
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced capacities of carbon nanosheets derived from functionalized bacterial cellulose as anodes for sodium ion batteries†
Tianyun Zhangabc,
Jiangtao Chenc,
Bingjun Yangc,
Hongxia Licd,
Shulai Leic and
Xin Ding *a
*a
aCollege of Textiles, Donghua University, Shanghai 201620, China. E-mail: xding@dhu.edu.cn; Tel: +86-21-67792663
bSchool of Mechanical and Electronical Engineering, Lanzhou University of Technology, Lanzhou 730050, China
cLaboratory of Clean Energy Chemistry and Materials, State Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
dSchool of Petrochemical Engineering, Lanzhou University of Technology, Lanzhou 730050, China
First published on 30th October 2017
Abstract
We studied carbon nanosheets prepared from bacterial cellulose (BC) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) oxidized-BC through carbonization at temperatures ranging from 900 to 1100 °C. Based on experimental results, we propose a comprehensive perspective of Na storage for BC based anode material. The pyrolysis TEMPO-oxidized BC delivers the highest capacity at annealing temperature of 1000 °C under different current densities, and displays excellent rate capability and cyclability. The superior electrochemical performance is attributed to the increasing interlayer distance, rich porous structure and oxygen-containing functional groups. The experimental studies reveal that the introduction of carboxyl is an effective strategy to enhance the specific capacity and cycling stability for Na-ion storage.
Introduction
Sodium ion batteries (SIBs) have been widely investigated for energy storage owing to the abundance distribution of Na resources in the earth. Numerous attempts have been made to find suitable anode materials for SIBs, including metal/alloys,1–3 metal oxide4–8/sulphide6,9–13/phosphide,14 and carbonaceous materials.15–20 Among them, carbonaceous materials display promising possibility in SIBs because of their high capacity, stability, abundant resource and safety.21,22 Attention has been paid to carbonaceous anode derived from cellulosic substances because of their porous and fibrous network structure.24–31 Various cellulosic materials, such as natural wood,32 bleached pulp,21 filter paper1,24 and bacterial cellulose (BC),26,29,33 have been used to fabricate carbon-based anode. In general, the reversible capacity of the carbon demonstrates considerable high capacity (200–300 mA h g−1).17,34–37 However, sodium storage performance of the carbon derived from different carbon precursor and thermal annealing processes are quite different because of the difference in the micro-structure.34As one in the families of nanocellulose, BC has attracted more attention owing to its ultrafine 3D fibrous network structure, high porosity and renewability, which represents a desirable material for energy-storage devices.31,38–41 However, the carbon derived from BC still faces major barriers, such as low specific capacity, poor rate capability and low initial coulombic efficiency (ICE).27 Significant efforts have been made to improve the electrochemical performance of carbon anode for SIBs. Among these, using heteroatoms doping in the carbon structure is a common approach to create a significant amount of defects, which were suggested to serve as additional active sites for sodium storage.42 Yu and co-workers31,33 fabricated nitrogen/oxygen and nitrogen/boron dual-doped carbon network using BC as template materials and showed an improved electrical conductivity and surface wettability. However, low ICE of ≈30% and 37% was observed because of increased high specific surface area of the materials. To increase the ICE of the carbon materials, Hu's group27 reported a method to decrease the surface area of the hard carbon derived from wood fiber by a TEMPO treatment. The treated wood fiber exhibited a much higher ICE (72%) compared with ICE = 25% by the porous carbon derived from pristine wood fibers. The increasing ICE by the TEMPO treatment was attributed to the denser structure and lower specific area induced. However, they neglected the influence of the oxygen-containing functional groups, which was suggested to play an important role in the rate performance and cycling stability because of the enhanced reaction kinetics and pseudocapacitive charge storage.43,44 Nor they pay attention to the sodium storage mechanism of the pristine and TEMPO-treated wood fibers. Without understanding the mechanism one would be less confident in developing a cellulose-based anode material with high specific capacity and cycling stability.
For the bulk carbon materials, the insertion and extraction of Na ions is much difficult because of sluggish diffusion kinetics of Na ion.43,45 Therefore, the fast redox reaction kinetics between Na ions and oxygen functional groups are beneficial to improve the sodium ion storage performance have been explored. According to Liu's work,43 the surface-reaction by tuning functional groups of carbon materials leaded to a high rate performance and cycling stability. In another work, Huang and co-worker45 fabricated oxygen-rich carbon materials that performed a fast surface redox reaction with reversible intercalation. They also showed that the anode with a larger interlayer distance and oxygen content resulted in the highest specific capacity and the best rate capability.
Carbon materials derived from BC have been investigated widely for application in SIBs.26,29,31 However, there are still a number of inadequacies in the study. First, it is insufficient to discover the relationship among the microstructure, specific surface area and functionalized surface of carbon derived from BC before and after the TEMPO treatment.26,29,31,33 Second, there are still uncertainties in understanding the mechanism of carbon anode derived from BC, although three possible mechanisms have been suggested in literature regarding the sodium storage in hard carbon materials, i.e. the mechanism of insertion–absorption, absorption–insertion and absorption–filling.37 Further, there are few reports on carbon anode prepared from BC and TEMPO-treated BC and the relationship between the structure and sodium storage performance.
We carbonized the pristine BC into carbon nanosheets under temperature between 900 and 1100 °C. Using the pyrolysis BC as carbon anode, the results showed that, with increasing annealing temperature, there was no advantage in improving electrochemical performance, including reversible capacity and ICE, compared with other carbon materials. Before thermal carbonization of BC, we conducted the TEMPO oxidization process,27 which is an effective method of introducing carboxylate functional groups on the surface of BC under mild conditions without any changes to the original crystallinity of BC.46,47 This work regarding the properties and performances of BC-based anode for SIBs investigated the sodium ion storage performance of functionalized carbon sheets through experiments at the first time, which provides some inspiration on the study of BC derived carbon for SIBs.
Experimental
Sample preparation
BC samples were prepared by cutting gel-like BC pellicles (Hainan Yide Food Industry Co., Ltd., China) into small pieces, pulped by a mechanical stirring and freeze drying. The samples of modified BC (MBC) were fabricated by TEMPO oxidation method.43 Then, both samples of BC and MBC were pyrolytic carbonized at 900, 1000, and 1100 °C for 2 hours in a tube furnace under argon ambient. The samples of carbon sheet from BC and MBC at different temperatures were denoted as p-BC900, p-BC1000, p-BC1100, p-MBC900, p-MBC1000 and p-MBC1100, respectively.Sample characterization
The morphology of the samples were characterized by field emission scanning electron microscopy (FESEM, JSM6701F) and transmission electron microscopy (TEM, JEOL 2100FEG). The structure was investigated by powder X-ray diffraction (XRD, Rigaku D/Max-2400, Cu-Kα radiation, λ = 0.15405 nm). The nitrogen adsorption–desorption isotherm measurements were performed on an ASAP 2020 volumetric adsorption analyzer (Micromeritics, USA) at 77 K. Raman spectrum (Horiba Jobin Yvon HR Evolution) was used to investigate the microstructure of the as-prepared samples. The surface chemical species of samples were examined on an X-ray photoelectron spectroscope (XPS, ESCALAB250x) using A1Kα radiation of 1486.6 eV as the excitation source.Electrochemical measurements
By mixing the prepared active material (the pyrolytic carbonized BC and MBC), acetylene black and polyvinylidenefluoride (PVDF) in a mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 and homogenizing in a N-methyl-2-pyrrolidinone (NMP) solvent, and then the slurry was coated on a copper foil as the working electrode for electrochemical measurements. The as-coated electrodes were dried at 110 °C for 12 hours in vacuum. The loading mass of the active material was about 2.0 mg cm−2. The electrode was assembled into a coin cell (CR2032) in an argon-filled glovebox using 1 M NaClO4 in a mixture of ethylene carbonate (EC) and propylene carbonate (PC) (1:1, in volume) as the electrolyte.48 A sodium foil was used as the counter electrode and Whatman GF/D glass fibers as separator. The discharge and charge tests were carried out on a CT2001A cell test instrument (Land Electronic Co., China) in a voltage range of 0.01–2.5 V at room temperature.25 Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) at a scan rate of 0.5 mV s−1 were recorded by an Autolab PGSTAT302N (Metrohm, Switzerland).
:1:1 and homogenizing in a N-methyl-2-pyrrolidinone (NMP) solvent, and then the slurry was coated on a copper foil as the working electrode for electrochemical measurements. The as-coated electrodes were dried at 110 °C for 12 hours in vacuum. The loading mass of the active material was about 2.0 mg cm−2. The electrode was assembled into a coin cell (CR2032) in an argon-filled glovebox using 1 M NaClO4 in a mixture of ethylene carbonate (EC) and propylene carbonate (PC) (1:1, in volume) as the electrolyte.48 A sodium foil was used as the counter electrode and Whatman GF/D glass fibers as separator. The discharge and charge tests were carried out on a CT2001A cell test instrument (Land Electronic Co., China) in a voltage range of 0.01–2.5 V at room temperature.25 Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) at a scan rate of 0.5 mV s−1 were recorded by an Autolab PGSTAT302N (Metrohm, Switzerland).
Results and discussion
Synthesis and characterization of samples
As a typical process, Scheme 1 shows the preparation of p-BC1000 and p-MBC1000 electrode. As seen from SEM image (Scheme 1a), BC is mainly composed of nanofibers and fibril bundles, and shows a dense 3D porous network structure. This structure is well maintained after the TEMPO treatment (Scheme 1b), but the length of fibers became shorter and the diameter finer after the treatment.46,49 The individualized nanofiber of sample MBC was about 20 nm in diameter and the width of the fiber bundles was about 50 nm. Compared with MBC, individualized nanofiber of BC was about 40 nm and the width bundles around 100 nm. This is because that the partial amorphous region among the bundles were dissolved after the TEMPO treatment.49 Furthermore, because of the introduction of carboxylate groups, MBC dispersed more homogeneous (Fig. S1†).50 After carbonization at 1000 °C under argon ambient, the carbon derived from BC and MBC were obtained and showed in Scheme 1d and e. The morphology of p-BC1000 is similar to that of p-MBC1000 with nanofibrils packed irregularly, forming sheet-like porous network. Further increasing the temperature to 1100 °C, the carbon sheets maintain (Fig. S2†). The unique open morphology of p-BC and p-MBC is beneficial to electrolyte penetration and ion diffusion.17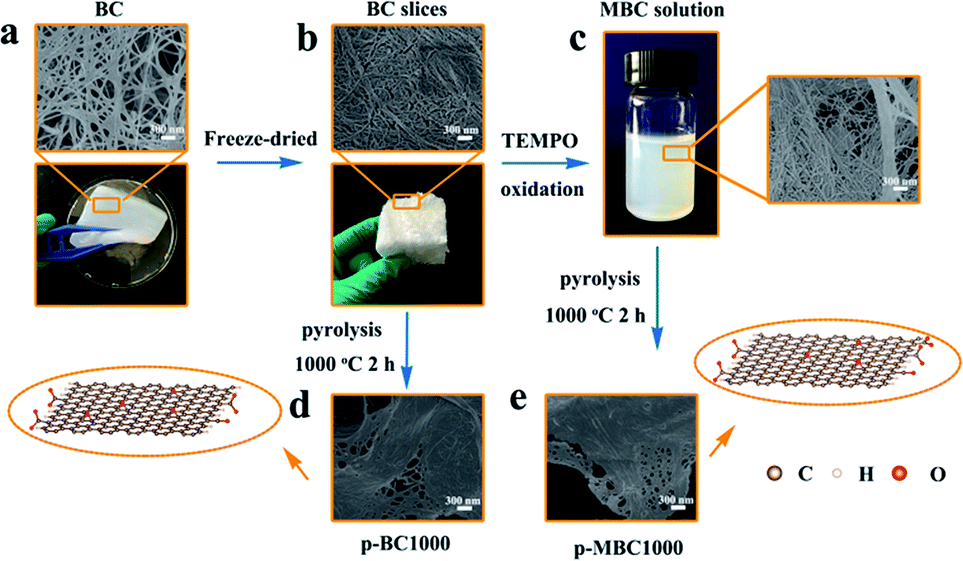 | ||
| Scheme 1 Fabrication process of p-BC1000 and p-MBC1000. | ||
Raman spectra and XRD patterns demonstrate that the carbon sheets are partially graphitic. Raman spectra show two characteristic bands centred at 1352 cm−1 (D-band) and at 1584 cm−1 (G-band).21 The ratio of ID/IG (the intensity ratio of D-band to G-band) listed in Table S1† shows that it is 1.31 for p-BC1000, indicating the presence of a disordered structure. For p-MBC900, p-MBC1000 and p-MBC1100, the ratios of ID/IG are 1.29, 1.33 and 1.33, respectively. The XRD pattern shows two broad peaks at about 24° and 43°, which indexed to the (002) and (100) planes, respectively (Fig. 1b and S3b†).15,51 The (002) peaks shift to higher diffraction angles with increasing carbonization temperature to 1100 °C, indicating that the average d-spacing of the graphene sheets decreases with an increasing relative ordering.52
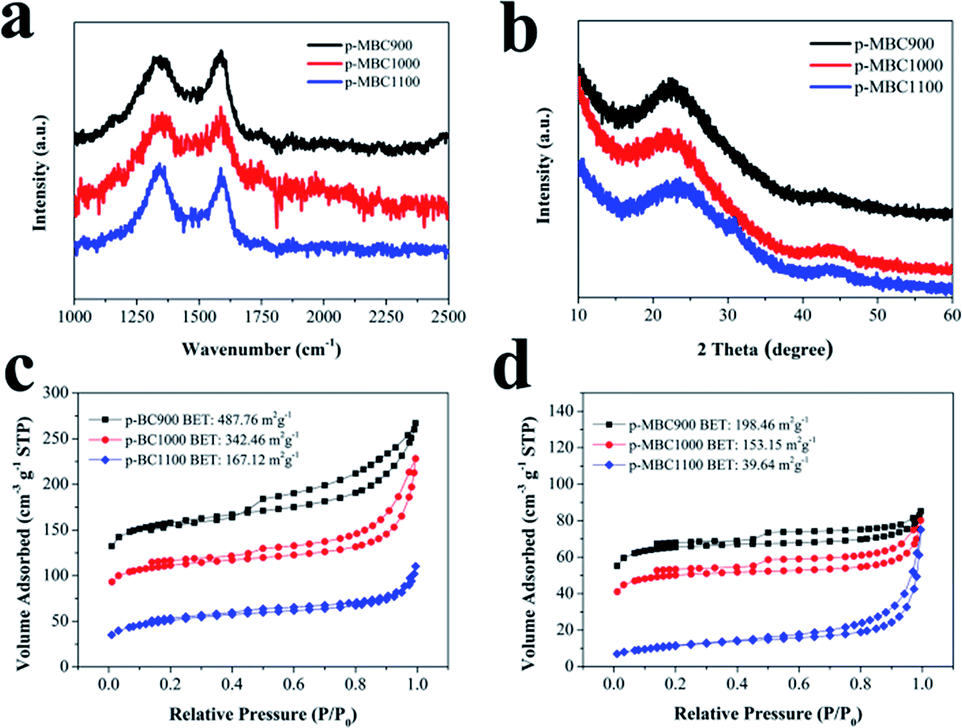 | ||
| Fig. 1 (a) Raman spectra of p-MBC; (b) XRD patterns of p-MBC; nitrogen adsorption–desorption isotherms of (c) p-BC and (d) p-MBC. | ||
As carbonization temperature increases, the BET specific surface area (SSA) of p-BCs and p-MBCs gradually decreased. It is noticed that the BET SSA of the modified samples (p-MBCs) is smaller than that of the samples derived from pristine BC (p-BCs) at the same carbonization temperature, with p-MBC1100 having the lowest SSA of 39.64 m2 g−1. Two reasons leading to the decreasing BET SSA after the TEMPO treatment. One is the enhanced oxygen functional groups after the treatment to fill the space of pores and the other is the destruction of the hydrogen bond and induction of negative charges on the surface of nanofibers.27
Fig. S3c and d† display that p-BCs and p-MBCs exhibit rich porous structures, consisting of a large number of micropores, mesopores and macropores. The total pore volume of p-MBC900, p-MBC1000 and p-MBC1100, listed in Table S1,† were 0.13, 0.12 and 0.09 cm3 g−1, respectively, indicating the decreasing number of micropores with increasing carbonization temperature. It is noted that the total pore volume of p-MBCs is smaller than that of p-BCs with the same carbonization temperature, owing to the blocking of some pores by surface functional groups.42,45
Fig. 2 displays the resolution TEM image of p-MBCs. The carbon samples prepared at 900, 1000 and 1100 °C show a gradual development of local graphite-like structure with the increasing temperature. The inter-layer distance in the crystalline region with a short-range ordered of carbon is 0.40, 0.39 and 0.36 nm, respectively, which were larger than that of crystalline graphite (0.335 nm). More HRTEM images of the obtained samples are presented in Fig. S4.† According to these images, they show that both amorphous carbon and short-range ordered crystalline pattern are identified, which further exhibit a feature of hard carbon materials described in a card model.35 In contrast, the inter-layer distance of p-BCs is smaller than that of p-MBC. The residual oxygen-containing functional groups on p-MBC plays a role in the lattice expansion.53
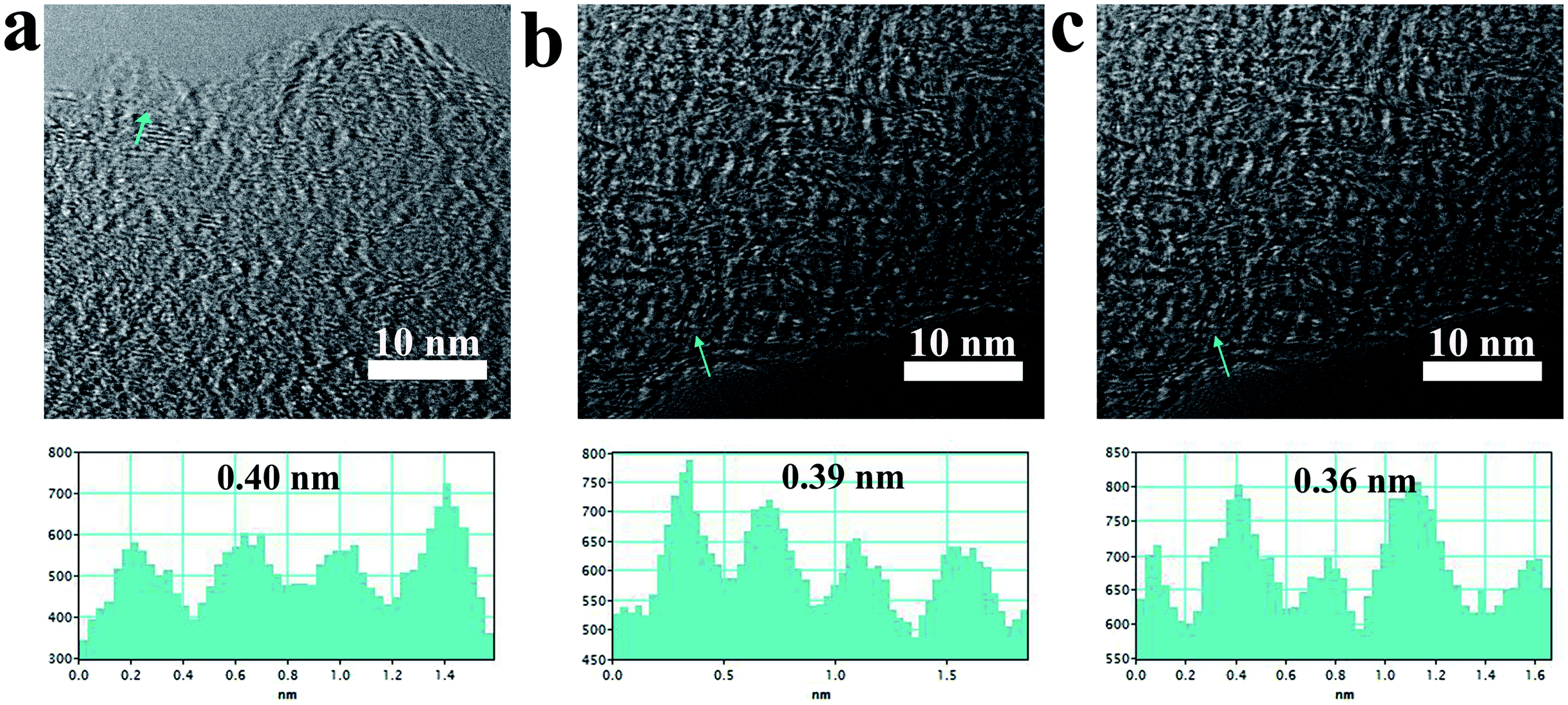 | ||
| Fig. 2 HRTEM images of (a) p-MBC900; (b) p-MBC1000; (c) p-MBC1100. | ||
The element species of p-MBCs and p-BCs were investigated by XPS. From the XPS survey (Fig. S5b†), it can be observed that samples of p-BC and p-MBC contain mainly carbon and oxygen with no other heteroatoms. The O 1s XPS of p-BC1000 and p-MBC1000 are shown in Fig. S5c and d.† The oxygen-containing functional groups presented are C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (531.3 eV), C–O (532.2 eV) and OC–O (533.2 eV).54–56 For p-BC1000, the O 1s spectrum reveals three peaks, assigned to CO (13.6%), C–O (56.5%) and OC–O (29.9%) bonds. For p-MBC1000, the contents of CO, C–O and OC–O are 14.1%, 38.8%, and 47.1%, respectively, containing higher concentration of the total of OC–O and CO than those of p-BC1000 (Fig. S5a†). Similar results can be observed in Fig. S5e and f† for the C 1s XPS of p-BC1000 and p-MBC1000, i.e. p-MBC1000 contains a higher oxygen content than its counterpart of p-BC1000. These oxygen-containing functional groups can result in the surface redox reaction with sodium ions, especially through –CO/–C–O–Na redox pair.23,42 It is important to realize that the functional groups on the surface of carbon nanosheets can enhance the wettability of electrolyte. Besides, the surface-driven reactions between Na+ and oxygen-containing groups can enhance the electrochemical performance such as ICE and capacities.43,57,58
O (531.3 eV), C–O (532.2 eV) and OC–O (533.2 eV).54–56 For p-BC1000, the O 1s spectrum reveals three peaks, assigned to CO (13.6%), C–O (56.5%) and OC–O (29.9%) bonds. For p-MBC1000, the contents of CO, C–O and OC–O are 14.1%, 38.8%, and 47.1%, respectively, containing higher concentration of the total of OC–O and CO than those of p-BC1000 (Fig. S5a†). Similar results can be observed in Fig. S5e and f† for the C 1s XPS of p-BC1000 and p-MBC1000, i.e. p-MBC1000 contains a higher oxygen content than its counterpart of p-BC1000. These oxygen-containing functional groups can result in the surface redox reaction with sodium ions, especially through –CO/–C–O–Na redox pair.23,42 It is important to realize that the functional groups on the surface of carbon nanosheets can enhance the wettability of electrolyte. Besides, the surface-driven reactions between Na+ and oxygen-containing groups can enhance the electrochemical performance such as ICE and capacities.43,57,58
Electrochemical performance
The electrochemical performance of p-BCs and p-MBCs served as SIBs anodes were investigated. Fig. 3a and b show the charge/discharge curves of p-BCs and p-MBCs for the first two cycles at a current density of 0.05 A g−1 in a voltage range of 0.01–2.5 V. An obvious discharge plateau at the low voltage range can be observed for the samples with the annealing temperature greater than 900 °C. It implies that there are different mechanism of sodium ion insertion between p-BCs and p-MBCs. The sodiation and desodiation capacities of p-BC1000 were 574 mA h g−1 and 219 mA h g−1, respectively, giving the ICE of 38%. Similarly, the ICEs of p-BC900 and p-BC1100 electrodes were 36% and 44%. Because carbon electrode with a high surface area would exhibit a relatively low ICE,27 it was then expected that, among all the samples, p-BC900 had the highest surface areas of 487.76 m2 g−1 and the lowest ICE of 36%. The large capacity loss of the initial charge capacity is attributed to the occurrence of an irreversible reaction, including the decomposition of the electrolyte and the formation of a solid electrolyte interphase (SEI) film.15,21 For p-MBCs, the samples exhibited better electrochemical performance than their counterparts. In the first Na insertion curve, p-MBC900 delivered a low discharge capacity of 310 mA h g−1 with an obvious sloping region and the ICE of 52% in the electrochemical performance curves. Importantly, p-MBC1000 showed a reversible sodium storage capacity of about 276 mA h g−1 with a high capacity in the plateau region and the ICE of 50%. The TEMPO-treated BCs exhibited higher ICEs compared with the pristine BCs, except for the samples prepared at high annealing temperature of 1100 °C. This lowest BET SSA of p-MBC1100 could induce a limitation to the reaction of active sites.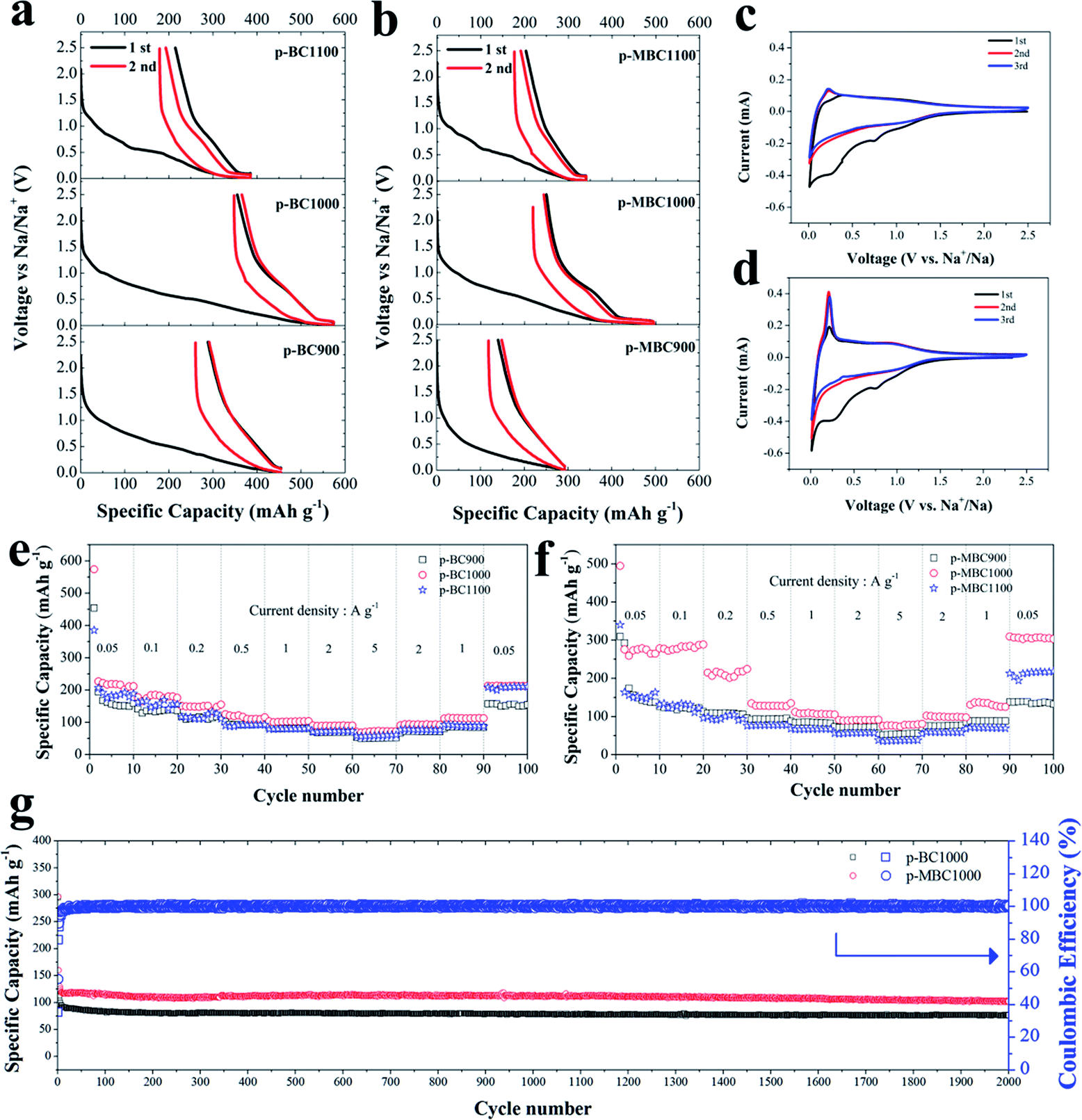 | ||
| Fig. 3 Electrochemical performance of p-BC and p-MBC electrodes in a voltage range of 0.01–2.5 V. Voltage profiles of (a) p-BC and (b) p-MBC at 0.05 A g−1; the CV curves of (c) p-BC900 and (d) p-MBC1000; rate capability of (e) p-BCx and (f) p-MBC from 0.05 A g−1 to 5 A g−1; (g) cycle performance of p-BC1000 and p-MBC1000 at a current density of 1 A g−1. | ||
From the charge/discharge curves of p-BCs and p-MBCs in Fig. 3c and d, we compared the CV curves for the first three cycles of p-BC900 and p-MBC1000 electrode at a scanning rate of 0.5 mV s−1. In the first cycle, a small reduction peak appeared at 0.8 V, which was associated with the irreversible reaction of the electrolyte with surface functional groups. Another sharp irreversible peak at ∼0.4 V can be assigned to the formation of a SEI layer.23,42,59 The two reduction peaks disappeared after two cycles and the curves were highly overlapped, indicating a good cyclic performance during sodium ion insertion and extraction. Moreover, the CV curves of these samples were of rectangular shape in the potential range of 1.0–2.5 V, indicating an ideal capacitor-like behaviour of sodium storage in carbon anode above 1 V. As shown in Fig. 3c and d, a pair of redox peaks at a lower potential (about 0.1 V versus Na/Na+) are observed, indicating the insertion–extraction of Na+ in the graphitic layers, which is similar to Li+ insertion into carbonaceous materials.15,60 However, the peak of p-MBC1000 near 0.1 V becomes sharper than that of p-BC900. In addition, compared to the CV curve of p-BC1000, it is clearly observed that there is a pair of weak redox peaks at about 1.1 V and the peak near 0.1 V also sharper for p-MBC1000, shown in Fig. S6b,† which can be ascribed to the surface redox reactions on the functionalized carbon electrode.23 Furthermore, the irreversible area of p-MBC1000 was smaller than that of p-BC1000, corresponding to a higher ICE, which is due to the formation of a SEI layer and the side reversible reactions of surface functional groups in the first discharge scan,23 also indicating the low surface area is critical for the stabilization of SEI layer.42
When evaluating the performance of an anode for SIBs, the rate capability is another critical property (Fig. 3e and f). For p-BCs, all of them show a good rate capability, indicating the improvement of the property with increasing carbonization temperature. When the current density is increased to 5 A g−1, p-BC1000 electrode can deliver a discharge capacity of 70 mA h g−1. When the rate returns to 0.05 A g−1 after cycling at different rates, the specific capacity of 213 mA h g−1 can be recovered for p-BC1000, demonstrating good rate performance. As shown in Fig. 3f, p-MBC shows a better rate performance compared with p-BC. The reversible capacities of p-MBC1000 are 276, 289, 224, 135, 105, 92 and 81 mA h g−1 at the current densities of 0.05, 0.1, 0.2, 0.5, 1, 2 and 5 A g−1, respectively. Moreover, when the current density is tuned back to 0.05 A g−1 again, the electrode is still able to recover a high capacity of 310 mA h g−1. As shown in Fig. S7,† p-MBC1000 exhibits a stable capacity of 232 mA h g−1 at 0.1 A g−1 over 500 cycles with coulombic efficiency of nearly 100%. Even at high density of 1 A g−1, a reversible capacity of 102 mA h g−1 after 2000 cycles is still delivered (Fig. 3g). As shown in Fig. S8,† a comparable stable capacity can still be obtained from galvanostatic 1st, 100th, 200th, 500th, 1000th and 2000th discharge/charge profiles.
Fig. 4 shows the EIS of p-BC1000 and p-MBC1000 half-cells after different cycles at 1 A g−1. For p-MBC1000 the charge transfer resistance Rct reaches its maximum after 10 cycles and then decreases gradually from cycle to cycle afterwards (Fig. 4a), but it is always smaller than that of p-BC1000 (Fig. 4b). This means that the oxygen functional groups on the carbon surface facilitate the formation of a stable SEI film, sodium ion insertion/extraction, and charge transfer at the electrode/electrolyte interface.45
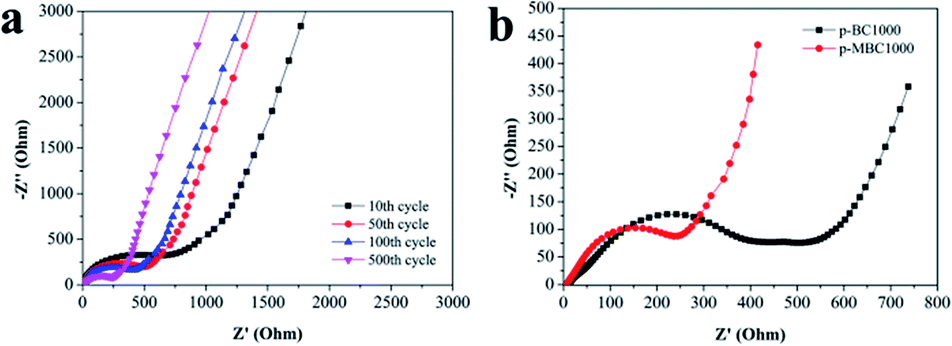 | ||
| Fig. 4 (a) The Nyquist plots of p-MBC1000 electrode after different cycles at a current density of 1 A g−1. (b) The Nyquist plots of p-BC1000 and p-MBC1000 electrodes after 500 cycles at a current density of 1 A g−1. | ||
As reported previously,47 the TEMPO treatment can introduce more oxygen-containing functional groups on the surface of BC. Although the majority of the functional groups have been removed after thermal treatment, a few of them can still retain and play a critical role in enhancing the specific capacity of the carbon materials for SIBs.42,45 The oxygen-containing functional groups result in the surface redox reaction between the carbon-oxygen functional groups and Na+, that is –CO + Na+ + e ↔ –C–O–Na, which is similar to that for Li+.23,43
To further understand the role played by the oxygen-containing functional groups, the capacity potential distribution of p-BCs and p-MBCs are compared. The capacity can be divided into two parts, slope capacity (capacity above 0.1 V vs. Na/Na+) and plateau capacity (capacity below 0.1 V vs. Na/Na+). As summarized in Fig. S9a,† the sloping region decreases and the plateau region increases for p-BC as the temperature increases which can be attributed to the change of the microstructure. For samples of p-MBC, the influence of annealing temperature on the capacity potential distribution shows similar trend as that of p-BC (Fig. S9b†). The capacity of p-BC1000 and p-MBC1000 at different rates and the separate capacities of plateau and sloping regions are presented in Fig. S9c and d.† It is likely that the high voltage plateau region capacity is related to Na ion adsorption on the surface, which should be related to the specific area, porous structure and surface functional groups. Obviously, the plateau capacity of p-MBC1000 is higher than that of p-BC1000, indicating that the plateau region can be ascribed to the insertion–extraction of sodium ions in the graphitic interlayers at a low current density. Therefore, the introduction of oxygen functional groups to carbon structure not only provides many surface reaction sites and defects for accommodation of Na ions, but also enlarges the interlayer distance of carbon.
Conclusions
In summary, we have reported the novel method to prepare functionalized carbon nanosheets derived from the pristine BC and TEMPO-treated BC. The carbon anodes synthesized by carbonization at different temperatures showed different electrochemical performance. The results show that the factors including BET SSA, porous structure, surface functional groups and interlayer distance play important roles in enhancing electrochemical performance of carbon anode for SIBs. The p-MBC1000 electrode with larger interlayer distance and abundant surface oxygen functional groups presents the highest specific capacity, the best rate capacity, the excellent cycling stability and the higher ICE. This study not only provides a better understanding of Na+ storage mechanisms in BC-based anodes but also offers a practical method to enhance the capacity of carbon anodes for SIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the plan of Natural Science Foundation of Gansu Province (Grant No. 1610RJYA019).Notes and references
- W. Chen and D. Deng, Carbon, 2015, 87, 70–77 CrossRef CAS.
- J. Ding, H. Zhou, H. Zhang, T. Stephenson, Z. Li, D. Karpuzov and D. Mitlin, Energy Environ. Sci., 2017, 10, 153–165 CAS.
- M.-S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- Y. H. Liu, X. Fang, M. Y. Ge, J. P. Rong, C. F. Shen, A. Y. Zhang, H. A. Enaya and C. W. Zhou, Nano Energy, 2015, 16, 399–407 CrossRef CAS.
- W. N. Ren, W. W. Zhou, H. F. Zhang and C. W. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 487–495 CAS.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- J. Lee, J. K. Lee, K. Y. Chung, H.-G. Jung, H. Kim, J. Mun and W. Choi, Electrochim. Acta, 2016, 200, 21–28 CrossRef CAS.
- M.-K. Kim, S.-H. Yu, A. Jin, J. Kim, I.-H. Ko, K.-S. Lee, J. Mun and Y.-E. Sung, Chem. Commun., 2016, 52, 11775–11778 RSC.
- J. Zhou, J. Qin, L. Guo, N. Zhao, C. Shi, E.-z. Liu, F. He, L. Ma, J. Li and C. He, J. Mater. Chem. A, 2016, 4, 17370–17380 CAS.
- J. Wang, J. Liu, H. Yang, D. Chao, J. Yan, S. V. Savilov, J. Lin and Z. X. Shen, Nano Energy, 2016, 20, 1–10 CrossRef CAS.
- Y. Wang, Q. T. Qu, G. C. Li, T. Gao, F. Qian, J. Shao, W. J. Liu, Q. Shi and H. H. Zheng, Small, 2016, 12, 6033–6041 CrossRef CAS PubMed.
- X. Guo, G. L. Cao, F. Ding, X. Y. Li, S. Y. Zhen, Y. F. Xue, Y. M. Yan, T. Liu and K. N. Sun, J. Mater. Chem. A, 2015, 3, 5041–5046 CAS.
- Y. Xiao, S. H. Lee and Y.-K. Sun, Adv. Energy Mater., 2017, 7, 1601329 CrossRef.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 CAS.
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- S. Q. Wang, L. Xia, L. Yu, L. Zhang, H. H. Wang and X. W. Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- J. Ding, H. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. Xu, B. Zahiri, X. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed.
- J. T. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. X. Dou and L. M. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- Z. Li, Z. Jian, X. Wang, I. A. Rodriguez-Perez, C. Bommier and X. Ji, Chem. Commun., 2017, 53, 2610–2613 RSC.
- F. Niu, J. Yang, N. Wang, D. Zhang, W. Fan, J. Yang and Y. Qian, Adv. Funct. Mater., 2017, 27, 1700522 CrossRef.
- Q. Zhao, Y. Huang and X. Hu, Electrochem. Commun., 2016, 70, 8–12 CrossRef CAS.
- W. Luo, J. Schardt, C. Bommier, B. Wang, J. Razink, J. Simonsen and X. L. Ji, J. Mater. Chem. A, 2013, 1, 10662–10666 CAS.
- K. L. Hong, L. Qie, R. Zeng, Z. Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. J. Fan, W. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 CAS.
- P. Zheng, T. Liu and S. W. Guo, Sci. Rep., 2016, 6, 35620–35626 CrossRef CAS PubMed.
- H. Zhu, F. Shen, W. Luo, S. Zhu, M. Zhao, B. Natarajan, J. Dai, L. Zhou, X. Ji, R. S. Yassar, T. Li and L. Hu, Nano Energy, 2017, 33, 37–44 CrossRef CAS.
- B. Dursun, T. Sar, A. Ata, M. Morcrette, M. Y. Akbas and R. Demir-Cakan, Cellulose, 2016, 23, 2597–2607 CrossRef.
- F. Shen, H. Zhu, W. Luo, J. Wan, L. Zhou, J. Dai, B. Zhao, X. Han, K. Fu and L. Hu, ACS Appl. Mater. Interfaces, 2015, 7, 23291–23296 CAS.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- Z. Zhang, J. Zhang, X. Zhao and F. Yang, Carbon, 2015, 95, 552–559 CrossRef CAS.
- M. Dahbi, T. Nakano, N. Yabuuchi, T. Ishikawa, K. Kubota, M. Fukunishi, S. Shibahara, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, Electrochem. Commun., 2014, 44, 66–69 CrossRef CAS.
- M. Wang, Z. Yang, W. Li, L. Gu and Y. Yu, Small, 2016, 12, 2559–2566 CrossRef CAS PubMed.
- F. Shen, W. Luo, J. Q. Dai, Y. G. Yao, M. W. Zhu, E. Hitz, Y. F. Tang, Y. F. Chen, V. L. Sprenkle, X. L. Li and L. B. Hu, Adv. Energy Mater., 2016, 6, 1600377 CrossRef.
- M. Wang, Y. Yang, Z. Yang, L. Gu, Q. Chen and Y. Yu, Adv. Sci., 2017, 4, 1600468 CrossRef PubMed.
- B. Zhang, C. M. Ghimbeu, C. Laberty, C. Vix-Guterl and J.-M. Tarascon, Adv. Energy Mater., 2016, 6, 1501588 CrossRef.
- V. Simone, A. Boulineau, A. de Geyer, D. Rouchon, L. Simonin and S. Martinet, J. Energy Chem., 2016, 25, 761–768 CrossRef.
- L. F. Xiao, Y. L. Cao, W. A. Henderson, M. L. Sushko, Y. Y. Shao, J. Xiao, W. Wang, M. H. Engelhard, Z. M. Nie and J. Liu, Nano Energy, 2016, 19, 279–288 CrossRef CAS.
- H. Hou, X. Qiu, W. Wei, Y. Zhang and X. Ji, Adv. Energy Mater., 2017, 1602898 CrossRef.
- L. F. Chen, Z. H. Huang, H. W. Liang, Q.-F. Guan and S.-H. Yu, Adv. Mater., 2013, 25, 4746–4752 CrossRef CAS PubMed.
- Y. Huang, M. Zheng, Z. Lin, B. Zhao, S. Zhang, J. Yang, C. Zhu, H. Zhang, D. Sun and Y. Shi, J. Mater. Chem. A, 2015, 3, 10910–10918 CAS.
- W. Wang, Y. Sun, B. Liu, S. Wang and M. Cao, Carbon, 2015, 91, 56–65 CrossRef CAS.
- S. F. Tong, M. B. Zheng, Y. Lu, Z. X. Lin, X. P. Zhang, P. He and H. S. Zhou, Chem. Commun., 2015, 51, 7302–7304 RSC.
- H. L. Wang, W. H. Yu, N. Mao, J. Shi and W. Liu, Microporous Mesoporous Mater., 2016, 227, 1–8 CrossRef.
- Y. Y. Shao, J. Xiao, W. Wang, M. Engelhard, X. L. Chen, Z. M. Nie, M. Gu, L. V. Saraf, G. Exarhos, J. G. Zhang and J. Liu, Nano Lett., 2013, 13, 3909–3914 CrossRef CAS PubMed.
- M. Y. Song, N. R. Kim, S. Y. Cho, H.-J. Jin and Y. S. Yun, ACS Sustainable Chem. Eng., 2017, 5, 616–624 CrossRef CAS.
- W. Chen, C. Chen, X. Xiong, P. Hu, Z. Hao and Y. Huang, Adv. Sci., 2017, 4, 1600500 CrossRef PubMed.
- W. Fan, K. Si-Seup, K. Chang-Doo, S. Yun-De and O. Il-Kwon, Smart Mater. Struct., 2014, 23, 074006–074016 CrossRef.
- S. S. Kim, J. H. Jeon, H. I. Kim, C. D. Kee and I.-K. Oh, Adv. Funct. Mater., 2015, 25, 3570 Search PubMed.
- A. Ponrouch, E. Marchante, M. Courty, J. M. Tarascon and M. R. Palacin, Energy Environ. Sci., 2012, 5, 8572–8583 CAS.
- Y. Jia, X. Zhai, W. Fu, Y. Liu, F. Li and C. Zhong, Carbohydr. Polym., 2016, 151, 907–915 CrossRef CAS PubMed.
- F. Wang, J.-H. Jeon, S. Park, C.-D. Kee, S.-J. Kim and I.-K. Oh, Soft Matter, 2016, 12, 246–254 RSC.
- P. Liu, Y. M. Li, Y. S. Hu, H. Li, L. Q. Chen and X. J. Huang, J. Mater. Chem. A, 2016, 4, 13046–13052 CAS.
- S. Qiu, L. Xiao, M. L. Sushko, K. S. Han, Y. Shao, M. Yan, X. Liang, L. Mai, J. Feng, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2017, 1700403 CrossRef.
- X.-F. Luo, C.-H. Yang, Y.-Y. Peng, N.-W. Pu, M.-D. Ger, C.-T. Hsieh and J.-K. Chang, J. Mater. Chem. A, 2015, 3, 10320–10326 CAS.
- L. L. Jiang, L. Z. Sheng, C. L. Long, T. Wei and Z. J. Fan, Adv. Energy Mater., 2015, 5, 771–780 Search PubMed.
- Z. Li, Z. W. Xu, H. L. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. H. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- Z. Xie, Z. Yu, W. Fan, G. Peng and M. Qu, RSC Adv., 2015, 5, 90041–90048 RSC.
- D. B. Xiong, X. F. Li, H. Shan, Y. Zhao, L. Dong, H. Xu, X. F. Zhang, D. J. Li and X. L. Sun, Electrochim. Acta, 2015, 174, 762–769 CrossRef CAS.
- N. Sun, H. Liu and B. Xu, J. Mater. Chem. A, 2015, 3, 20560–20566 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10118j |
| This journal is © The Royal Society of Chemistry 2017 |