM and TEM images. The calculated simulation results show an electric field distribution that indicates an enhancement of light absorption for the top and cross-section views of the nanostructure. Fig. 6a shows that the electric field was formed around the vertex points of the patterned Au nanostructure, which was oriented along the direction of polarization. In Fig. 6b, the electric field was confined prominently along the gold edge between two adjacent Au triangles. In particular, a strong electric field was formed at the interface between the Au and Co3O4 due to the higher refractive index of the Co3O4.41 For the patterned Au nanostructure without Co3O4, there was no electric field on the top surface of the Au nanostructure and the electric field was formed only along the side of the Au nanostructure (Fig. S6 in ESI†). This interesting LSPR confined at the Au–Co3O4 interface can support the plausible hot electron transfer mechanism, indicating that the generated hot electrons at the Au edge or the Au–Co3O4 interface were transferred to the Co3O4 over the Schottky barrier and can easily transfer to the Co3O4 surface owing to their long electron mean free path affecting the catalytic activity on the cobalt oxide.
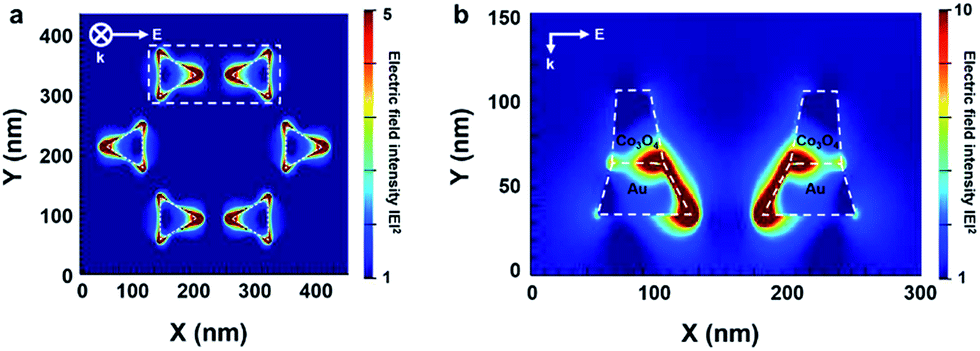 | ||
| Fig. 6 FDTD calculations of the electric field intensity from LSPR on the patterned Co3O4–Au nanostructure: (a) top view and (b) cross-section view. | ||
CO molecules are known to be well adsorbed on Co3O4, especially on the Co3+ site, and the mechanism for CO oxidation over Co3O4 is believed to the Mars–van Krevelen type in which CO molecules adsorb on the Co3O4 surface and react with lattice oxygen at Co3O4 followed by O2 filling in the oxygen vacancy of the Co3O4.32,42 We can explain that the enhancement of catalytic activity when under light irradiation is attributed to the Co3O4 surface becoming negatively charged by hot electrons from LSPR excitation of the triangular Au nanostructure. Previous studies used ambient-pressure X-ray photoelectron spectroscopy (APXPS) and near edge X-ray adsorption fine structure spectroscopy (NEXAFS) to show that the main reaction intermediate for CO oxidation on Cu-based catalysts is adsorbed CO2δ−.43 Maximoff et al. shows that partially charged oxygen (Oδ−) can react with CO and produce CO2δ− (i.e. the partially negatively charged reaction intermediate), and finally produce CO2 and generate chemicurrent.44 Calaza et al. reported that electrons are shuttled from the ultrathin MgO(001) film grown on Ag(001) to Au islands, thus inducing the formation of a negative CO2 radical that reacts to form an oxalate.45 There are also earlier studies showing that the catalytic activity of CO oxidation on Pt catalysts supported on F-doped TiO2 was enhanced by O activation because of electron spillover from the highly n-type TiO2.22 The mechanism for electron spillover is still not clear; it was postulated that electrons in the highly doped TiO2 can activate lattice oxygen in the TiO2, which is then followed by CO oxidation with adsorbed CO at the Pt/TiO2 interface in a Mars–van Krevelen type mechanism.46 In our studies, hot electrons at negatively charged Co3O4 can transfer to the lattice oxygen at Co3O4 and produce CO2 through the reaction intermediate (CO2δ−), O (Co3O4) + δe− → Oδ− (Co3O4), CO(g) + Oδ− (Co3O4) → CO2δ−(ad) → CO2(g); or transfer to the reaction intermediate (CO2δ−) and produce gas-phase CO2, CO(g) + O (Co3O4) → CO2(ad), CO2(ad) + δe− → CO2δ−(ad) → CO2(g). Thus, hot electrons from LSPR excitation of the triangular Au nanostructure can easily transfer to the Co3O4 surface because of their long electron mean free path, and the negatively charged species can transfer to lattice oxygen at Co3O4 or adsorbed CO2 to generate negatively charged oxygen (Oδ−) or adsorbed CO2 (CO2δ−), which play important roles for CO2 production from CO and oxygen via the Mars–van Krevelen type mechanism.
Another possible mechanism for explaining the enhancement of catalytic activity by LSPR of the triangular Au nanostructure under light irradiation is the partial reduction of Co3+ to Co2+. Marimuthu et al. reported that selectivity for propylene oxide in propylene epoxidation increases on the Cu2O shell–metallic Cu core structure when under light irradiation. They found that Cu2O was reduced to metallic Cu, which is attributed to LSPR of the metallic copper under visible light irradiation using an in situ UV-vis extinction experiment.47 Kim et al. demonstrated the reduction of Fe3+ ions bound to a cyanide/Ag nanoparticle system induced by plasmonically generated hot electrons from the Ag nanoparticles.48 Also, Li et al. reported the selectivity of benzyl alcohol oxidation to benzaldehyde on a gold core@ceria shell catalyst under visible light irradiation that resulted from reducing Ce4+ to Ce3+ by hot electron transfer from the LSPR of the gold core to the CeO2 shell.30 Shen and co-workers found that Co3O4 nanorods with predominantly exposed [110] planes are very catalytically active for CO oxidation at low temperature, giving 100% CO conversion because of the presence of active Co3+ species at the surface. This unexpected catalytic reactivity of Co3O4 nanorods for CO oxidation is explained in the following mechanism: Co3O4 has a spinel structure containing Co3+ in an octahedral coordination and Co2+ in a tetrahedral coordination. In these surface atomic configurations, there are only Co2+ cations on the [001] and [111] planes, which are almost inactive, while the [110] plane is mainly composed of Co3+ cations, which are regarded as the active site for CO oxidation. Thus, CO only adsorbs on the surface Co3+ site and oxidation of the adsorbed CO then occurs by abstracting the surface oxygen that was coordinated with the Co3+ cations. The Co3+ cations are partially reduced to Co2+ and, finally, the partially reduced cobalt oxide can be re-oxidized by a gas-phase oxygen molecule to become active Co3+ cations again.33 The reaction between the adsorbed CO on Co3+ and the lattice oxygen is the overall rate-determining step in CO oxidation.42 Broqvist et al. performed a first-principles density functional theory (DFT) study regarding the CO oxidation mechanism at the Co3O4(110) surface and found that the reduction of Co3+ to Co2+ (Co3+ + e− → Co2+) in Co3O4(110) is crucial for the O abstraction step for CO2 formation.49 Here, in our patterned Co3O4–Au inverse catalysts, if a negatively charged hot electron from LSPR excitation on Au is transferred to the cobalt oxide, reduction of the cobalt oxide can occur, which is a decisive step for CO oxidation.
To find the exact mechanism for the enhancement of catalytic activity at the cobalt oxide by hot electron transfer from the Au, theoretical DFT studies or in situ spectroscopic and microscopic experiments (e.g. ambient-pressure techniques) are needed for these catalysts under light irradiation. But our studies here on these reactive oxide/plasmonic noble metal inverse catalysts can connect the enhancement of catalytic activity to LSPR excitation from the metal, having possible applications for surface plasmon-based oxide/metal hybrid nanocatalysts or catalytic nanodiodes because of the long electron mean free path of the oxide materials.
4 Conclusions
In summary, Co3O4 is known as a reactive oxide material for CO oxidation. Triangular noble metal nanostructures fabricated using the NSL technique exhibit higher SERS activities than flat noble metals. We used cobalt oxide as the catalytic portion and a triangular Au nanostructure as the SPR source for the patterned Co3O4/Au nanostructure. The patterned Co3O4/Au inverse catalyst nanostructures were prepared using the NSL technique with SiO2 nanospheres as the mask for metal deposition by electron beam evaporation. FDTD simulations showed that a particularly strong electric field was formed at the Au–Co3O4 interface because of the higher refractive index of the Co3O4. CO oxidation on the patterned Co3O4/Au nanostructure was carried out with and without light to find the relationship between hot electron transfer to the reactive oxide from LSPR excitation on the triangular Au nanostructure and the change in catalytic activity for CO oxidation. The patterned Co3O4–Au inverse catalysts showed 25–50% higher catalytic activity for CO oxidation when under light irradiation than in the dark. The reversibility of the plasmonic hot carrier effect on catalytic activity was confirmed by observation of two catalytic activities with the light turned on and off, respectively. The change in catalytic activity implies that hot carriers generated from LSPR excitation of the Au nanostructure by absorption of photons on the Au metal can transfer to the Co3O4, and thus reversibly enhance the catalytic activity for CO oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute for Basic Science (IBS) [IBS-R004-A2-2017-a00].Notes and references
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567 CrossRef CAS PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205 CrossRef CAS PubMed.
- W. H. Hung, M. Aykol, D. Valley, W. Hou and S. B. Cronin, Nano Lett., 2010, 10, 1314–1318 CrossRef CAS PubMed.
- D. B. Ingram and S. Linic, J. Am. Chem. Soc., 2011, 133, 5202–5205 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- I. H. El-Sayed, X. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS PubMed.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- J. Endriz and W. Spicer, Phys. Rev. Lett., 1970, 24, 64 CrossRef CAS.
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702–704 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- Y. K. Lee, C. H. Jung, J. Park, H. Seo, G. A. Somorjai and J. Y. Park, Nano Lett., 2011, 11, 4251–4255 CrossRef CAS PubMed.
- F. Wang and N. A. Melosh, Nat. Commun., 2013, 4, 1711 CrossRef PubMed.
- E. M. Hicks, S. Zou, G. C. Schatz, K. G. Spears, R. P. Van Duyne, L. Gunnarsson, T. Rindzevicius, B. Kasemo and M. Käll, Nano Lett., 2005, 5, 1065–1070 CrossRef CAS PubMed.
- E. Vesseur, R. De Waele, H. Lezec, H. Atwater, F. García de Abajo and A. Polman, Appl. Phys. Lett., 2008, 92, 083110 CrossRef.
- K. Li, L. Clime, B. Cui and T. Veres, Nanotechnology, 2008, 19, 145305 CrossRef PubMed.
- H. Ko, S. Singamaneni and V. V. Tsukruk, Small, 2008, 4, 1576–1599 CrossRef CAS PubMed.
- K. An, S. Alayoglu, N. Musselwhite, S. Plamthottam, G. r. m. Melaet, A. E. Lindeman and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 16689–16696 CrossRef CAS PubMed.
- L. R. Baker, G. Kennedy, M. Van Spronsen, A. Hervier, X. Cai, S. Chen, L.-W. Wang and G. A. Somorjai, J. Am. Chem. Soc., 2012, 134, 14208–14216 CrossRef CAS PubMed.
- L. R. Baker, A. Hervier, H. Seo, G. Kennedy, K. Komvopoulos and G. A. Somorjai, J. Phys. Chem. C, 2011, 115, 16006–16011 CAS.
- S. Tauster, S. Fung, R. Baker and J. Horsley, Science, 1981, 211, 1121–1125 CAS.
- S. M. Kim, H. Lee, K. C. Goddeti, S. H. Kim and J. Y. Park, J. Phys. Chem. C, 2015, 119, 16020–16025 CAS.
- S. M. Kim, S. J. Lee, S. H. Kim, S. Kwon, K. J. Yee, H. Song, G. A. Somorjai and J. Y. Park, Nano Lett., 2013, 13, 1352–1358 CrossRef CAS PubMed.
- S. M. Kim, D. Park, Y. Yuk, S. H. Kim and J. Y. Park, Faraday Discuss., 2013, 162, 355–364 RSC.
- J. Y. Park, S. M. Kim, H. Lee and I. I. Nedrygailov, Acc. Chem. Res., 2015, 48, 2475–2483 CrossRef CAS PubMed.
- H. Lee, I. I. Nedrygailov, C. Lee, G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 2340–2344 CrossRef CAS PubMed.
- F. Yang, J. Graciani, J. Evans, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, J. Am. Chem. Soc., 2011, 133, 3444–3451 CrossRef CAS PubMed.
- B. Li, T. Gu, T. Ming, J. Wang, P. Wang, J. Wang and J. C. Yu, ACS Nano, 2014, 8, 8152–8162 CrossRef CAS PubMed.
- G. Jernigan and G. Somorjai, J. Catal., 1994, 147, 567–577 CrossRef CAS.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, J. Catal., 2009, 267, 121–128 CrossRef CAS.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- W. Song, A. S. Poyraz, Y. Meng, Z. Ren, S.-Y. Chen and S. L. Suib, Chem. Mater., 2014, 26, 4629–4639 CrossRef CAS.
- C.-J. Jia, M. Schwickardi, C. Weidenthaler, W. Schmidt, S. Korhonen, B. M. Weckhuysen and F. Schüth, J. Am. Chem. Soc., 2011, 133, 11279–11288 CrossRef CAS PubMed.
- M. Tabatabaei, A. Sangar, N. Kazemi-Zanjani, P. Torchio, A. Merlen and F. o. Lagugné-Labarthet, J. Phys. Chem. C, 2013, 117, 14778–14786 CAS.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- W. Huang, W. Qian and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 18881–18888 CrossRef CAS PubMed.
- S. K. Cushing, J. Li, J. Bright, B. T. Yost, P. Zheng, A. D. Bristow and N. Wu, J. Phys. Chem. C, 2015, 119, 16239–16244 CAS.
- L. R. Baker, A. Hervier, G. Kennedy and G. A. Somorjai, Nano Lett., 2012, 12, 2554–2558 CrossRef CAS PubMed.
- Z. W. Seh, S. Liu, M. Low, S. Y. Zhang, Z. Liu, A. Mlayah and M. Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef CAS PubMed.
- H.-F. Wang, R. Kavanagh, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, J. Catal., 2012, 296, 110–119 CrossRef CAS.
- B. Eren, C. Heine, H. Bluhm, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2015, 137, 11186–11190 CrossRef CAS PubMed.
- S. N. Maximoff and M. P. Head-Gordon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11460–11465 CrossRef CAS PubMed.
- F. Calaza, C. Stiehler, Y. Fujimori, M. Sterrer, S. Beeg, M. Ruiz-Oses, N. Nilius, M. Heyde, T. Parviainen and K. Honkala, Angew. Chem., Int. Ed., 2015, 54, 12484–12487 CrossRef CAS PubMed.
- J. Y. Park, L. R. Baker and G. A. Somorjai, Chem. Rev., 2015, 115, 2781–2817 CrossRef CAS PubMed.
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 339, 1590–1593 CrossRef CAS PubMed.
- K. Kim, S. H. Lee, J.-Y. Choi and K. S. Shin, J. Phys. Chem. C, 2014, 118, 3359–3365 CAS.
- P. Broqvist, I. Panas and H. Persson, J. Catal., 2002, 210, 198–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of the Au nanostructure (Fig. S1). SEM image of triangular Au nanostructure after sonication (Fig. S2). Sample temperature measured by temperature controller with and without light at 523 K (Fig. S3). SEM images (Fig. S4) and XPS spectra (Fig. S5) of the patterned Co3O4/Au inverse catalyst before and after CO oxidation. FDTD simulations on the patterned Au nanostructure (Fig. S6). See DOI: 10.1039/c7ra10450b |
| This journal is © The Royal Society of Chemistry 2017 |