DOI:
10.1039/C7RA10303D
(Paper)
RSC Adv., 2017,
7, 50657-50662
On-site separation of Cr(VI) and Cr(III) in natural waters by parallel cartridge ion-exchange columns†
Received
16th September 2017
, Accepted 23rd October 2017
First published on 1st November 2017
Abstract
A simple, fast, portable, and solvent-free method is developed for field separation of Cr(VI) and Cr(III) in natural waters. The method involves passing a water sample through parallel cation- and anion exchange resin cartridge columns at the field site. In a matter of seconds, all the Cr(III) is retained on the cation-exchange column and Cr(VI) passes to the effluent, while the Cr(VI) is quantitatively retained on the anion-exchange resin and Cr(III) passes to the effluent. The two collected solutions are preserved and determined later in the laboratory using inductively coupled plasma mass spectrometry (ICP-MS) or any other elemental analysis technique sufficiently sensitive to measure the Cr concentrations of interest. Cr(VI) (or Cr(III)) can be separated from Cr(III) (or Cr(VI)) at the pH range 1–10 at Cr(III)/Cr(VI) (or Cr(VI)/Cr(III)) concentration ratios as high as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (or 500). The limits of detection for Cr(III) and Cr(VI) are 0.03 and 0.01 μg L−1, respectively. Repeatability expressed as relative standard deviation is determined to be 7.1% for Cr(VI) and 5.7% for Cr(III), using a drinking water sample containing 1.14 μg L−1 Cr(VI) and 0.72 μg L−1 Cr(III). The parallel cartridge ion-exchange resin columns can be used continuously for 40 drinking water samples without any regeneration or cleaning procedure. The accuracy is validated by analysing Cr(III) and Cr(VI) in three water standard reference materials (GBW080257, GBW080403, and GBW080404). Furthermore, the proposed method is applied to the on-site separation of Cr(III) and Cr(VI) from natural waters.
000 (or 500). The limits of detection for Cr(III) and Cr(VI) are 0.03 and 0.01 μg L−1, respectively. Repeatability expressed as relative standard deviation is determined to be 7.1% for Cr(VI) and 5.7% for Cr(III), using a drinking water sample containing 1.14 μg L−1 Cr(VI) and 0.72 μg L−1 Cr(III). The parallel cartridge ion-exchange resin columns can be used continuously for 40 drinking water samples without any regeneration or cleaning procedure. The accuracy is validated by analysing Cr(III) and Cr(VI) in three water standard reference materials (GBW080257, GBW080403, and GBW080404). Furthermore, the proposed method is applied to the on-site separation of Cr(III) and Cr(VI) from natural waters.
Introduction
Chromium (Cr) present in the environment appears mainly as a result of human activities such as electroplating, leather tanning, metal finishing, and the textile industry.1,2 The bioavailability and toxicity of Cr depend upon its chemical form; Cr(III) is a biologically important species that plays essential roles in the metabolic pathways of glucose and lipids as well as protein metabolism, whereas Cr(VI) is carcinogenic, mutagenic, and is implicated in stomach and skin cancers.3,4 Cr(VI) concentration limits for drinking water stipulated by the World Health Organization (WHO), the US Environmental Protection Agency (EPA), and the Chinese government are 50, 10, and 6 μg L−1, respectively.5–7 The California EPA has gone as far as to set a public health goal for Cr(VI) in drinking water of 0.02 μg L−1 in order to protect the health of residents at a lifetime risk level of “one-in-a-million”.6
The most commonly performed analytical strategy for Cr monitoring involves two steps: (1) collection of water samples at the field site followed by storage and transportation it to the laboratory, and (2) analysis using atomic spectrometry after separation by one of a number of online or offline separation approaches, such as solid-phase extraction, liquid–liquid extraction, cloud-point extraction, or co-precipitation.8–14 However, accurate speciation of Cr in natural water bodies is difficult, mainly due to matrix complexity and the instability of Cr redox species during sample collection, transport, and analysis.15 For example, Meeravali et al.16 found that the concentration of Cr(III) increased by 135% and that of Cr(VI) decreased by 75% within 24 h when lake water samples were collected and stored at 4 °C. This demonstrates that the concentrations of chemical species in particular redox states are inherently unstable in water and subject to change by re-equilibration upon reaction with other dissolved components.17 Thus, a method for Cr speciation that ensures that the final results reflect the original states of the Cr species is required.
Field speciation methods or techniques for immediate on-site separation (followed by laboratory analysis) could help prevent inter-conversion of Cr species and provide more reliable results. To this end, Yang et al.18 developed a fully automated portable analyser consisting of a micro-sequential injection lab-on-value system and a miniature USB2000 spectrophotometer for field speciation of Cr. Based on the chromogenic reaction of Cr(VI) with diphenylcarbazide (DPC), the Cr(VI) content in an original sample and the total Cr after oxidation were quantified using a micro-column reactor containing bismuthate-immobilized silica. The feasibility of this method was validated using three wastewater samples. However, its relatively poor detection limit (5.6 μg L−1 for Cr(VI)) does not meet the requirements of most natural water samples (0.1–10 μg L−1 levels). Furthermore, a sensitive speciation method based on field separation with dispersive liquid–liquid micro-extraction (M-MM-DLLME) for sequential and simultaneous separation and pre-concentration of Cr(VI) and Cr(III) was developed by Kumar et al.16 The low volumes of the final extracted phase (0.6 mL for Cr(VI) or Cr(III)) are easily transported to the laboratory at room temperature for quantification using electrothermal atomic absorption spectrometry (ETAAS). This on-site separation method does not require storage and transportation of samples at 4 °C. However, the separation procedure is complicated and its application to real samples is difficult.16 Moreover, Romero et al.19 used multilayer grapheme membranes as the pre-concentration platform for selective absorption of Cr(VI) from natural waters followed by quantitation using a portable total reflection X-ray fluorescence spectroscopy apparatus. In addition, a field analysis method has been developed based on the separation of Cr(VI) and Cr(III) using two tandem small resin-loaded extraction disks and analysing them directly using diffuse reflectance spectroscopy (DRS).20 When the aqueous sample is pushed through the disks, the Cr(VI) is retained on the top anion-exchange disk and Cr(III) is extracted by the second cation-exchange disk. The concentration on each disk is several-hundred fold higher than that of the original sample.20 Although the detection capacity is limited by the DRS measuring method (sub-mg L−1 levels), this solvent-free and simple method introduced the possibility of field sampling and separation techniques for Cr species, avoiding the sampling and preservation operations needed when using centralized analytical techniques.
Consequently, this study describes a simple method for on-site separation of Cr(VI) and Cr(III) from natural waters using parallel cation- and anion-exchange column cartridges. The cation-exchange column retains Cr(III), while Cr(VI) passes through and is collected in the effluent. Conversely, the anion-exchange column retains Cr(VI) and elutes the Cr(III). The final solutions are analysed later in a laboratory using inductively coupled plasma mass spectrometry (ICP-MS). The optimization of the technique, evaluation of its analytical performance, and its application to the determination of Cr(VI) and Cr(III) in natural and drinking water samples are described.
Experimental
On-site separation assembly and procedure
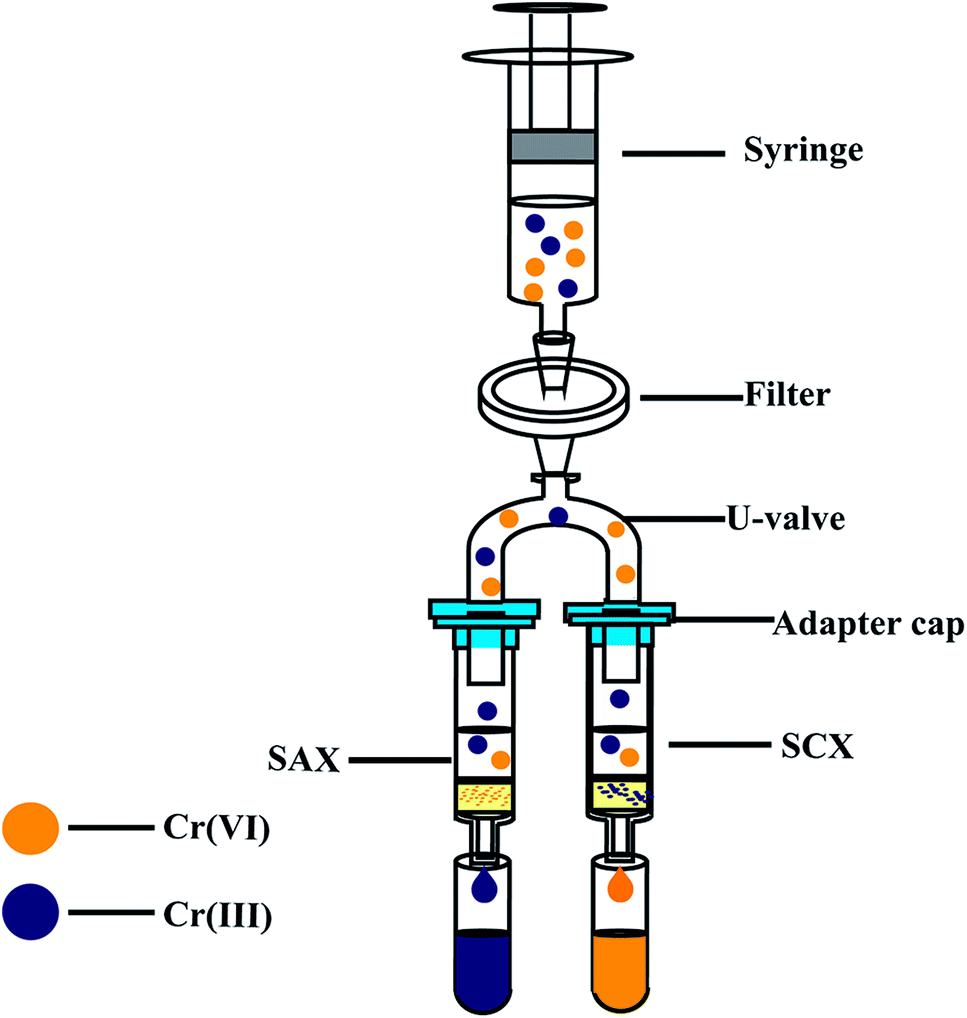
As shown in Fig. 1, the on-site assembly consists of a cation ion-exchange column (SCX, 500 mg, 55 μm particle size, Phenomenex, USA), an anion ion-exchange column (SAX, 500 mg, 55 μm particle size, Phenomenex, USA), a disposable syringe (5 mL), a syringe filter (cellulose acetate, 0.45 μm), a U-valve, a suitable adapter cap, and two centrifuge tubes (5 mL). The structures of the SCX and SAX resins are shown in Fig. S1.† Before use, the SCX and SAX resin columns were activated with 10% H2SO4 for 3 h and 5% NaCl for 4 h, respectively. A 4.0 mL sample is aspirated into a disposable plastic syringe. After purging of all air, the sample is slowly forced through the filter and split into the SCX and SAX columns. In a matter of seconds, the Cr(III) is retained on the cation-exchange resin and Cr(VI) passes into the first centrifuge tube, meanwhile the Cr(VI) is retained on the anion-exchange resin and Cr(III) passes into the second centrifuge tube. The two collected solutions (each 2.0 mL) are preserved and analysed later in the laboratory.
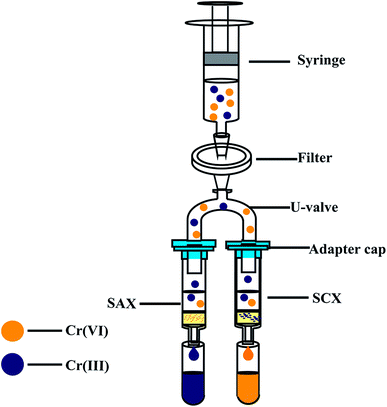 |
| | Fig. 1 Schematic of Cr(VI) and Cr(III) separation equipment. SCX: strong cation-exchange resin column (500 mg, 3 mL), SAX: strong anion-exchange resin column (500 mg, 3 mL). | |
Instrumentation, reagents, and standards
A 7700x ICP-MS (Agilent, USA) with a collision reaction cell (CRC) was used for determining the separated solutions in the laboratory. Helium was introduced at an optimized flow rate of 4.0 mL min−1 into the CRC to remove the ions (i.e., 40Ar12C+) that interfere with 52Cr+ determination. Ion chromatography (IC, DIONEX ICS-900, Thermo Fisher, USA) coupled with ICP-MS (7700x)21 was used to certify the accuracy of the proposed method. ICP-optical emission spectrometry (ICP-OES, IRIS, Thermo Fisher, USA) was employed to check the matrix elements in the water samples. High-purity water (18.2 MΩ cm−1) used for the preparation of all the blanks, samples, and standards was obtained from a water purification system (Millipore, France). Analytical-grade HNO3 and H2SO4 were purchased from Alfa Aesar Ltd. (Tianjin, China). Standard solutions of Cr(III) and Cr(VI), single element stock solutions (including K, Na, Ca, Mg, Fe, Ba, and Al), anion solutions (including F−, Cl−, NO3−, and SO42−), and three water standard reference materials (SRMs, GBW080257, GBW080403, and GBW080404) were purchased from the National Institute of Metrology of China (NIMC, China).
Results and discussion
Separation of Cr(VI) and Cr(III)
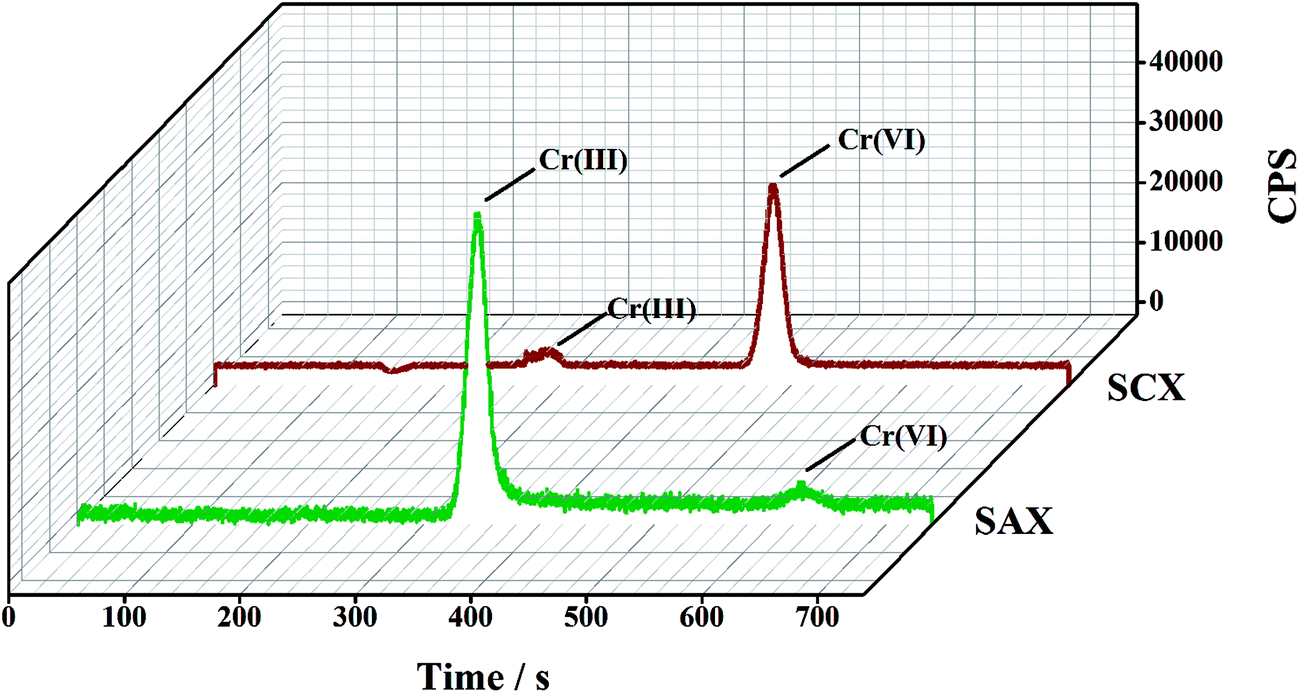
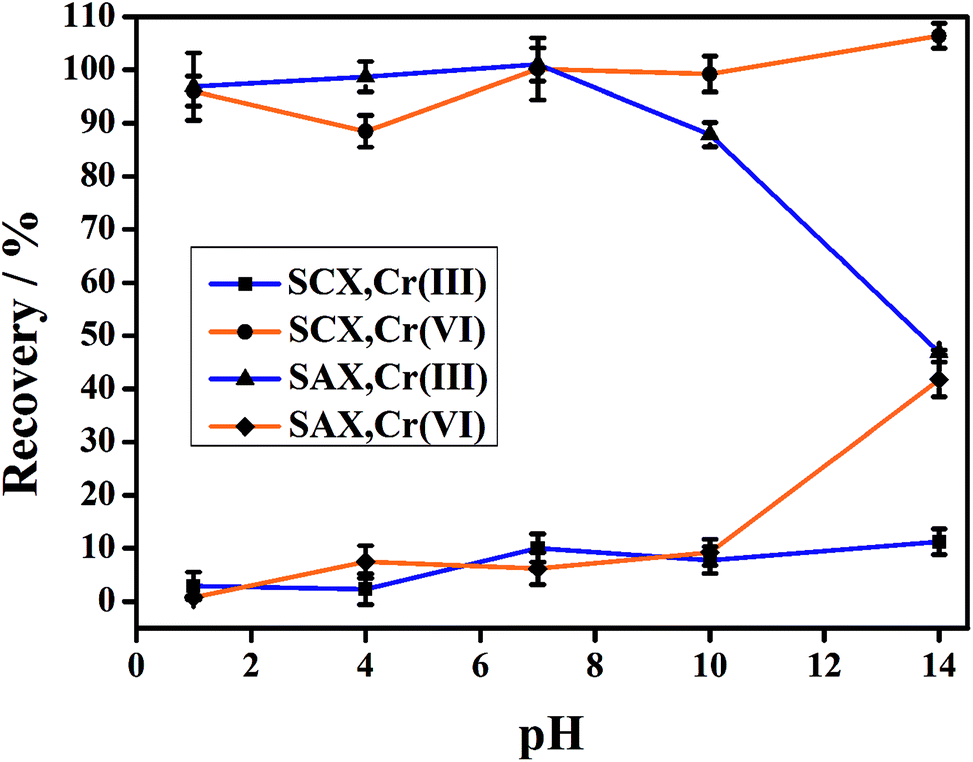
Owing to the opposite charges of Cr(III) and Cr(VI) species, cationic Cr(III) has a strong affinity for the cation-exchange resin, while anionic Cr(VI) passes into the effluent. Conversely, the anion-exchange resin quantitatively retains the anionic Cr(VI) species and completely elutes the Cr(III). The feasibility of this method was determined in an initial experiment, in which a mixed solution of Cr(III) and Cr(VI) (2.0 μg L−1 for each species in 1% HNO3 solution) was directly injected to the parallel-cartridge ion-exchange columns and the Cr(III) and Cr(VI) levels in the effluents were determined by ICP-MS to be 1.94 and 1.97 μg L−1, respectively. The results of IC-ICP-MS analysis confirmed that only Cr(VI) species were present in the SCXs effluent and only Cr(III) species was present in the SAXs effluent (Fig. 2). The prerequisite for separation by ion-exchange resin is that all the targets are present in their ionic states, such as Cr(VI) as negatively-charged hydrogen chromate (HCrO4−) or chromate (CrO42−) and Cr(III) as positively-charged Cr3+, CrOH2+, or Cr(OH)2+.15 The Cr ionic state is strongly dependent on the solution pH value;22 thus, the effect of sample pH on the separation of Cr(III) and Cr(VI) was investigated. Fig. 3 shows that the recoveries of Cr(III) and Cr(VI) are both in the range 90–100% for solution pH values between 1 and 10. A high pH (>11) leads to poor recovery of Cr(III). Therefore, the sample pH need not be pre-adjusted by acid or base, because this wide range of pH (1–10) is common to almost all natural waters.23
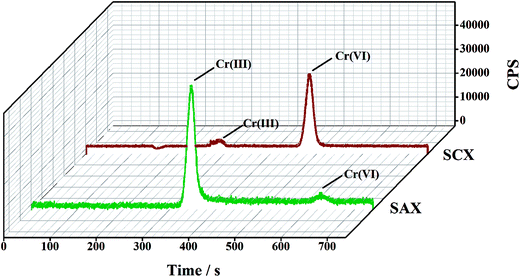 |
| | Fig. 2 A typical IC-ICP-MS spectrum for the two solutions collected after separation by the parallel cartridge ion-exchange columns (2.0 μg L−1 for each Cr species). | |
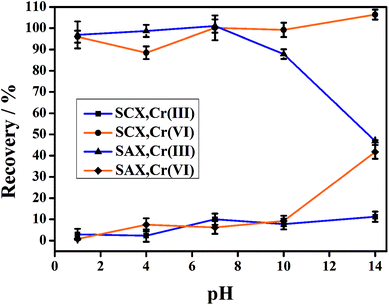 |
| | Fig. 3 Effect of sample pH on the separation of Cr(VI) and Cr(III) using the parallel cartridge ion-exchange columns (10 μg L−1 for each Cr species). | |
Interfering effects
In the ion-exchange resin method, interference ions can compete with the target ions (Cr(VI) or Cr(III)) for absorption to the resin surface or direct oxidization or reduction. Thus, in order to assess potential interference, the tolerability of the method frequently encountered ions was studied by spiking solutions containing Cr(VI) and Cr(III) at 20 μg L−1 each. Table 1 shows that Cr species (other than the target species) having concentrations at least 500 times than that of the target Cr species can be separated. The recovery of Cr(III) is 107% in the presence of 10 mg L−1 CrO42− and that of Cr(VI) is 90.6% in the presence of 10 mg L−1 Cr3+. Nearly all the investigated ions have a negligible effect on the recovery of Cr(III) by the SAX column. Except for in the presence of Fe2+ and Fe3+, the recoveries of Cr(VI) range from 89.4 to 106% when the ratios of concentration of foreign species to that of the target ion are in the range 5000–100000 (Table 1). However, this method appears to provide accurate Cr(VI) recovery (85.5%) in the presence of Fe2+ at ∼250-fold concentration (5.0 mg L−1 Fe2+), beyond which the recoveries decrease dramatically with increasing Fe2+ content. This may be due to the reduction of Cr(VI) to Cr(III) by Fe2+ and subsequent co-precipitation with (or adsorbing of) Cr. Although the tolerance limits for Fe2+ and Fe3+ are relatively low, their concentrations in common water samples does not exceed 1.0 mg L−1 (Table S1†). In addition, the nature water contains negatively charged colloids, including humic substances, kaolin, hydrated silica and their mixtures with hydrated iron(III) oxide,24 which may sorb Cr(III) and be partly retained on the anion-exchange resins.25 However, the concentration of these Cr(III) associated with negatively charged colloids is less than 10% of Cr(VI) in natural waters.26 In fact, no oxidation or reduction occurs (i.e., interconversion of Cr species) during this separation because no acid/base or solvent are required to adjust sample pH or elute the target, respectively.
Table 1 Tolerance concentration ratios for foreign ions on the separation of Cr species, n = 3
| Foreign species |
Concentration ratios |
Recovery (%) |
| Cr(III) |
Cr(VI) |
| The concentration of each ion was 100 mg L−1. The concentration of each ion was 500 mg L−1. |
| Cr3+ |
500 |
— |
90.6 ± 0.6 |
| CrO42− |
500 |
107 ± 1 |
— |
| K+, Na+, Ca2+, Mg2+a |
20000 |
95.6 ± 0.1 |
106 ± 1 |
| Fe2+ |
100 |
99.1 ± 0.6 |
95.5 ± 0.1 |
| Fe2+ |
250 |
99 ± 1 |
85.5 ± 2.5 |
| Fe2+ |
50000 |
97.5 ± 0.9 |
1.2 ± 0.6 |
| Fe3+ |
100 |
99.3 ± 0.7 |
92.8 ± 1.9 |
| Fe3+ |
250 |
99.2 ± 1.0 |
86.7 ± 3.0 |
| Fe3+ |
50000 |
97.8 ± 3.2 |
0.4 ± 0.5 |
| Al3+ |
50000 |
99.2 ± 0.3 |
89.4 ± 1.3 |
| Ba2+ |
50000 |
99.3 ± 0.1 |
95.1 ± 0.3 |
| F−, Cl−, SO42−, NO3−b |
100000 |
88.4 ± 2.0 |
97.2 ± 1.3 |
Analytical performance
The limits of detection (LODs) for Cr(VI) and Cr(III), define as three times the relative deviation of 11 consecutive measurements of the blank solutions, are 0.01 and 0.03 μg L−1, respectively. Table 2 shows that the LODs of the proposed method ars better than those of other reported field separation methods18–20 and are comparable with the IC-ICP-MS results (0.04 μg L−1 for Cr(VI) and 0.01 μg L−1 for Cr(III)).27 The relative standard deviation (RSDs) for Cr(VI) and Cr(III), calculate from the relative deviation of three consecutive measurements of a drinking water (containing 1.14 μg L−1 Cr(VI) and 0.72 μg L−1 Cr(III)). The accuracy of the proposed method was first validated by analysing three water SRMs. The results for GBW080257, GBW080403, and GBW080404 are 0.16, 4.44, and 88.83 μg L−1, respectively, for Cr(VI) and 0.45, 0.74, 9.49 μg L−1, respectively, for Cr(III), which are in good agreement with the reference values measured by IC-ICP-MS. Although no certified values are available for Cr(VI), no differences are observed between the values detected by this method and those obtained by IC-ICP-MS for Cr(VI) and Cr(III) (Table 3). In addition, for a drinking water sample spiked with Cr(VI) and Cr(III) at different levels, the recoveries for the Cr species in the ranges 98.3–108.5% and 99.3–115.2%, respectively (Table 4).
Table 2 Comparison of the current method with other reported field speciation methods for Cr(III) and Cr(VI)
| Separation procedure, determination method |
LOD (μg L−1) |
Separation time (s) |
Sample pH |
Continuous use capacity |
Solvent-free |
| Two parallel cartridge ion-exchange columns, ICP-MS (this work) |
Cr(III): 0.03, Cr(VI): 0.01 |
<10 |
1–10 |
Yes, >40 times |
Yes |
| Two resin-loaded extraction disks, portable diffuse reflectance spectroscopy (DRS)20 |
Cr(III): 1000, Cr(VI): 100 |
<10 |
3 |
No |
No |
| Multilayer grapheme membranes, total reflection X-ray fluorescence (TXRF)19 |
Selective extraction Cr(VI): 0.08 |
>20 min |
1 |
No |
No |
| Micro-sequential injection lab-on-valve, a miniature USB2000 spectrophotometer18 |
Selective extraction Cr(VI): 5.6 |
Not available |
0.6 mol L−1 H2SO4 |
No |
No |
| Micelle dispersive liquid–liquid microextraction, ETAAS16 |
Cr(III): 0.0005, Cr(VI): 0.0006 |
Not available |
2 |
No |
No |
Table 3 Analytical results for Cr species in SRMs (μg L−1), n = 3
| SRMsa |
This method |
IC-ICP-MS |
Certified value, total Cr |
| Cr(III) |
Cr(VI) |
Cr(III) |
Cr(VI) |
| Dilution of 1000-fold for the original SRM. |
| GBW (E)080403 |
0.45 ± 0.08 |
0.16 ± 0.04 |
0.40 ± 0.11 |
0.17 ± 0.02 |
0.50 |
| GBW (E)080404 |
0.74 ± 0.03 |
4.44 ± 0.01 |
0.62 ± 0.09 |
4.45 ± 0.08 |
5.00 |
| GBW (E)080257 |
9.49 ± 1.18 |
88.83 ± 0.13 |
11.59 ± 0.07 |
89.02 ± 0.26 |
100.00 |
Table 4 Recoveries (%) of Cr(VI) and Cr(III) in drinking water samples, n = 3
| Species |
Tested values (μg L−1) |
Spiked values (μg L−1) |
Found values (μg L−1) |
Recovery (%) |
| Cr(VI) |
1.14 ± 0.07 |
0.94 |
2.16 ± 0.12 |
108.5 |
| 4.42 |
5.49 ± 0.31 |
98.5 |
| 9.52 |
10.5 ± 0.77 |
98.3 |
| Cr(III) |
0.72 ± 0.03 |
1 |
1.76 ± 0.11 |
105.1 |
| 4.43 |
5.15 ± 0.24 |
99.3 |
| 10.35 |
11.18 ± 0.54 |
115.2 |
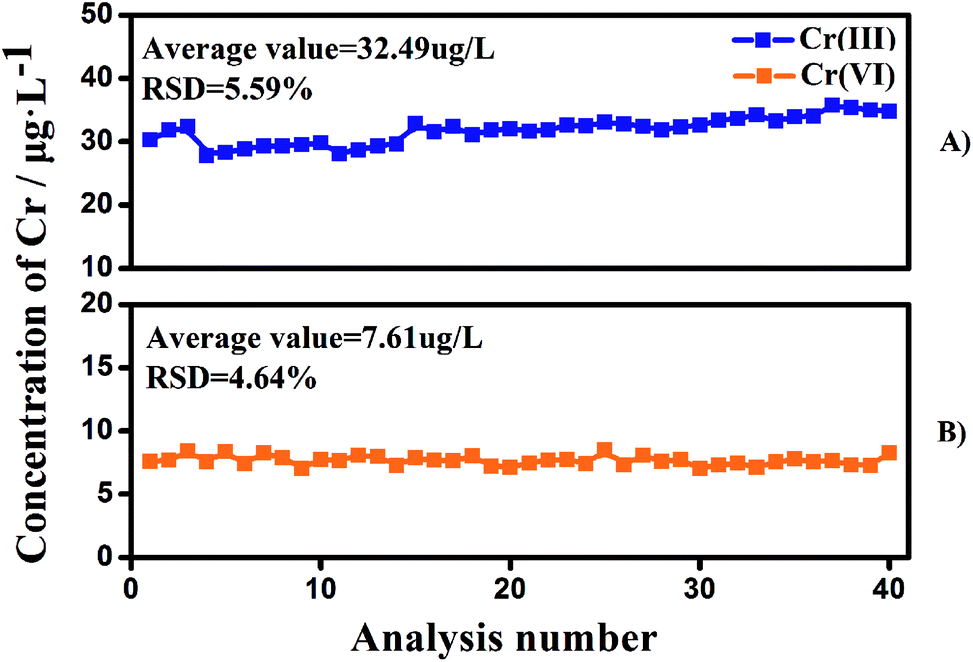
To check the column capacity of this method, a groundwater sample was passed continuously through the ion-exchange columns and the two effluents were collected (Fig. 4). Without any regeneration or cleaning steps, the values obtained for the Cr species for the 40th injection (7.6 μg L−1 for Cr(VI) and 32.6 μg L−1 for Cr(III)) are the same as those obtained for the first injection (7.5 μg L−1 for Cr(VI) and 31.2 μg L−1 for Cr(III)). Thus, the capacity of the resin column (500 mg) allows at least 40 samplings without any cleaning step for typical natural waters. Larger amounts of cation and anion exchange resins are recommended for water samples with high concentrations of dissolved organic carbon and/or having high ionic strength.
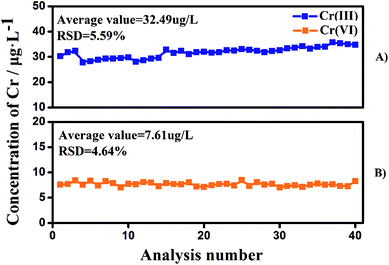 |
| | Fig. 4 The stability of the proposed method for Cr(VI) and Cr(III) separation without any regeneration or cleaning steps. Ground water was used in this experiment. | |
Application to the natural water samples
This on-site separation method was used to monitor the Cr(VI) and Cr(III) concentrations of 12 natural water samples, which are collected from lakes of neighboring a chrome salt factory in Huangshi (Fig. S2†). Table 5 shows that the Cr(VI) contents range from 0.17 to 4.25 μg L−1 and the Cr(III) contents range from <LOD to 1.01 μg L−1. Previous data28 (Table 5) reveal that the concentrations of Cr(VI) at the same sites almost a decade ago were 3- to 180-times to those measured recently.
Table 5 Speciation of Cr in natural water samples (μg L−1), n = 3
| Samples |
This study |
Reported value28 |
| Cr(III) |
Cr(VI) |
Cr(VI) |
| <0.03 μg L−1 (LOD of this method). Not reported. |
| S1 |
N.D.a |
0.60 ± 0.07 |
—b |
| S2 |
0.28 ± 0.03 |
0.98 ± 0.11 |
8 |
| S3 |
1.01 ± 0.03 |
2.46 ± 0.21 |
8 |
| S4 |
0.9 ± 0.02 |
2.92 ± 0.14 |
6 |
| S5 |
N.D. |
0.80 ± 0.08 |
— |
| S6 |
N.D. |
0.95 ± 0.13 |
— |
| S7 |
0.07 ± 0.02 |
1.12 ± 0.12 |
— |
| S8 |
0.10 ± 0.02 |
0.17 ± 0.01 |
— |
| S9 |
0.67 ± 0.06 |
4.25 ± 0.13 |
— |
| S10 |
0.33 ± 0.04 |
1.90 ± 0.17 |
6 |
| S11 |
N.D. |
0.95 ± 0.12 |
174 |
| S12 |
N.D. |
2.74 ± 0.26 |
113 |
Conclusions
Very fast (<10 s) on-site separation of Cr(VI) and Cr(III) was achieved by passing a water sample through parallel ion-exchange resin column cartridges. Anionic Cr(VI) was obtained from the effluent of the cation-exchange column, while Cr(III) was collected from the effluent of the anion-exchange column. The amounts of each Cr species can be obtained later by ICP-MS (or any other elemental analysis instrument with sufficient sensitivity) in the laboratory. This solvent-free method avoids inter-conversion of the Cr species and provides a routine protocol for the determination of Cr(VI) and Cr(III) in natural waters.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the China Scholarship Council, the National Natural Science Foundation of China (No. 41521001, 41773013, 41572335, and 41772370), the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (No. CUG170102), the Research Funds for Engineering Research Center of Nano-Geo Materials of Ministry of Education (NGM2016KF007), and the Research Program of State Key Laboratory of Biogeology and Environmental Geology of China.
References
- M. Chebeir and H. Liu, Environ. Sci. Technol., 2016, 50, 701–710 CrossRef CAS PubMed.
- C. Pan, L. D. Troyer, J. G. Catalano and D. E. Giammar, Environ. Sci. Technol., 2016, 50, 13502–13510 CrossRef CAS PubMed.
- K. Pytlakowska, J. Anal. At. Spectrom., 2016, 31, 968–974 RSC.
- U.S. Environmental Protection Agency, Toxicological review of hexavalent chromium, Washington, DC, 2015, http://www.epa.gov/iris/toxreviews/0144tr.pdf Search PubMed.
- World Health Organization, Guidelines for Drinking Water Quality, WHO Press, World Health Organization, Geneva, 4th edn, 2011 Search PubMed.
- http://www.ct.gov/dph/lib/dph/drinking_water/pdf/Hexavalent_Chromium_in_Drinking_Water.pdf.
- People's Republic of China Ministry of Health, Standards for drinking water quality, GB 5749-2006, revised in 2006.
- S. Cuello, J. Entwisle, J. Benning, C. Liu, S. Coburn, K. G. McAdam, J. Braybrook and H. Goenaga-Infante, J. Anal. At. Spectrom., 2016, 31, 1818–1829 RSC.
- I. Gaubeur, M. A. Aguirre, N. Kovachev, M. Hidalgo and A. Canals, J. Anal. At. Spectrom., 2015, 30, 2541–2547 RSC.
- F. Hernandez, P. Jitaru, F. Gormant, L. Noel and T. Guerin, Food Chem., 2018, 240, 183–188 CrossRef CAS PubMed.
- N. Unceta, M. Astorkia, Z. Abrego, A. Gomez-Caballero, M. A. Goicolea and R. J. Barrio, Talanta, 2016, 154, 255–262 CrossRef CAS PubMed.
- B. Markiewicz, I. Komorowicz and A. Sajnog, Talanta, 2015, 132, 814–828 CrossRef CAS PubMed.
- K. Pyrzynska, TrAC, Trends Anal. Chem., 2012, 32, 100–112 CrossRef CAS.
- K. Pyrzynska, Int. J. Environ. Anal. Chem., 2012, 92, 1262–1275 CrossRef CAS.
- J. W. Ball and R. B. McCleskey, Talanta, 2003, 61, 305–313 CrossRef CAS PubMed.
- N. N. Meeravali, K. Madhavi and S. J. Kumar, J. Anal. At. Spectrom., 2016, 31, 1582–1589 RSC.
- S. Comber and M. Gardner, J. Environ. Monit., 2003, 5, 410–413 RSC.
- M. Yang, J.-X. Li and J.-H. Wang, Talanta, 2007, 72, 1710–1716 CrossRef CAS PubMed.
- V. Romero, I. Costas-Mora, I. Lavilla and C. Bendicho, RSC Adv., 2016, 6, 669–676 RSC.
- S. A. Steiner, M. D. Porter and J. S. Fritz, J. Chromatogr. A, 2006, 1118, 62–67 CrossRef CAS PubMed.
- H. Cui, W. Guo, L. L. Jin, Q. H. Guo and S. H. Hu, Anal. Methods, 2017, 9, 1307–1312 RSC.
- F. Seby, S. Charles, M. Gagean, H. Garraud and O. F. X. Donard, J. Anal. At. Spectrom., 2003, 18, 1386–1390 RSC.
- B. Markiewicz, I. Komorowicz, A. Sajnog, M. Belter and D. Baralkiewicz, Talanta, 2015, 132, 814–828 CrossRef CAS PubMed.
- M. Hiraide, M. Ishii and A. Mizuike, Anal. Sci., 1988, 4, 605–609 CrossRef CAS.
- P. A. Sule and J. D. Ingle Jr, Anal. Chim. Acta, 1996, 326, 85–93 CrossRef CAS.
- M. Hiraide and A. Mizuike, Fresenius' Z. Anal. Chem., 1989, 335, 924–926 CrossRef CAS.
- E. Leese, J. Morton, P. H. E. Gardiner and V. A. Carolan, J. Anal. At. Spectrom., 2016, 31, 924–933 RSC.
- B.-J. Hu, T. Ma, C.-F. Liu and L. Li, Saf. Environ. Eng., 2007, 14, 20–23 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10303d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Qinghai Guo*ac,
Lanlan Jina,
Zhifu Liua and
Shenghong Hu
*ab,
Qinghai Guo*ac,
Lanlan Jina,
Zhifu Liua and
Shenghong Hu