
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical properties of NiCoO2 synthesized by hydrothermal method
Xianwei Wang *,
Weixia Li†
,
Xiaoer Wang†,
Jingjie Zhang†,
Lingyun Sun†,
Chang Gao,
Jun Shang,
Yanchun Hu and
Qianqian Zhu
*,
Weixia Li†
,
Xiaoer Wang†,
Jingjie Zhang†,
Lingyun Sun†,
Chang Gao,
Jun Shang,
Yanchun Hu and
Qianqian Zhu
Laboratory of Functional Materials, College of Physics and Materials Science, Henan Normal University, Henan Key Laboratory of Photovoltaic Materials, Xinxiang 453007, China. E-mail: xwwang2000@163.com
First published on 1st November 2017
Abstract
NiCoO2 microspheres were successfully synthesized via an easy hydrothermal method, followed by an annealing process at 350 °C under a nitrogen atmosphere. The X-ray diffraction results show that a single phase NiCoO2 was synthesized. The results of electrochemical measurements show that the NiCoO2 exhibits a high specific capacitance of 760 and 470 F g−1 at current densities of 1 and 16 A g−1, respectively, and the specific capacitance retention is 40.9% after 5000 cycles at the current density of 16 A g−1. This reveals the great potential of NiCoO2 as an electrode material for high-performance supercapacitors.
1. Introduction
Clean energy conversion and storage devices have attracted great attention. Currently, supercapacitors, as a clean energy conversion and storage technology, have drawn intensive research attention in the past several years due to their high power densities, short charging times, safe operation mode and long cycle life.1–6 The electrode material, as an important component of a supercapacitor, plays a key role to gain high supercapacitor performances.7 Many kinds of materials have been considered as the electrode materials for supercapacitors, and they can be divided into three main kinds of electrode materials:8 carbonaceous materials,9 transition metal oxides10 and conducting polymer materials.11 However, many carbonaceous materials show low specific capacitance, and the conducting polymer materials exhibit poor electrical conductivity, leading to limited practical applications of these electrode materials for supercapacitors.12,13 Therefore, it is important to search for novel electrode materials, which show high electrical conductivity, large specific capacitance and high cyclic stability. Thus transition metal oxides as electrode materials have been widely investigated because of their alternative redox properties and high theoretical specific capacitance values.14 Especially, some transition metal oxides, such as MnO2,15 Co3O4 (ref. 16) and NiO17,18 have attracted great interest for use as supercapacitor electrode materials because of their low cost, natural abundance and environmental friendliness. Moreover, the nickel-cobalt binary metal oxides, such as NiCo2O4 (ref. 19 and 20) and NiCoO2 (ref. 21–23) have been also studied due to their higher electrical conductivity and specific capacitance than those of the single component nickel or cobalt oxides.Recently, much research on the preparation and electrochemical properties of NiCo2O4 has been reported.24–29 Especially, some artificial nanostructures show excellent properties.30–34 Two types of homogeneous NiCo2O4 nanosheet@NiCo2O4 nanorod and heterogeneous NiCo2O4 nanosheet@NiO nanoflake hierarchical core–shell arrays were synthesized via facile solution methods followed by a simple thermal treatment, and high specific capacitances of 1925 and 2210 F g−1 were achieved by NiCo2O4@NiCo2O4 and NiCo2O4@NiO at 0.5 A g−1, respectively.35 However, research on NiCoO2 as an electrode material for supercapacitors is rare.2,19,21,36–39 Du et al. prepared NiCoO2 single-crystalline nanoparticles with a specific capacitance of 184 F g−1 at 1 A g−1.40 NiCoO2 was synthesized based on the templates and used for supercapacitors.2,21,36–39
In this study, we successfully synthesized micro-spheres NiCoO2 by a simple hydrothermal method, followed by calcining at 350 °C under a nitrogen atmosphere. The NiCoO2 exhibited a specific capacitance of 760 F g−1 at a current density of 1 A g−1 in three electrode measurements. Especially, the specific capacitance retains approximately 40.9% of its maximum value after 5000 cycles at the current density of 16 A g−1.
2. Experimental
2.1 Preparation of NiCoO2
0.474 g NiCl2·6H2O, 0.474 g CoCl2·6H2O and 2.0 g urea were dissolved in 30 mL distilled water with magnetic stirring for 30 min at room temperature to form a clear solution. Then, the obtained solution was transferred into a 50 mL Teflon-lined stainless steel autoclave and maintained at 120 °C for 12 h. After cooling down to room temperature, the precipitate was centrifuged and washed with distilled water and ethanol. Then, the obtained precipitate was dried at 60 °C for 6 h. Finally, the dried precipitate was calcined at 350 °C for 3 h under nitrogen atmosphere.2.2 Characterization
The crystal structure was characterized by X-ray diffraction (XRD, Bruker D8 discover) using Cu Kα radiation at 40 kV and 40 mA. The morphology was examined by field emission scanning electron microscopy (FE-SEM, Zeiss SUPRA 40) and transmission electron microscopy (TEM, JEM-2100). X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi) was used to characterize valence states of the sample. The specific surface area and pore size distribution of the sample were performed by Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption and Barrett–Joyner–Halenda (BJH) methods (Autosorb-iQ). The electrochemical characterizations were carried out in a CS 350 electrochemical workstation (CorrTest, Wuhan) at room temperature using 3 M KOH solution as electrolyte solution. A three-electrode system was used in the measurement. Nickel foam coated with electroactive materials was used as the working electrode. Platinum and Hg/HgO electrode (1 M NaOH solution) were served as the counter electrode and the reference electrode, respectively. The working electrode was fabricated by mixing the as-prepared products, carbon blacks and polytetrafluoroethylene (PTFE) with a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 to form homogeneous paste, which was coated onto nickel foam with the area of 1 cm × 1 cm. After drying in a vacuum oven at 50 °C for 5 h to remove the solvent, the nickel foam was then pressed at 10 MPa for 2 min by bead machine. The mass of active materials loading on the nickel foam with the area of 1 cm × 1 cm was about 3–5 mg.
:20:10 to form homogeneous paste, which was coated onto nickel foam with the area of 1 cm × 1 cm. After drying in a vacuum oven at 50 °C for 5 h to remove the solvent, the nickel foam was then pressed at 10 MPa for 2 min by bead machine. The mass of active materials loading on the nickel foam with the area of 1 cm × 1 cm was about 3–5 mg.
Cyclic voltammetry (CV) tests were performed from −0.1 V to 0.8 V at a scan rate of 5, 10, 20, 40, 60 and 100 mV s−1. Galvanostatic charge–discharge (GCD) tests were performed at a current density of 1, 2, 4, 6, 10 and 16 A g−1 with the potential window from 0 V to 0.55 V. Electrochemical impedance spectroscopy (EIS) measurements were measured at a frequency range from 105 Hz to 0.01 Hz with an amplitude of 3 mV.
3. Results and discussion
3.1 Structure characterization
Fig. 1 shows the XRD pattern of the samples calcined at 350 °C under nitrogen atmosphere. The diffraction pattern showed six peaks at 36.80°, 42.82°, 61.79°, 73.99°, 77.99° and 93.22°, corresponding to (111), (200), (220), (311), (222) and (400) of NiCoO2 phase (JCPDS no. 10-0188), respectively. No impurity peaks were observed in the pattern, confirming the single pure phase of the obtained product.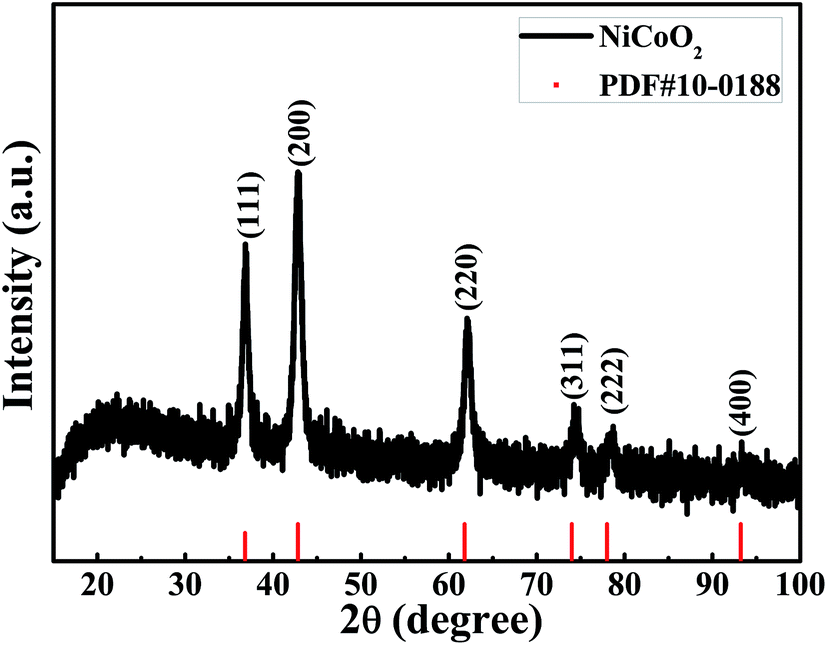 | ||
| Fig. 1 XRD patterns of the NiCoO2 samples. | ||
The elemental composition and the valence states of the Ni and Co in the sample were investigated by XPS, and the results are presented in Fig. 2. Fig. 2(a) shows the survey spectrum of the sample. It clearly showed the peaks of Ni, Co, O and C elements. The C element can be attributed to the exposure of air. Fig. 2(b) shows the spectrum of Ni 2p, which was composed of two spin–orbit doublets and two shakeup satellites. As reported in ref. 41 there are four easily discernible features in Ni 2p spectrum, they are the Ni 2p3/2 main peak and its satellite, and the Ni 2p1/2 main peak and its satellite, respectively. The fitting peaks at 854.2 and 856.0 eV are corresponding to Ni 2p3/2, whereas the peaks at 871.8 and 873.7 eV are corresponding to Ni 2p1/2. The peaks Sat.1 at 861.2 eV and Sat.2 at 879.1 eV are ascribed to the two shakeup satellites of Ni main peaks. The peak of Ni 2p3/2 at 854.2 eV is ascribed to local screening from lattice oxygen adjacent to the Ni 2p core hole.41 And the existence of peak at 856.0 eV is prevalent, suggesting a higher hydroxylation of NiO in the composite oxide surface,42 which can be further confirmed by the O 1s spectrum. The binding energy peaks of Ni 2p1/2 are consistent with that of Ni 2p3/2. The fitting results show the appropriate binding energies of Ni 2p, indicating that the divalent oxide state of Ni was existed in sample.41 Fig. 2(c) shows the core level spectrum of Co 2p. The fitting peak at 780.1 eV and 797.0 eV is corresponding to Co 2p3/2 and Co 2p1/2, respectively, and these two peaks could be indexed to Co2+.43,44 In addition, the shake-up satellite peak Sat.1 located at 785.8 eV has a separation energy of ∼5.2 eV with Co 2p3/2 at 780.1 eV, while the shake-up satellite peak Sat.2 located at 802.2 eV has a separation energy of ∼5.2 eV with Co 2p1/2 at 797.0 eV. The energy gap between the Co 2p main peak and the satellite peaks is related to the oxidation states, and the energy gaps are about 6.0 eV and 9–10 eV for Co2+ and Co3+.45 The results prove that the divalent oxide state of Co was existed in sample. The high resolution spectrum for O 1s peak in Fig. 2(d) decomposes into two oxygen contributions. The fitting peak located at 529.4 eV (O1) are typical of the metal–oxygen bonds (M = Ni or Co).46 The component O2 located at 531.1 eV indicates the presence of surface hydroxyl groups.41,42,47,48 According to the XPS analysis, these results show that the NiCoO2 samples contain Ni2+ and Co2+, the atomic percentage of Ni, Co and O is 23.11, 23.49 and 53.4%, respectively. The calculated atomic ratio of Ni to Co and O is close to 1:1:2, which is on the verge of the stoichiometry of NiCoO2.
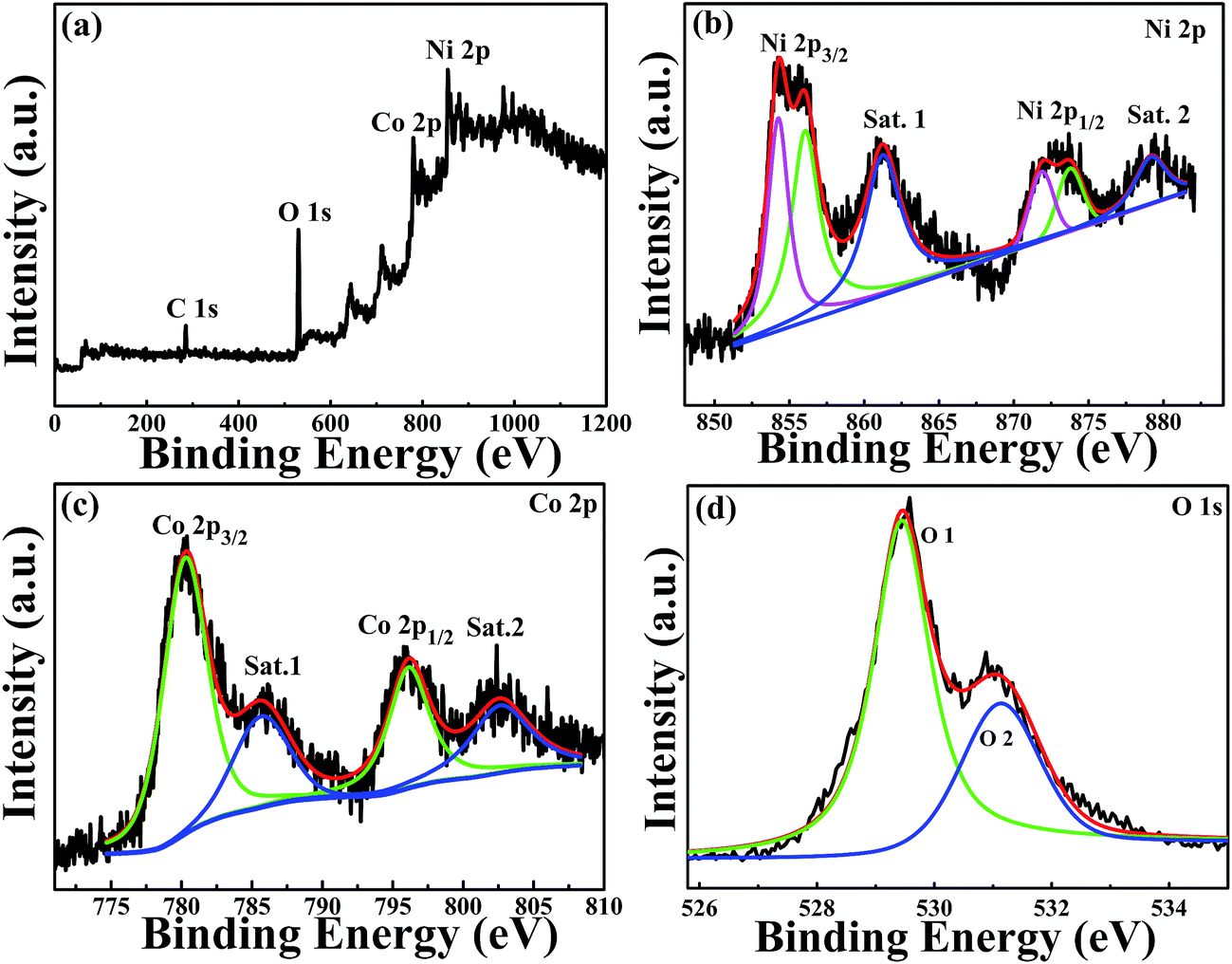 | ||
| Fig. 2 XPS spectrum of (a) survey spectrum, (b) Ni 2p spectrum, (c) Co 2p spectrum and (d) O 1s spectrum for the NiCoO2 samples. | ||
The morphologies of NiCoO2 sample were characterized by SEM and TEM. As shown in Fig. 3(a), NiCoO2 sample showed the microspheres with different size in the low-magnification SEM image of sample. The diameter of the sphere varied from 1 μm to 6 μm. Furthermore, the microspheres are assembled by a large amount of nanowires according to the high-magnification SEM images as shown in Fig. 3(b). To identify the distribution of element in sample, the element mappings of Ni, Co and O were examined by energy dispersive spectroscopy (EDS). Fig. 3(c) is a typical microsphere of sample, and the element mapping of Ni, Co and O were included. It can be seen clearly that Ni, Co and O elements are uniformly distributed in the microsphere, and that the ratio of three elements is around 1:1:2, which corresponded with the composition of NiCoO2. Fig. 3(d)–(f) show TEM images of the samples. Pompon shape was observed in samples, and this is consistent with the morphology results of SEM. The high-magnification TEM image revealed that the nanowires in pompon were composed of nanoparticles. Fig. 3(e) selected area electron diffraction (SAED) pattern showed ring patters, which revealed that the nanowire in pompon has a polycrystalline structure. These rings could be assigned to the (111), (200), (220), (311), (222) and (400) crystal planes of NiCoO2, which is consistent with the observation of XRD results. The lattice spacing of 0.208 nm is attributed to the (200) crystal plane of NiCoO2 sample, as shown in the HRTEM image of Fig. 3(f).
 | ||
| Fig. 3 (a) Low-magnification SEM, (b) high-magnification SEM, (c) energy dispersive spectroscopy (EDS) element mapping images of the NiCoO2 sample and the Ni, Co and O distribution, (d) low-magnification TEM, (e) high-magnification TEM (inset showing the selected area electron diffraction ring) and (f) HRTEM image of NiCoO2 sample. | ||
The results of XRD, TEM, EDS and XPS confirm that single pure phase of NiCoO2 with pompon shape was obtained in present research, and the pompon with different diameter is composed of nanowires, which were composed of NiCoO2 nanoparticles.
The porous structure of sample was evaluated by Brunauer–Emmett–Teller (BET) N2-adsorption–desorption analysis. The N2 adsorption–desorption isotherm and pore size distribution were shown in Fig. 4. The BET specific surface area is about 88.3 m2 g−1. As can be seen from Fig. 4, the typical type IV hysteresis loop can be observed in the region of P/P0 from 0.6 to 1.0, which illustrates the existence of a mesoporous structure in sample.43,49 The pore size distributions, which were shown in insert of Fig. 4, were calculated by the Barrett–Joyner–Halenda (BJH) method. It shows that sharp peaks are located at 2.7 nm and 11.0 nm, illustrating a few mesoporous were presented in NiCoO2 samples. Such results are consistent with the results of SEM and TEM results. The rich internal mesoporous structure could increase the contact area between electrode and electrolyte, leading to more sufficient active sites for redox reaction and efficient transport of electrons and ions.43,49 Therefore, the high surface area and mesoporous structure of NiCoO2 sample are benefic for application in supercapacitor.43,49,50
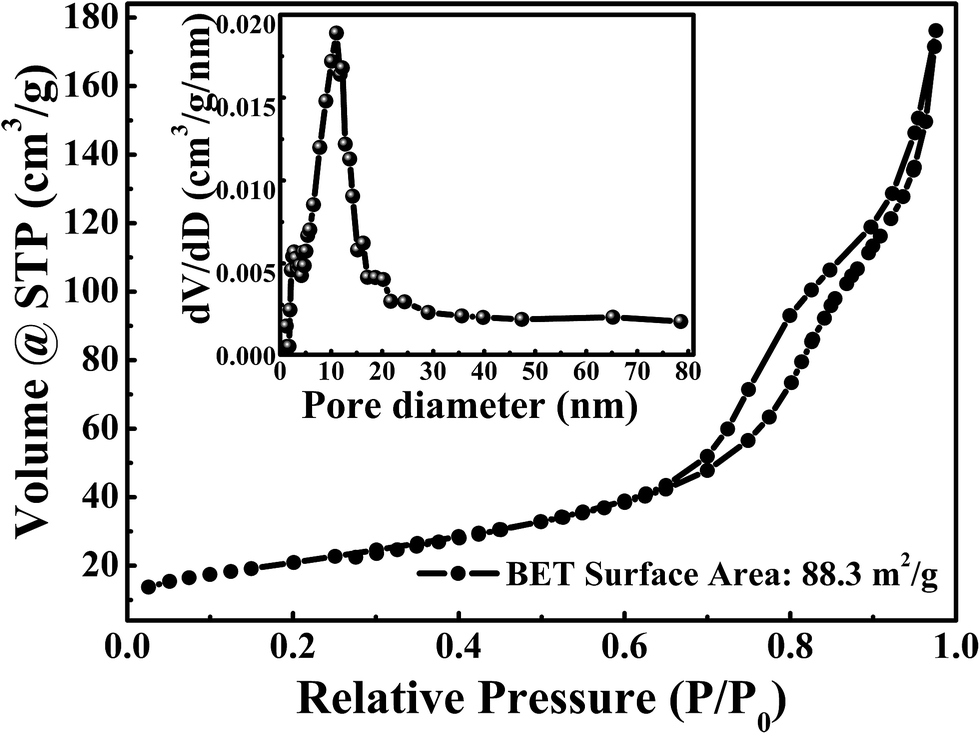 | ||
| Fig. 4 The N2 adsorption–desorption isotherms of the NiCoO2. The inset is the pore size distributions are calculated by the Barrett–Joyner–Halenda (BJH) method. | ||
3.2 Electrochemical characterization
Fig. 5(a) shows the CV curves of the NiCoO2 micro pompon at different scan rates from 5 mV s−1 to 100 mV s−1 within potential range from −0.1 V to 0.8 V (vs. Hg/HgO) in 3 M KOH solution. All the curves show typical faradic reaction peaks, corresponding to the M–O/M–O–OH (M represents Ni and Co ions).39,51,52 With the increase of scan rate, a shift toward higher and lower potentials was observed for the oxidation and reduction peaks, respectively. As shown in inset of Fig. 5(b), this shift leading to the increase of the potential difference between redox peaks. It indicated that the irreversible degree of the quasi-reversible reaction increased with the increase of scan rate. The shift is mainly related to the internal resistance of the electrode and the polarization caused by high scan rate.53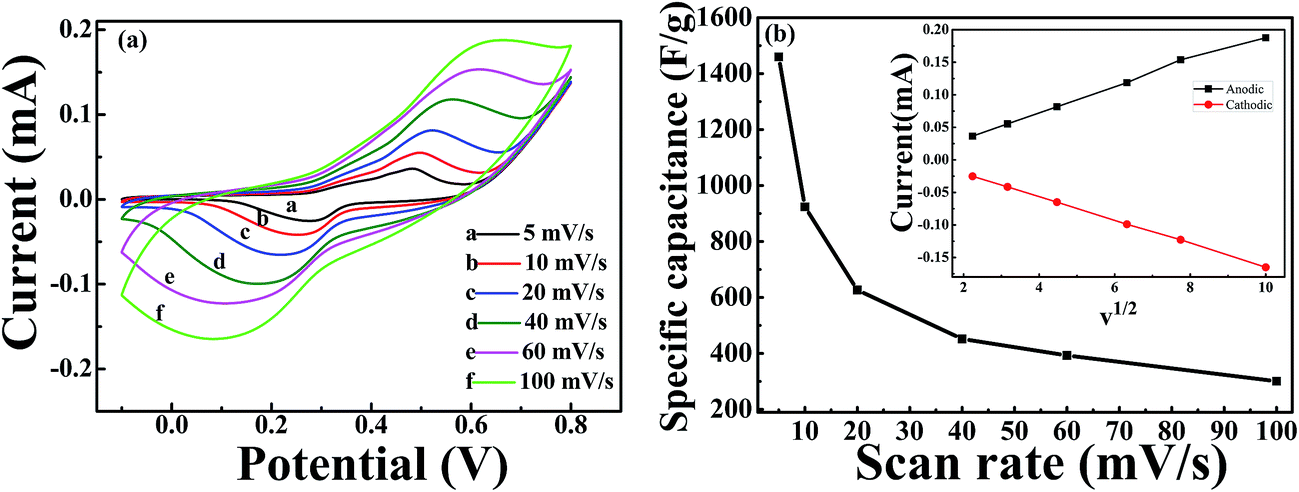 | ||
| Fig. 5 (a) CV curves under different scan rates of 5, 10, 20, 40, 60, 100 mV s−1 for NiCoO2 samples in the potential window ranged from −0.1 to 0.8 V; (b) the specific capacitance of NiCoO2 sample under different scan rates of 5, 10, 20, 40, 60 and 100 mV s−1. The inset in (b) is the plots of anodic and cathodic peak current against the square root of the scan rate. | ||
The specific capacitance of samples can be calculated by using the following equation:
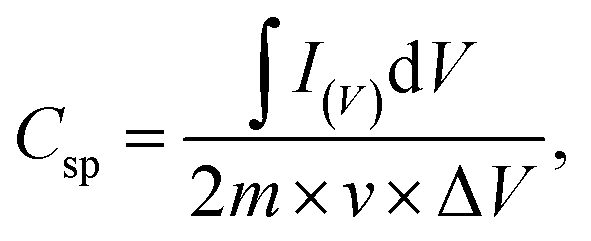 | (1) |
As shown in Fig. 6(a), the GCD curves of NiCoO2 electrodes were measured at different current densities of 1, 2, 4, 6, 10 and 16 A g−1 in 3 M KOH solution in the voltage range of 0–0.55 V (vs. Hg/HgO). The nonlinear curve indicated the pseudocapacitance behavior, and the potential plateaus corresponded with the redox reaction, which was consistent with the CV curves in Fig. 5(a). The specific capacitance could be calculated from the galvanostatic charge–discharge curves according to the following equation:
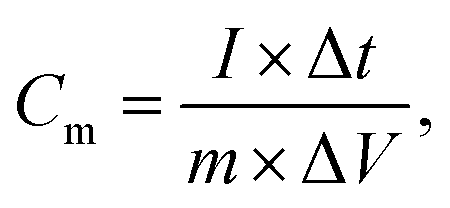 | (2) |
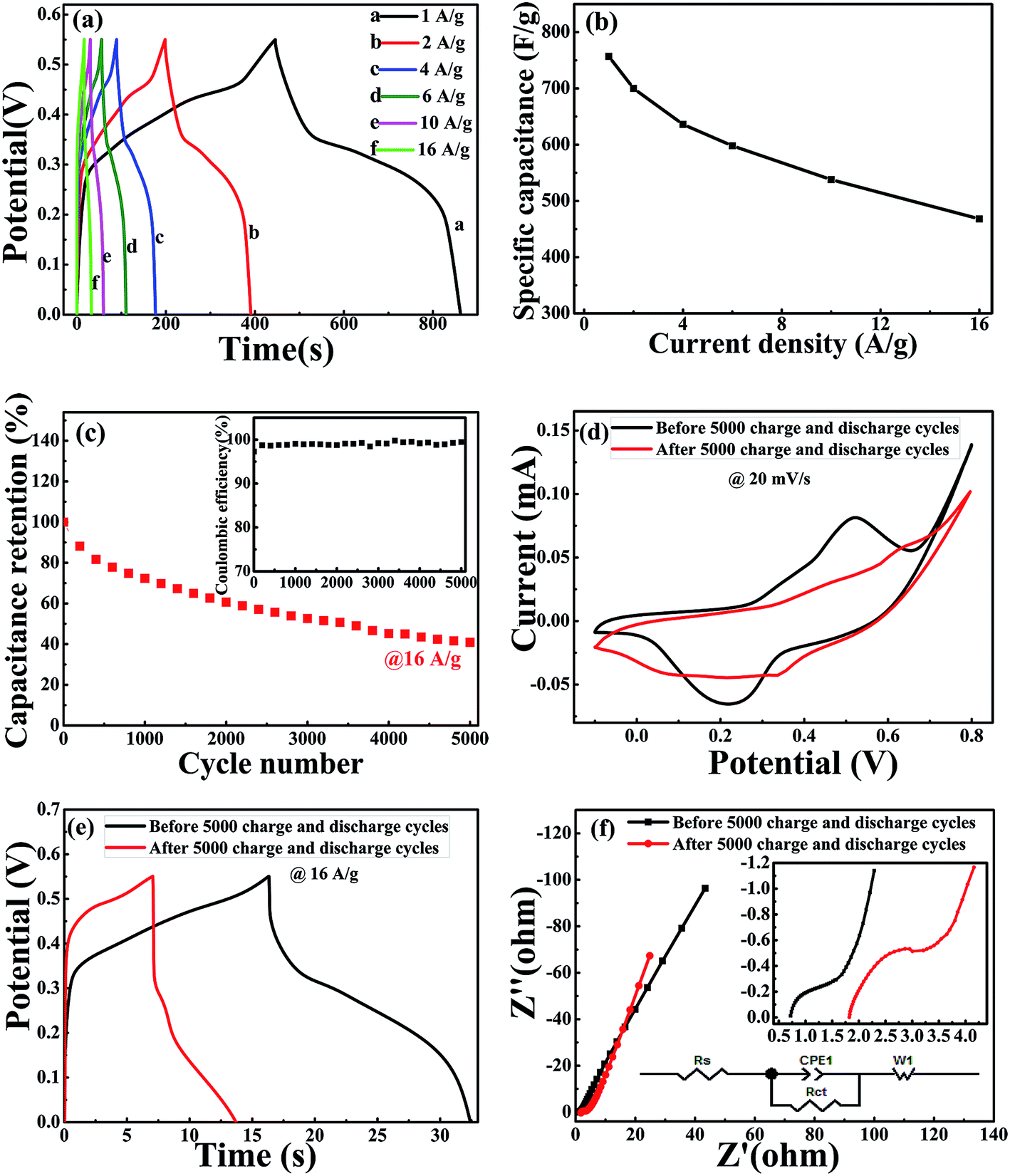 | ||
| Fig. 6 (a) Galvanostatic charge–discharge curves are within the potential range of 0 to 0.55 V at various current densities from 1 A g−1 to 16 A g−1, (b) specific capacitance at various current densities from 1 A g−1 to 16 A g−1. (c) Cycling performance of the NiCoO2 electrode at 16 A g−1, the insert shows the corresponding coulombic efficiency of NiCoO2 during cycling test. (d) CV and (e) GCD curves of NiCoO2 electrode before and after 5000 charge–discharge cycles. (f) Nyquist plots obtained for NiCoO2 samples at the frequency ranging from 0.01 Hz to 100 KHz in 3 M KOH. Insets: the local amplification of the Nyquist plots at high-frequency. | ||
The electrochemical impedance spectroscopy (EIS) was used to evaluate the electrical resistance responses of the NiCoO2 electrode materials. Fig. 6(f) shows the Nyquist plots of NiCoO2 before and after 5000 charge–discharge cycle measurement. The EIS spectra could be fitted according to the equivalent circuit diagram inserted in Fig. 6(f). Rs is the internal resistances of the electrode, including the total resistances of the electrode resistance, the bulk electrolyte resistance and the resistance at electrolyte–electrode interface,56 and Rct is the charge-transfer resistances, corresponding to the semicircle in the high-frequency region.57,58 The fitted Rs and Rct values are very small, and the low values are beneficial for long-cycle stability. After 5000 cycles, the Rs value increased from 0.71 Ω to 1.81 Ω, and the Rct value increased from 0.45 Ω to 0.84 Ω. The linear part at low frequency region representative of the typical Warburg resistance (W1),59 and the CPE1 represents double layer capacitance caused by the rough surface of NiCoO2 samples and inhomogeneous distribution of active sites.37 For an ideal supercapacitor, the Nyqusit plot should be a vertical line at a low frequency, which is parallel to the imaginary impedance axis.34 The slope of line increased from 2.32 to 2.67. The curve becomes more vertical in the low-frequency range after 5000 cycles, indicating that the electro active material becomes more capacitive after long-term cycling.60 EIS results further indicate that NiCoO2 sample with micro pompon structure show low internal and charge-transfer resistance, and stable electrochemical stability.
4. Conclusions
In summary, NiCoO2 micro pompon sample were successfully prepared by a facile hydrothermal method. The micro pompon was assembled with nanowire, and the nanowire is composed with NiCoO2 nanoparticles. Electrochemical measurement results display that the micro-sphere NiCoO2 materials have the remarkable electrochemical behavior. The specific capacitance was 760 F g−1 at a current density of 1 A g−1. The specific capacitance showed small loss under the current density increasing, and it was only 38% when the current density increased from 1 to 16 A g−1. In addition, the NiCoO2 micro pompon with low internal and charge-transfer resistance exhibited fine long-cycle stability, around 40.9% of its initial specific capacitance was maintained after 5000 charge–discharge cycles. The excellent electrochemical properties make the NiCoO2 micro pompon sample be the promising candidate electrodes materials of supercapacitor in practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the National Natural Science Foundation of China (No. 51402091, No. 51601059, No. 11304082 and No. 11404102), the scientific research foundation for new introduced doctors in Henan Normal University (No. 11114), and the National University Student Innovation Program (No. 201410476037).References
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- X. Xu, H. Zhao, J. K. Zhou, R. Xue and J. Gao, J. Power Sources, 2016, 329, 238–246 CrossRef CAS.
- W. Chen, C. Xia and H. N. Alshareef, ACS Nano, 2014, 8, 9531–9541 CrossRef CAS PubMed.
- M. Jana, S. Saha, P. Samanta, N. C. Murmu, N. H. Kim, T. Kuila and J. H. Lee, J. Mater. Chem. A, 2016, 4, 2188–2197 CAS.
- D. Li, Y. Gong, M. Wang and C. Pan, Nano-Micro Lett., 2017, 9, 16 CrossRef.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- X. L. Guo, X. Y. Liu, X. D. Hao, S. J. Zhu, F. Dong, Z. Q. Wen and Y. X. Zhang, Electrochim. Acta, 2016, 194, 179–186 CrossRef CAS.
- X. Yang, K. Xu, R. Zou and J. Hu, Nano-Micro Lett., 2016, 8, 1–8 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- H. B. Wu, H. Pang and X. W. Lou, Energy Environ. Sci., 2013, 6, 3619–3626 CAS.
- L. Nyholm, G. NyströM, A. Mihranyan and M. StrøMme, Adv. Mater., 2011, 23, 3751–3769 CAS.
- M. Jana, S. Saha, P. Samanta, N. C. Murmu, J. H. Lee and T. Kuila, Mater. Chem. Phys., 2015, 151, 72–80 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- H. Gao, G. Wang, M. Yang, L. Tan and J. Yu, Nanotechnology, 2012, 23, 015607 CrossRef PubMed.
- Y. Sun, Z. Fang, C. Wang, K. R. Ariyawansha, A. Zhou and H. Duan, Nanoscale, 2015, 7, 7790–7801 RSC.
- L. Hou, C. Yuan, L. Yang, L. Shen, F. Zhang and X. Zhang, RSC Adv., 2011, 1, 1521–1526 RSC.
- G. Chen, H. Guan, C. Dong, X. Xiao and Y. Wang, J. Phys. Chem. Solids, 2016, 98, 209–219 CrossRef CAS.
- Q. Sun and S. Bao, Nano-Micro Lett., 2013, 5, 289–295 CrossRef.
- Y. Zhang and Z. Guo, Mater. Chem. Phys., 2016, 171, 208–215 CrossRef CAS.
- M. Kuang, Y. X. Zhang, T. T. Li, K. F. Li, S. M. Zhang, G. Li and W. Zhang, J. Power Sources, 2015, 283, 270–278 CrossRef CAS.
- X. Xu, H. Zhou, S. Ding, J. Li, B. Li and D. Yu, J. Power Sources, 2014, 267, 641–647 CrossRef CAS.
- X. Xu, B. Dong, S. Ding, C. Xiao and D. Yu, J. Mater. Chem. A, 2014, 2, 13069–13074 CAS.
- X. Xu, S. Chen, C. Xiao, K. Xi, C. Guo, S. Guo, S. Ding, D. Yu and R. V. Kumar, ACS Appl. Mater. Interfaces, 2016, 8, 6004–6010 CAS.
- Q. Wang, X. Wang, B. Liu, G. Yu, X. Hou, D. Chen and G. Shen, J. Mater. Chem. A, 2013, 1, 2468–2473 CAS.
- Y. Li, M. Zhang, D. Pan, Y. Wang, J. Xie, Z. Yan and J. Jing, J. Alloys Compd., 2017, 722, 903–912 CrossRef CAS.
- W. Jiang, F. Hu, S. Yao, Z. Sun and X. Wu, Mater. Res. Bull., 2017, 93, 303–309 CrossRef CAS.
- M. Yao, N. Wang, J. Yin and W. Hu, J. Mater. Sci.: Mater. Electron., 2017, 28, 11119–11124 CrossRef CAS.
- K. Xu, X. Yang, J. Yang and J. Hu, J. Alloys Compd., 2017, 700, 247–251 CrossRef CAS.
- S. Han, L. Lin, K. Zhang, L. Luo, X. Peng and N. Hu, Mater. Lett., 2017, 193, 89–92 CrossRef CAS.
- M. Shahraki, S. Elyasi, H. Heydari and N. Dalir, J. Electron. Mater., 2017, 46, 1–7 CrossRef.
- Y. Zhang, Y. Zhang, D. Zhang and L. Sun, Dalton Trans., 2017, 46, 9457–9465 RSC.
- H. Xin, Z. Xu, Y. Liu, W. Li and Z. Hu, J. Alloys Compd., 2017, 711, 670–676 CrossRef CAS.
- T. Han, Int. J. Electrochem. Sci., 2017, 12, 4724–4732 CrossRef CAS.
- V. Venkatachalam, A. Alsalme, A. Alghamdi and R. Jayavel, Ionics, 2017, 23, 977–984 CrossRef CAS.
- W. Zhou, D. Kong, X. Jia, C. Ding, C. Cheng and G. Wen, J. Mater. Chem. A, 2014, 2, 6310–6315 CAS.
- X. W. Wang, X. E. Wang, Y. P. Liu, Y. Y. Kong, L. Y. Sun, Y. C. Hu and Q. Q. Zhu, J. Mater. Sci.: Mater. Electron., 2017, 1–7, DOI:10.1007/s10854-017-7364-x.
- Y. Chen, Y. Xu, J. Wei, L. Tan and J. Yu, J. Mater. Chem. A, 2015, 3, 7121–7131 Search PubMed.
- Y. Ma, P. Liu, Q. Liu, Z. Chen, D. Qu and J. Shi, RSC Adv., 2016, 6, 74643–74648 RSC.
- J. Liang, K. Xi, G. Tan, S. Chen, T. Zhao, P. R. Coxon, H. K. Kim, S. Ding, Y. Yang and R. V. Kumar, Nano Energy, 2016, 27, 457–465 CrossRef CAS.
- W. Du, Y. Gao, Q. Tian, D. Li, Z. Zhang, J. Guo and X. Qian, J. Nanopart. Res., 2015, 17, 1–12 CrossRef CAS.
- M. A. Peck and M. A. Langell, Chem. Mater., 2013, 24, 4483–4490 CrossRef.
- K. Wang, Z. Zhang, X. Shi, H. Wang, Y. Lu and X. Ma, RSC Adv., 2014, 5, 1943–1948 RSC.
- Y. Lei, J. Li, Y. Wang, L. Gu, Y. Chang, H. Yuan and D. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 1773–1780 CAS.
- X. Leng, Y. Shao, L. Wu, S. Wei, Z. Jiang, G. Wang, Q. Jiang and J. Lian, J. Mater. Chem. A, 2016, 4, 10304–10313 CAS.
- B. Babakhani and D. G. Ivey, Electrochim. Acta, 2011, 56, 4753–4762 CrossRef CAS.
- M. Li, J. P. Cheng, J. Wang, F. Liu and X. B. Zhang, Electrochim. Acta, 2016, 206, 108–115 CrossRef CAS.
- Y. Yang, M. Zhou, W. Guo, X. Cui, Y. Li, F. Liu, P. Xiao and Y. Zhang, Electrochim. Acta, 2015, 174, 246–253 CrossRef CAS.
- M. Li, J. P. Cheng, F. Liu and X. B. Zhang, Electrochim. Acta, 2015, 178, 439–446 CrossRef CAS.
- X. Liu, J. Zhao, Y. Cao, W. Li, Y. Sun, J. Lu, Y. Men and J. Hu, RSC Adv., 2015, 5, 47506–47510 RSC.
- X. W. Wang, D. L. Zheng, P. Z. Yang, X. E. Wang, Q. Q. Zhu, P. F. Ma and L. Y. Sun, Chem. Phys. Lett., 2016, 667, 260–266 CrossRef.
- X. W. Wang, Q. Q. Zhu, X. E. Wang, H. C. Zhang, J. J. Zhang and L. F. Wang, J. Alloys Compd., 2016, 675, 195–200 CrossRef CAS.
- X. W. Wang, X. E. Wang, H. C. Zhang, Q. Q. Zhu, D. L. Zheng and L. Y. Sun, Key Eng. Mater., 2017, 727, 698–704 CrossRef.
- G. Zhang and X. W. Lou, Adv. Mater., 2013, 25, 976–979 CrossRef CAS PubMed.
- G. Zhang, W. Li, K. Xie, F. Yu and H. Huang, Adv. Funct. Mater., 2013, 23, 3675–3681 CrossRef CAS.
- S. Peng, L. Li, C. Li, H. Tan, R. Cai, H. Yu, S. Mhaisalkar, M. Srinivasan, S. Ramakrishna and Q. Yan, Chem. Commun., 2013, 49, 10178–10180 RSC.
- M. Fan, B. Ren, L. Yu, D. Song, Q. Liu, J. Liu, J. Wang, X. Jing and L. Liu, Electrochim. Acta, 2015, 166, 168–173 CrossRef CAS.
- C. Yuan, X. Zhang, Q. Wu and G. Bo, Solid State Ionics, 2006, 177, 1237–1242 CrossRef CAS.
- Q. Y. Shan, X. L. Guo, F. Dong and Y. X. Zhang, Mater. Lett., 2017, 202, 103–106 CrossRef CAS.
- D. Du, Z. Hu, Y. Liu, Y. Deng and J. Liu, J. Alloys Compd., 2014, 589, 82–87 CrossRef CAS.
- H. Chen, J. Jiang, Y. Zhao, L. Zhang, D. Guo and D. Xia, J. Mater. Chem. A, 2014, 3, 428–437 Search PubMed.
Footnote |
| † The four authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |