
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoregulated phase-separable rhodium nanoparticle catalyst for selective hydrogenation of α,β-unsaturated aldehydes and ketones†
Xiuru Xue ,
Mingming Niu,
Yicheng Xu and
Yanhua Wang*
,
Mingming Niu,
Yicheng Xu and
Yanhua Wang*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, PR China. E-mail: yhuawang@dlut.edu.cn; Fax: +86 411 84986033; Tel: +86 411 84986033
First published on 30th October 2017
Abstract
Thermoregulated phase-separable Rh nanoparticles (abbreviated as TPS-Rhnano) were found to be efficient catalysts for selective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in α,β-unsaturated aldehydes and ketones with a selectivity of >99% at >99% conversion. More importantly, the catalyst could be easily separated by simple phase separation and reused directly eight times without evident loss in activity and selectivity.
C bonds in α,β-unsaturated aldehydes and ketones with a selectivity of >99% at >99% conversion. More importantly, the catalyst could be easily separated by simple phase separation and reused directly eight times without evident loss in activity and selectivity.
Soluble transition-metal nanoparticles have drawn much attention because of their high catalytic activity and excellent selectivity. However, the direct separation and recovery of these nanoparticles from the products in homogeneous catalytic reactions have always been technical problems that have needed to be solved for the past few decades.1–4 Recently, based on our discovery that the polyether chain-based quaternary ammonium salt ionic liquids [CH3(OCH2CH2)nN+Et3][CH3SO3−] (ILPEG, n = 12, 16, 22) exhibit electrosteric stabilization for nanoparticles and a distinctive critical solution temperature (CST) property in toluene and n-heptane mixtures, we have established thermoregulated phase-separable catalytic system (TPSCS) involving soluble transition-metal nanoparticles.5 The basic principle of TPSCS was described in Fig. 1. When temperature is lower than the CST, the lower ILPEG stabilized nanoparticle catalyst phase is immiscible with the upper mixed solvents phase containing substrate. Excitingly, once the reaction temperature is increased beyond the CST, the two-phase system becomes transparent and homogeneous which allows the reaction runs smoothly. After reaction, the homogeneous system returns into two-phase again at the temperature lower than CST. Therefore, the lower nanoparticle catalyst phase can be separated easily from the upper product phase by simple phase separation, and the catalyst can be reused in the next cycle. Obviously, such a catalytic system takes advantages of both classical homogeneous and heterogeneous systems. Till now, the TPSCS has been applied successfully for the hydrogenation of olefins,6 selective hydrogenation of 1,5-cyclooctadiene,5 Heck reaction7 and hydroaminomethylation of 1-octene8 with high catalytic efficiency and good recyclability.
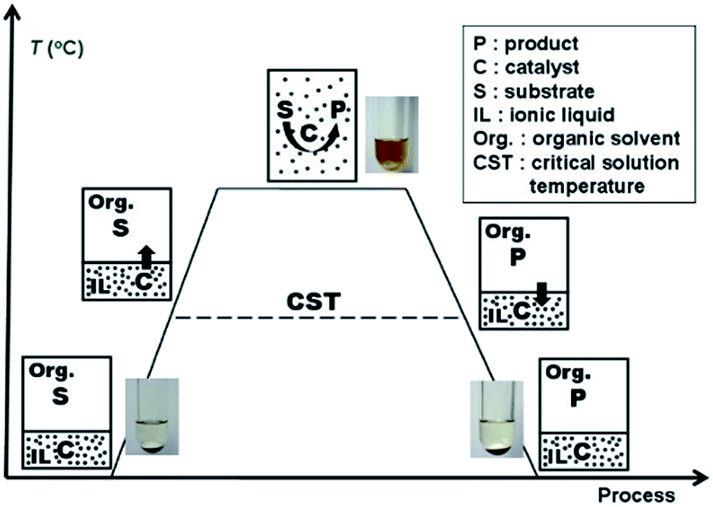 | ||
| Fig. 1 Schematic diagram of thermoregulated phase-separable catalytic system. | ||
The selective hydrogenation of CC or CO bond in α,β-unsaturated aldehydes and ketones to the corresponding saturated aldehydes/ketones or unsaturated alcohols has broad applications in the synthesis of many drugs, medical intermediates and fine chemicals.9–11 Although various catalyst systems for this selective hydrogenation reaction have been extensively reported, until now only a few catalytic systems affording the CC bond hydrogenation product with good selectivity and recyclability were reported.12–19 Encouraged by our pervious exciting results of TPSCS and with the aim to expand the application scopes of this system, in this paper, TPS-Rhnano was explored firstly for selective hydrogenation of α,β-unsaturated aldehydes and ketones.
TPS-Rhnano catalyst was prepared by hydrogen reduction of RhCl3·3H2O using ILPEG (n = 22) as a stabilizer (see ESI† for details). Fig. 2 shows the UV-visible absorption spectra of the solution of RhCl3 and ILPEG (n = 22) before and after reduction. Apparently, after reduction the characteristic absorption peak of Rh (III) at 520 nm disappeared completely, which indicated the formation of Rh0.20 Further analysis by TEM revealed that the Rh nanoparticles stabilized by ILPEG (n = 22) dispersed very well with an average particle size of 1.7 ± 0.2 nm (Fig. 3a). For comparison, TOAB (tetraoctyl ammonium bromide) with electrostatic stabilization,21–24 MPEG-1000 (polyethylene glycol monomethylether 1000) with steric stabilization25–27 and the mixture of TOAB and MPEG-1000 with electrosteric stabilization were used separately as a stabilizer for Rh nanoparticles under the same conditions. The results showed that Rh black could be seen by the naked eye when TOAB acted independently as stabilizer. Meanwhile, the nonuniform dispersion or even obvious aggregation was observed for nanoparticles prepared from MPEG-1000 or the mixture of TOAB and MPEG-1000 (Fig. 3b and c). Therefore, the formation of homogeneous-dispersed Rh nanoparticle catalyst stabilized by ILPEG (n = 22) was attributed to the perfect combination of electrostatic and steric stabilizations. Furthermore, all HRTEM images of Rh nanoparticles prepared by different stabilizers showed that the lattice fringes spacing of 0.20 nm were close to the (111) facet of Rh nanoparticles.
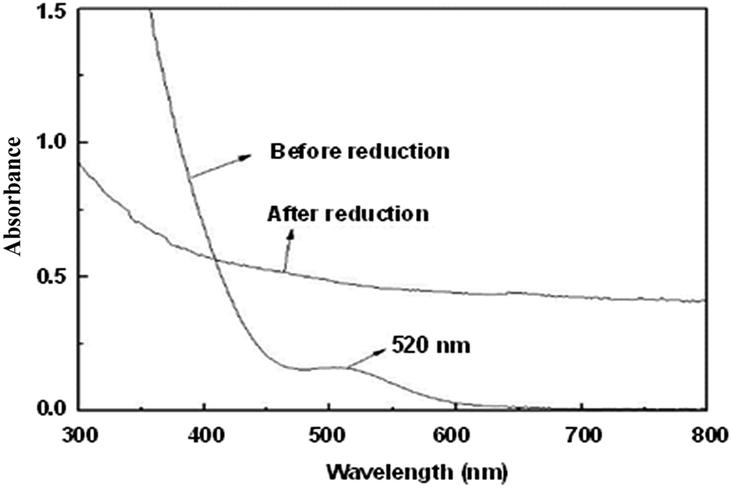 | ||
| Fig. 2 UV-visible absorption spectra of the solution of RhCl3 and ILPEG (n = 22) before and after reduction. | ||
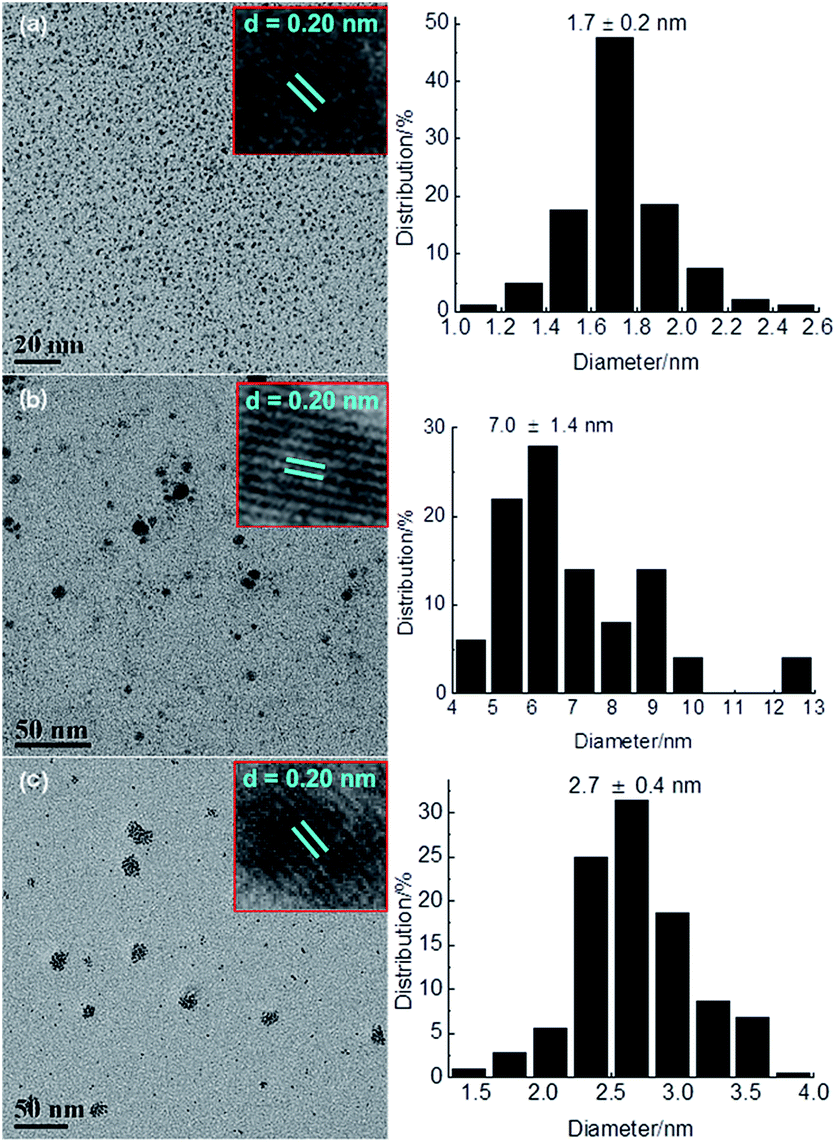 | ||
| Fig. 3 TEM images and particle size histograms of Rh nanoparticles prepared by different stabilizers: (a) electrosteric stabilization by ILPEG (n = 22); (b) steric stabilization by MPEG-1000; (c) electrosteric stabilization by the mixture of TOAB and MPEG-1000 (200 particles). | ||
For the TPS-Rhnano-catalyzed α,β-unsaturated aldehydes and ketones, cinnamaldehyde was chosen as a representative substrate to evaluate the catalyst performance and by varying temperature, hydrogen pressure, reaction time and molar ratio of cinnamaldehyde to Rh to identify the optimum reaction conditions (Table 1). The effect of reaction temperature was explored firstly in the range of 30–70 °C. When temperature was increased from 30 to 60 °C, the conversion of cinnamaldehyde increased from 12% to 99%, while the selectivity of the hydrogenation product of CC bond remained >99% all the time (entries 1–4). With a further increase in temperature to 70 °C, the selectivity towards the hydrogenation product of CC bond dropped to 94% although almost total conversion was observed (entry 5). And the by-product was proved to be the hydrogenation product of both CC and CO bonds. Further optimisation was the role of hydrogen pressure on the reaction and the results indicated that the conversion of cinnamaldehyde improved obviously with increasing hydrogen pressure from 0.1 to 1.0 MPa and 99% conversion was achieved at 1.0 MPa (entries 4, 6 and 7). When the hydrogen pressure was further increased to 1.5 MPa, the conversion reached >99% (entries 4 vs. 8). It was clear that prolonging the reaction time was in favour of improving the conversion of cinnamaldehyde (entries 4 and 9–11). In addition, we found that the conversion of cinnamaldehyde declined apparently when the molar ratio raised from 500 to 2000 (entries 4 and 12–14). For the sake of product separation and catalyst recycling, the best molar ratio of cinnamaldehyde to Rh was chosen to be 500 in our studies. It's worth mentioning that the selectivity towards the hydrogenation product of CC bond in cinnamaldehyde was totally unaffected by hydrogen pressure, reaction time and the molar ratio of cinnamaldehyde to Rh. Under the optimized reaction conditions, the conversion of cinnamaldehyde and the selectivity of the CC bond hydrogenation product were all >99%. The TOF was 245 h−1.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | T (°C) | PH2 (MPa) | t (h) | S/C (mol mol−1) | Conv.b (%) | Sel.c (%) | TOFd (h−1) |
| a Reaction conditions: ILPEG 0.3 g (containing 2.6 × 10−3 mmol rhodium), toluene (3.5 g), n-heptane (0.7 g). The miscibility temperature of the system is 60 °C.b Determined by GC with cyclohexane (0.2 g) as internal standard.c Selectivity of the CC bond hydrogenation product, the only by-product was the hydrogenation product of both CC and CO bonds.d Turnover frequency (TOF): moles of main product per mole of rhodium per hour. |
|||||||
| 1 | 30 | 1.0 | 2.0 | 500 | 12 | >99 | 30 |
| 2 | 40 | 1.0 | 2.0 | 500 | 48 | >99 | 119 |
| 3 | 50 | 1.0 | 2.0 | 500 | 72 | >99 | 178 |
| 4 | 60 | 1.0 | 2.0 | 500 | 99 | >99 | 245 |
| 5 | 70 | 1.0 | 2.0 | 500 | >99 | 94 | 233 |
| 6 | 60 | 0.1 | 2.0 | 500 | 9 | >99 | 22 |
| 7 | 60 | 0.5 | 2.0 | 500 | 38 | >99 | 94 |
| 8 | 60 | 1.5 | 2.0 | 500 | >99 | >99 | 245 |
| 9 | 60 | 1.0 | 0.5 | 500 | 22 | >99 | 218 |
| 10 | 60 | 1.0 | 1.0 | 500 | 65 | >99 | 322 |
| 11 | 60 | 1.0 | 1.5 | 500 | 91 | >99 | 300 |
| 12 | 60 | 1.0 | 2.0 | 1000 | 87 | >99 | 431 |
| 13 | 60 | 1.0 | 2.0 | 1500 | 65 | >99 | 483 |
| 14 | 60 | 1.0 | 2.0 | 2000 | 54 | >99 | 535 |
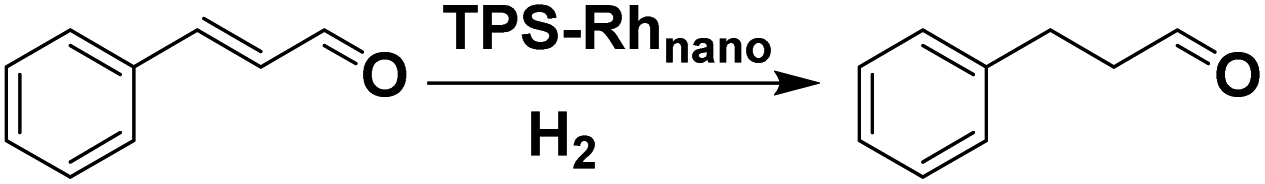
The reusability is a significant parameter in evaluation of a catalyst, especially for transition metal nanoparticle catalysts. Therefore, the reusability of TPS-Rhnano catalyst for selective hydrogenation of cinnamaldehyde was examined under the above-mentioned optimum conditions. After reaction, the lower TPS-Rhnano catalyst phase was simply separated from the product phase by simple phase separation and reused directly in the next catalytic cycle. As shown in Fig. 4, the TPS-Rhnano catalyst could be used eight times without evident loss in conversion and selectivity. Comparison of the TEM images of Rh nanoparticles between newly prepared and after eight cycles revealed that the particle size increased slightly from original 1.7 to current 2.2 nm (Fig. 3a vs. Fig. 5). In addition, the oxidation state and recyclability of the TPS-Rhnano catalyst basically remained before and after the catalytic reactions judging from XRD and XPS (Fig. S1 and S2, see the ESI† for details). Moreover, ICP-AES analysis of the upper organic phase after the first run indicated that the leaching of Rh was only 0.05 wt%, and from the second run on the leaching of Rh was lower than the detection limit. These results again confirmed that the TPS-Rhnano catalyst had good stability, catalytic activity, selectivity and recyclability.
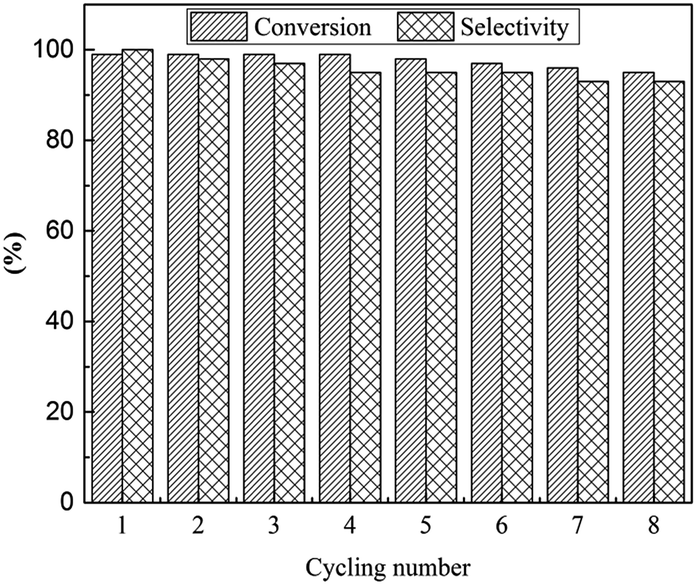 | ||
| Fig. 4 Recyclability of the TPS-Rhnano catalyst for selective hydrogenation of cinnamaldehyde. | ||
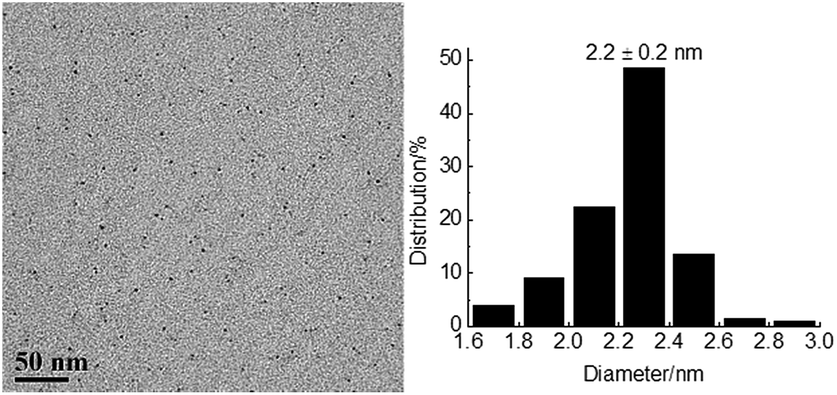 | ||
| Fig. 5 TEM image and particle size histogram of Rh nanoparticles after eight catalytic cycles (200 particles). | ||
In order to evaluate the generality of the above TPS-Rhnano catalyst, the selective hydrogenation of different α,β-unsaturated aldehydes and ketones, such as crotonaldehyde, benzylideneacetone and 2-cyclohexen-1-one, were also explored. Under the optimized reaction conditions, all substrates afforded their corresponding hydrogenation products of CC bond with >99% conversion and >99% selectivity (Table 2).
| Entry | Substrate | Product | Conv. (%) | Sel. (%) | TOF (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: ILPEG 0.3 g (n = 22, containing 2.6 × 10−3 mmol Rh), toluene 3.5 g, n-heptane 0.7 g; T = 60 °C, PH2 = 1.0 MPa, t = 2.0 h, substrate/Rh = 500, the miscibility temperature of the system is 60 °C. The conversion was determined by GC with cyclohexane (0.2 g) as internal standard; the selectivity was the corresponding hydrogenation product of CC bond. |
|||||
| 1 | 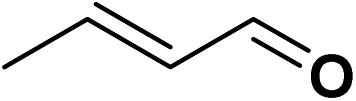 |
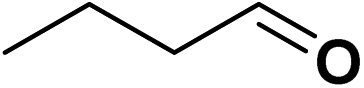 |
>99 | >99 | 245 |
| 2 | 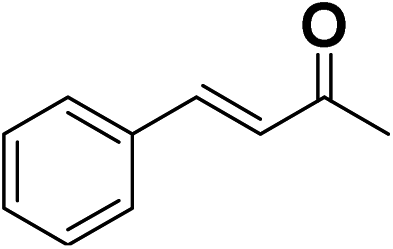 |
 |
>99 | >99 | 245 |
| 3 | 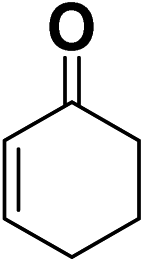 |
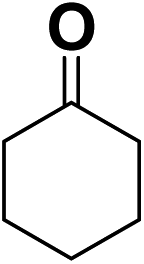 |
>99 | >99 | 245 |
Conclusions
In summary, the TPS-Rhnano catalyst was employed firstly for the selective hydrogenation of α,β-unsaturated aldehydes and ketones. Excellent selectivity (>99%) at the site of CC bond with conversion of >99% was achieved. More excitingly, the catalyst could be easily separated from the product by simple phase separation and reused directly for eight times without evident loss in activity and selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21173031, 21373039).Notes and references
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS PubMed.
- N. Koukabi, E. Kolvari, M. A. Zolfigol, A. Khazaei, B. S. Shaghasemi and B. Fasahati, Adv. Synth. Catal., 2012, 354, 2001 CrossRef CAS.
- B. Karimi, F. Mansouri and H. M. Mirzaei, ChemCatChem, 2015, 7, 1736 CrossRef CAS.
- Y. Xu, Y. Wang, Y. Zeng, Y. Song, J. Zhao, J. Jiang and Z. Jin, Chin. J. Catal., 2012, 33, 1871 CrossRef CAS.
- Y. Zeng, Y. Wang, J. Jiang and Z. Jin, Catal. Commun., 2012, 19, 70 CrossRef CAS.
- Y. Zeng, Y. Wang, Y. Xu, Y. Song, J. Jiang and Z. Jin, Catal. Lett., 2013, 143, 200 CrossRef CAS.
- Y. Xu, Y. Wang, K. Li, M. Niu, Y. Zeng, J. Jiang and Z. Jin, J. Nanosci. Nanotechnol., 2013, 13, 5048 CrossRef CAS PubMed.
- M. Bartok and A. Molnar, The Chemistry of Double-bonded Functional Groups, Wiley, New York, 1997 Search PubMed.
- D. D. Falcone, J. H. Hack and R. J. Davis, ChemCatChem, 2016, 8, 1074 CrossRef CAS.
- M. Tamura, D. Yonezawa, T. Oshino, Y. Nakagawa and K. Tomishige, ACS Catal., 2017, 7, 5103 CrossRef CAS.
- N. M. Callis, E. Thiery, J. L. Bras and J. Muzart, Tetrahedron Lett., 2007, 48, 8128 CrossRef CAS.
- S. Morrissey, I. Beadham and N. Gathergood, Green Chem., 2009, 11, 466 RSC.
- V. R. Landaeta, F. López-Linares, R. Sánchez-Delgado, C. Bianchini, F. Zanobini and M. Peruzzini, J. Mol. Catal. A: Chem., 2009, 301, 1 CrossRef CAS.
- X. Ni, B. Zhang, C. Li, M. Pang, D. Su, C. T. Williams and C. Liang, Catal. Commun., 2012, 24, 65 CrossRef CAS.
- M. Niu, Y. Wang, W. Li, J. Jiang and Z. Jin, Chin. J. Catal., 2013, 34, 674 CrossRef CAS.
- Y. Zhang, X. Yang, Y. Zhou, G. Li, Z. Li, C. Liu, M. Bao and W. Shen, Nanoscale, 2016, 8, 18626 RSC.
- W. Li, Y. Wang, P. Chen, M. Zeng, J. Jiang and Z. Jin, Catal. Sci. Technol., 2016, 6, 7386 CAS.
- S. Doherty, J. G. Knight, T. Backhouse, E. Abood, H. Alshaikh, I. J. S. Fairlamb, R. A. Bourne, T. W. Chamberlain and R. Stones, Green Chem., 2017, 19, 1635 RSC.
- W. Tu, H. Liu and K. Y. Liew, J. Colloid Interface Sci., 2000, 229, 453 CrossRef CAS PubMed.
- V. Cimpeanu, M. Kočevar, V. I. Parvulescu and W. Leitner, Angew. Chem., Int. Ed., 2009, 48, 1085 CrossRef CAS PubMed.
- N. Podoliak, D. Bartczak, O. Buchnev, A. G. Kanaras and M. Kaczmarek, J. Phys. Chem. C, 2012, 116, 12934 CAS.
- L. M. Rossi, L. L. R. Vono, M. A. S. Garcia, T. L. T. Faria and J. A. Lopez-Sanchez, Top. Catal., 2013, 56, 1228 CrossRef CAS.
- Y. Yu, B. W. Goodfellow, M. R. Rasch, C. Bosoy, D.-M. Smilgies and B. A. Korgel, Langmuir, 2015, 31, 6924 CrossRef CAS PubMed.
- H. Bönnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2001, 2455 CrossRef.
- Z. Sun, Y. Wang, M. Niu, H. Yi, J. Jiang and Z. Jin, Catal. Commun., 2012, 27, 78 CrossRef CAS.
- M. Niu, Y. Wang, P. Chen, D. Du, J. Jiang and Z. Jin, Catal. Sci. Technol., 2015, 5, 4746 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10351d |
| This journal is © The Royal Society of Chemistry 2017 |