
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic copper-catalyzed azide–alkyne cycloaddition click reaction with Cu(II) coordination polymer†
Xiangyang Guo,
Le Zeng,
Zhe Wang,
Tiexin Zhang ,
Cheng He and
Chunying Duan*
,
Cheng He and
Chunying Duan*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: cyduan@dlut.edu.cn
First published on 15th November 2017
Abstract
A two dimensional Cu(II) coordination sheet has been developed. By incorporating well-designed chromophore linkers into the coordination polymer, the long conjugated system caused the maximum UV-vis absorption wavelength red shift to the visible area (up to 550 nm). Adjacent sheets were assembled in an ABAB fashion via π⋯π stacking and C–H⋯π interactions to form a three-dimensional structure. The interlayer stacking mode is important because its liability allows for exfoliation of the material which results in the dispersion of the active sites and the enhancement of the visible-light trapping capacity. Using this copper(II) coordination polymer, a series of click reactions were successfully conducted under household light irradiation in the air at room temperature. And 1,4-substituted triazoles were the single products in good yields. Moreover, the catalyst can be easily removed from the reaction mixture.
1. Introduction
The copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is a hot research subject in click chemistry.1–3 This reaction has been widely used in organic synthesis, medicinal chemistry, and polymer science.4–7 Recently, this particularly powerful ligation reaction has been applied to biolabeling in vivo, due to its high degree of specificity and the biocompatibility of both starting materials. In previous work, a large variety of copper-related catalysts which provide Cu(I) species have been used for the important reaction. Such as the copper halide compounds, some improved Cu(I)-organic complexes, simple Cu(II) salts together with reducing agents and nano-structured solid catalysts.8–15 However, the oxide species existing in the reaction system will influence the effect of Cu(I) catalysts and homogeneous catalytic systems are inconvenient with regards to the separation and purification of the product. As we all know, although copper is an essential element for humans, large and acute doses can have harmful, even fatal, effects.16 In this case and due to the toxic properties of copper species in vivo, low loading of copper catalyst or mild reaction conditions, such as heterogeneous photocatalysis systems, should be applied.17–19Coordination polymers are crystalline solids generated by the association of metal-containing secondary building units and multi-topic organic linkers through strong bonds.20–22 Their function and application can be predicted through predesigned ligand and the selective active catalytic sites. Because of their unique attributes, low density, open structures with periodic dual compositions, and amenability to bottom-up assembly into a desired framework, these frameworks have emerged as very promising materials for heterogeneous catalysis, gas storage, drug delivery and photocatalysis over the past few decades.23–30 So far, considerable work has been done on their application in catalytic field using functional linkers or active metal nodes of frameworks. And there have been only a few amusing researches about application of photocatalysis in organic synthesis using copper(II) coordination polymers.31–34 Only a tiny amount of studies has been reported of using Cu(I) or the reduced Cu(II) coordination polymers as heterogeneous catalysts for ‘click’ reactions.35–39 It is a challenge that use Cu(II) coordination polymers as photocatalyst for CuAAC reaction.
In this work, we report the synthesis of a two-dimensional (2D) Cu(II) coordination polymer Cu(C33H19NO6S3) (denoted as Cu–TCTA); where H3TCTA is 5,5′,5′′-(nitrilotris(benzene-4,1-diyl))tris(thiophene-2-carboxylic acid); as the photocatalyst facilitates the important reaction (Scheme 1).40 The introduction of the thiophene units into the triphenylamine which results in the increase of the conjugation. Compared with 4,4′,4′′-tricarboxytriphenyl amine (H3TCA, 390 nm), the maximum UV-vis absorption wavelength of H3TCTA (450 nm) shifted toward long wavelength (Fig. S1a, ESI†). And the first oxidation potential is progressively shifted to more negative values with increasing size of the conjugated backbone.41 By incorporate the well-designed chromophore linkers (H3TCTA) into the coordination polymer, Cu–TCTA exhibits a broad solid-state UV-vis absorption up to 550 nm (Fig. S1b, ESI†). It can be used to catalyse the click reaction under household light irradiation in air with co-catalyst. The catalytic activity was then demonstrated by a serious of cycloaddition reactions.
 | ||
| Scheme 1 Synthetic procedure of the Cu–TCTA and the design of CuAAC reaction under irradiation. | ||
2. Results and discussion
2.1 Synthesis and characterization of Cu–TCTA
The two-dimension coordination polymer of Cu–TCTA (CCDC no. 1536834†) was synthesized through solvothermal reaction of Cu(NO3)2·3H2O and H3TCTA at 100 °C for 3 days produced oval-shaped crystals in the mixture of DMF and ethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), producing a yield of 57%. Elemental analyses and powder X-ray diffraction (XRD) analysis indicated the pure phase of its bulk sample. Single-crystal X-ray diffraction studies reveal that Cu–TCTA crystallizes in the monoclinic with space group P21/c. As shown in Fig. 1a, the asymmetric unit consists of one Cu(II) ion connected by one ligand and one coordinated DMF molecule. The copper atom is surrounded by four oxygen atoms from carboxyl groups of four different H3TCTA ligands and one oxygen atom from DMF molecule to attain a five-coordinated square pyramidal geometry. The four carboxyl groups from four different ligands bridging Cu(1) and Cu(1A) (symmetry codes: A, 1 − x, 2 − y, −2 − z) forming the dinuclear paddle-wheel units. The Cu(II)–O distances and the bond angles of the adjacent donors around Cu(II) centre are in the range from 1.943(9)–2.158(9) Å and 87.9(4)–167.2(4)°, respectively. Each dinuclear paddle-wheel cluster is connected to four different deprotonated ligands to form two-dimensional layer structures. One of the three carboxyl groups of the ligands is uncoordinated (Fig. 1b). It is unlike the similar ligands.42,43
:2), producing a yield of 57%. Elemental analyses and powder X-ray diffraction (XRD) analysis indicated the pure phase of its bulk sample. Single-crystal X-ray diffraction studies reveal that Cu–TCTA crystallizes in the monoclinic with space group P21/c. As shown in Fig. 1a, the asymmetric unit consists of one Cu(II) ion connected by one ligand and one coordinated DMF molecule. The copper atom is surrounded by four oxygen atoms from carboxyl groups of four different H3TCTA ligands and one oxygen atom from DMF molecule to attain a five-coordinated square pyramidal geometry. The four carboxyl groups from four different ligands bridging Cu(1) and Cu(1A) (symmetry codes: A, 1 − x, 2 − y, −2 − z) forming the dinuclear paddle-wheel units. The Cu(II)–O distances and the bond angles of the adjacent donors around Cu(II) centre are in the range from 1.943(9)–2.158(9) Å and 87.9(4)–167.2(4)°, respectively. Each dinuclear paddle-wheel cluster is connected to four different deprotonated ligands to form two-dimensional layer structures. One of the three carboxyl groups of the ligands is uncoordinated (Fig. 1b). It is unlike the similar ligands.42,43
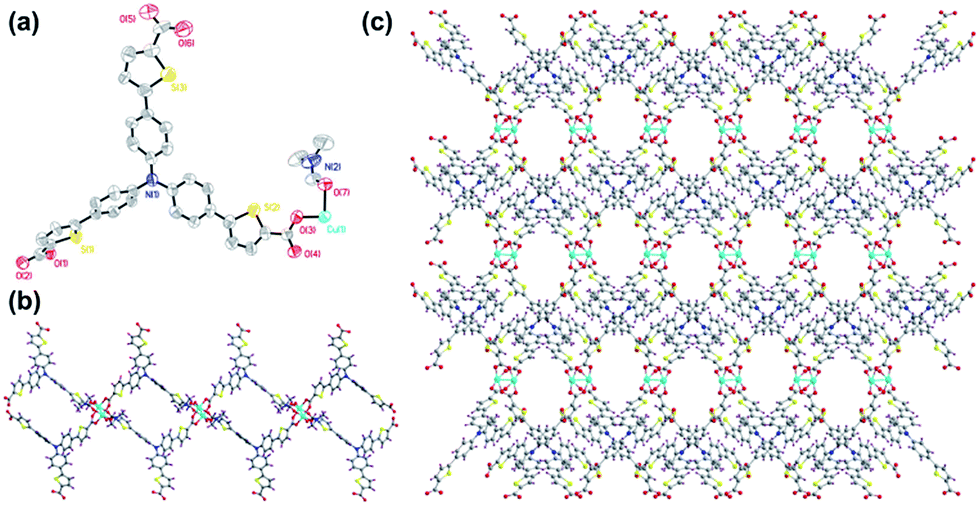 | ||
| Fig. 1 (a) The ORTEP drawing of asymmetric unit of Cu–TCTA, the atoms are represented by 30% probability thermal ellipsoids; (b) perspective view of the 2D structure along the b axis and (c) along the c axis. Colour code: Cu, turquoise; S, yellow; N, blue; O, red; C, grey; H, violet. The lattice and coordinating solvents are omitted for clarity. | ||
Since H3TCTA has enormous flexibility as a supramolecular linker, it can be involved not only in coordination and hydrogen-bonds via its carboxyl groups, but also the uncoordinated arms of the ligands could form aromatic π⋯π stacking interactions.44–46 As shown in Fig. 2a, the distance of the thiophene rings is 4.16 Å between the adjacent sheets. And the thiophene rings also have the C–H⋯π interactions with benzene rings of the another sheet. The distance and angle of C30–H30A⋯π is 3.39 Å and 143.7°, respectively. Adjacent sheets were assembled in an ABAB fashion via π⋯π stacking and C–H⋯π interactions to form a three-dimensional structure (Fig. 2b). When viewed along the c axis, each layer exhibits a large rhombic cages with a height of 17.27 Å and width of 34.48 Å (Fig. 1b). The packed structure exhibits 9.7 × 10.3 Å2 one-dimension channels along the c axis after removed the coordinating DMF molecules (Fig. 1c). The interlayer stacking mode is important because of its liability allows for exfoliation of the material which results in the dispersion of the active sites and the enhancement of visible-light trapping capacity.47,48 The effective free volume of Cu–TCTA was estimated to be 31.4% after removed the lattice solvent and the coordinating DMF by PLATON software.49
 | ||
| Fig. 2 (a) The interpenetration structure between 2D layers. (b) The 3D stacking structure of Cu–TCTA view along the b axis. | ||
The introduction of the thiophene units results in the increase of the conjugation. As shown in Fig. 3a, the distances between the nitrogen-centred atom of TCTA fragment and the copper atoms of adjacent layers range from 7.72 to 11.05 Å, which is shorter than the coordinated copper atoms ranging from 11.35 to 12.33 Å. These shorter distances benefit Cu–TCTA forming the intermolecular charge transfer salts. Hence the copper nodes have certain properties of copper(I), leading to the framework presenting a weak fluorescence (Fig. 2b).50,51
 | ||
| Fig. 3 (a) The distance of nitrogen centre of TCTA with copper cations in interpenetration structure. (b) Family of emission spectra of Cu–TCTA (0.05% in weight) in CHCl3 suspension upon addition of 2a, 1a and TEA, excitation at 390 nm. | ||
Besides, the long π-conjugated system reduces the rate of back electron transfer and makes charge separation easy to control. And the first oxidation potential is progressively shifted to more negative values with increasing size of the conjugated backbone. Solid state cyclic voltammetry of Cu–TCTA measurements exhibited (Fig. 4a) reversible redox band centred at 187 mV (vs. SCE) relative to the CuII/CuI redox couple and also an irreversible oxidative peak at 1.07 V (vs. SCE) relative to TCTA+/TCTA. The transition energies (E0−0) of H3TCTA is 2.70 eV, which is lower than H3TCA (E0−0 = 3.25 eV) (Fig. 4b). The redox potential of the excited-state H3TCTA+/H3TCTA* couple was determined to be −1.63 V on the basis of transition energies.52,53
 | ||
| Fig. 4 (a) Solid state cyclic voltammetry of Cu–TCA (blue line) and Cu–TCTA (red line), scan rate: 100 mV s−1. (b) Normalized absorption (red line) and emission spectrum (black line) of H3TCA (dash line) and H3TCTA (solid line) in DMF, excitation at 350 nm and 390 nm, respectively. | ||
2.2 Photocatalytic properties of Cu–TCTA
To probe the catalytic activity of Cu–TCTA for the CuAAC, a series of experiments were performed to optimize the reaction conditions. Trimethylamine (TEA) was used as the electron donor (ED), instead of preventing copper(I) oxidation for most the azide–alkyne cycloaddition reaction. And the reaction of (azidomethyl) benzene (1a) and phenylacetylene (2a) was initially investigated with 28 W household light as light source at room temperature. Firstly, the reaction was tested with different solvents with 0.5 mol% photocatalytic loading (Table S2, ESI†). The reactions were monitored by means of 1H NMR spectroscopy. And conversions were determined by integration of a particular signal from a proton in one of the starting molecules (4.3 ppm) and the corresponding proton in the product (5.5 ppm).54 After standing the reaction mixture for several days, the colourless rod like crystal was obtained. The single crystal diffraction demonstrated that the product was 1,4-substituted triazole (3a) (Fig. S8, ESI†). There were relatively low yields in the solution of acetonitrile or ethanol under irradiation. Moderate catalytic activity was obtained for THF. When CDCl3 was used as the solvent, the yield of 3a was near 94% under irradiation and only 8.4% under dark after six hours. CDCl3 was selected as the optimal solvent. Control experiments in the absence of TEA or in the presence of H3TCTA were also performed, but just minimal amount of 3a was detected under irradiation and no click reaction was detected under dark. In conclusion, the suitable reaction conditions: Cu–TCTA (0.5 mol%), azide/alkyne/amine (0.5 mmol:0.5 mmol:0.5 mmol), room temperature, 6 h, was chosen to study the catalytic activity.
In order to research the mechanism of photocatalytic CuAAC click reaction. The luminescence properties of Cu–TCTA was investigated when the substrates and co-catalyst were gradually added to the suspension of Cu–TCTA in CHCl3. Cu–TCTA exhibited a weak luminescence band at 500 nm upon excitation at the wavelength of 390 nm. The luminescence was quenched upon addition of 2a. Then the intensity of the emission at 500 nm was no significant change along the addition of 1a. But the intensity was dramatically increase with TEA added into the mixture (Fig. 3b). By analogy with previous reports, a stepwise mechanism based on the observations for the photocatalytic reaction with Cu(II)-coordination polymers is proposed. It begins the Cu(II)-coordination polymers was reduced into Cu(I)-coordination polymers under irradiation, then the process as same as most azide–alkyne cycloaddition CuAAC reaction mechanism which was proposed by Fokin and co-workers from their DFT calculation.2
A detailed kinetic study of the reaction between 1a and 2a was conducted (Table S3, ESI†) to study the catalytic activity of Cu–TCTA. There was a very low conversion in the first hour. Two hours later, most of the substrates were converted into product, and almost quantitatively converted (yield, 94.3%) after six hours. When the catalytic reaction was stopped after 3 h, the supernatant was obtained by centrifugation and filtration. Just a 6.7% transformation was observed when subjected to standard conditions for another 3 h (Fig. S5, ESI†). These results clearly demonstrate that the azide–alkyne cycloaddition reaction majorly occurred in the heterogeneity fashion.15,55,56
After the reaction, Cu–TCTA was easily isolated from the reaction mixture by centrifugation. On the basis of the scanning electron microscopy and transmission electron microscopy images, the nanoscale thin layers were in situ exfoliated from the single-crystal blocks of 2D coordination polymers (Fig. 5a–c). The exfoliation was postulated to be due to the interlayer π⋯π stacking by the comprehensive effects of mechanical shearing forces during stirring, weak interactions relevant to the solvents and other compounds, and interlayer charge repulsions during the photoredox cycles. In addition, the indexing of the X-ray powder diffraction patterns of the recovered catalyst indicated that the integrity of the 2D coordination polymers was maintained during the reaction (Fig. 5d).57
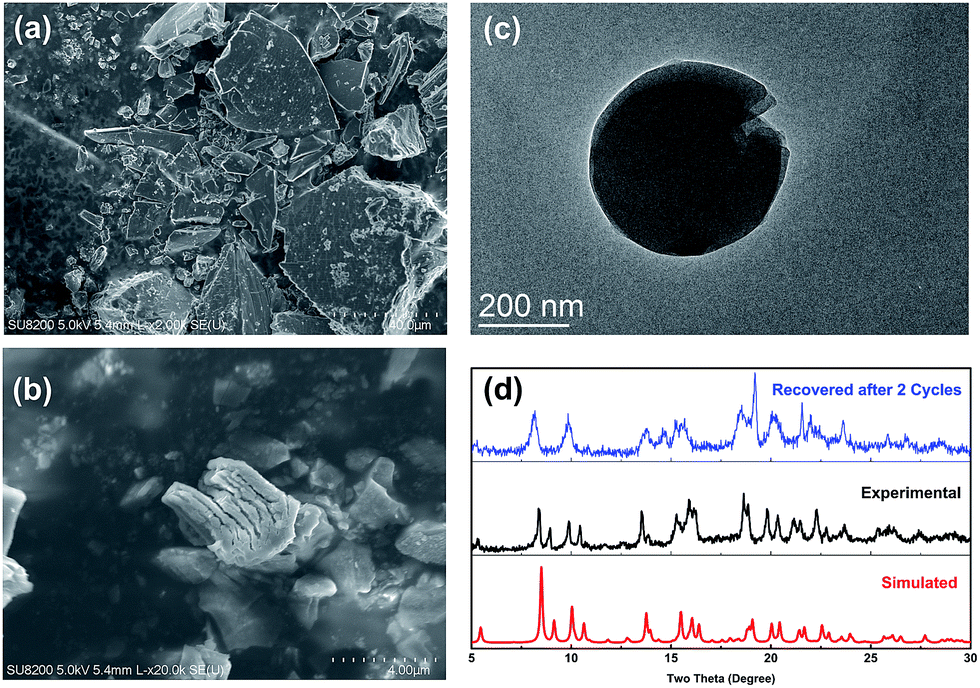 | ||
| Fig. 5 (a) SEM image of Cu–TCTA; (b) SEM image of Cu–TCTA after catalysed; (c) TEM image of exfoliated thin layers and (d) simulated (red) and experimental (black) XRD patterns of the Cu–TCTA crystal and the experimental pattern of the catalyst recovered after 2 cycles of catalysis (blue). | ||
Control experiment with Cu–TCA and MOF-150 was conducted under the same conditions. There was only 2% conversion for MOF-150 and trace amount of product for Cu–TCA (Fig. S7, ESI†). Dye uptake experiment (Fig. S2, ESI†) and IR spectrum (Fig. S3, ESI†) of Cu–TCTA soaked with azide and alkyne revealed that the photocatalytic azide–alkyne cycloaddition reaction was the surface reaction on the catalyst. In order to demonstrated the surface catalytic reaction, the as-synthesized Cu–TCTA was used to conduct click reaction. The yield was only 7% at the same conditions (Fig. S7, ESI†). With the size of the microcrystals reduced to a few micrometres or nanometres by grinding Cu–TCTA crystals, the reaction of 1a with 2a has a dramatic increase conversion (>90%) than the as-synthesized catalyst, which size are hundred micrometres (Fig. S6, ESI†).
To evaluate the scope of this new Cu–TCTA catalysed process further, reaction of benzyl azide and 4-methylbenzyl azide with several aliphatic and aromatic terminal alkynes substituted by electron-donating/electron-withdrawing groups were conducted (Table 1). In all the cases, a single product 1,4-disubstituted 1,2,3-triazoles were obtained. And the substrates bearing electron-donating group such as methoxyl group (entries 3 and 8) or electron-withdrawing group such as cyan group (entries 5 and 10) at the para position of the phenyl ring are all suitable substrates.
| Entry | Azide | Alkyne | Yield (%) |
|---|---|---|---|
| a Reaction conditions: Cu–TCTA (0.5 mol%), azide/alkyne/amine (0.5 mmol:0.5 mmol:0.5 mmol), room temperature, household light irradiation, 6 h. Yields were calculated by 1H NMR analysis in CDCl3. |
|||
| 1 |  |
 |
93.5 |
| 2 |  |
 |
65.8 |
| 3 |  |
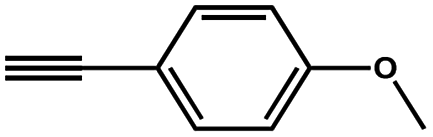 |
67.1 |
| 4 |  |
 |
90.9 |
| 5 |  |
 |
78.1 |
| 6 |  |
 |
77.0 |
| 7 |  |
 |
81.3 |
| 8 |  |
 |
66.7 |
| 9 |  |
 |
42.7 |
| 10 |  |
 |
54.3 |
Recyclability is an essential feature of any catalyst considered for use.58 The reusability of the catalyst was tested for the reaction of 1a and 2a in CDCl3 in air upon household light irradiation for 6 h. Though solid Cu–TCTA could be easily isolated from the reaction suspension by filtration, it could only be subsequently reused two times because of the unavoidable loss of small quantities of catalyst, with only slight decrease in its reactivity (yield, 90.2%). We also conducted the extensive experiment that using large excesses of 1a (10 mmol), 2a (10 mmol) and Cu–TCTA (2.5 μmol) while added 140 μL TEA every 12 hours. A small aliquot of the supernatant reaction mixture was analyzed by 1H NMR to calculate the reaction yields. After 48 h reaction, the yield was 58.5%. It should be noted that such a considerable amount indicates that Cu–TCTA can be used into the laboratory practice.
Compared to previous works, our system does not require UV irradiation nor heating while still acquiring the same high product yields with similarly low catalyst loading. As it is a heterogeneously catalysed reaction it is easy to separate the catalyst from the mixture. However, there are still some problems such as poor recyclability and the currently unavoidable loss of catalyst.35–38
3. Experimental section
3.1 Reagents and instrumentation
All chemicals were of reagent-grade quality, obtained from commercial sources and used without further purification. The crystallographic data were collected on a Bruker SMART APEX CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using the SMART and SAINT programs. Elemental analyses of C, H and N were performed using a Vario EL III elemental analyzer. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded using KBr pellets on a JASCO FT/IR-430. Powder X-ray diffraction (PXRD) measurements were obtained on a Rigaku SmartLab XRD instrument with a sealed Cu tube (λ = 1.54178 Å). Thermogravimetric analyses were performed on a Mettler-Toledo TGA/SDTA851 instrument and recorded under N2 or air after 14 equilibrations at 100 °C followed by a ramp of 10 °C min−1 up to 800 °C. 1H NMR was measured on a Varian INOVA-400 spectrometer with chemical shifts reported as ppm (in DMSO-d6 or CDCl3, TMS as internal standard). The solid UV-vis spectra were recorded on a Hitachi U-4100 UV-VIS-NIR Spectrophotometer. Liquid UV-vis spectra were performed on a TU-1900 spectrophotometer. The fluorescent spectra were measured on Edinburgh FS920 Spectrometers. Transmission electron microscopy (TEM) images were collected on a Tecnai F30 operated at 300 kV. The excitation and emission slits were both 3 nm wide. Scanning electron microscopy (SEM) images were taken with a HITACHI UHR FE-SEM SU8200 microscope.3.2 General methods
Solid-state voltammograms were measured by using a carbon-paste working electrode, and a well-ground mixture of each bulk sample and carbon paste (graphite and mineral oil) was set in the channel of a glass tube and connected to a copper wire. A platinum wire with a 0.5 mm diameter counter electrode and saturated calomel reference electrode were used. Measurements were performed by using a three electrode system in 0.1 M KNO3 aqueous solution at a scan rate of 100 mV s−1.Dye-uptaking experiments were displayed by soaking Cu–TCTA (7.8 mg) in a CH3CN solution of methylene blue dye (24 mM, 2 mL) overnight. The resulting crystalline powders were washed thoroughly with methanol until the solution became colorless. The washed samples were digested by concentrated hydrochloric acid in CH3CN/methanol (v/v = 1:1), and the resultant clear solution with light olivine color was diluted to 10 mL. The concentration of methylene blue was determined by comparing the UV-vis absorption with a standard curve.
3.3 Synthetic procedures
3.4 Catalysis details
:0.5 mmol:0.5 mmol)) was added. The reaction mixture was stirred and irradiated with household light at room temperature and under air for 6 h. The solid catalyst was separated by filtration. The reactions were monitored by means of 1H NMR spectroscopy. And conversions were determined by integration of a particular signal from a proton in one of the starting molecules (4.3 ppm) and the corresponding proton in the product (5.5 ppm).4. Conclusions
In a summary, a new 2D copper(II) coordination polymer has been prepared using elaborately designed ligand of thiophenyl triphenylamine carboxylic acid derivatives. The long conjugated system caused the maximum UV-vis absorption wavelength red shift to the visible area and reduced the recombination velocity of electrons and holes. Using this copper(II) coordination polymer, a series of click reactions were successfully conducted under the household light irradiation in air with the presence of trimethylamine. The 1,4-substituted triazoles were the single products and have good yields. And it features high catalytic activity, rapid dynamics of the substrate to product transformation. Moreover, the catalyst can be easily removed from the reaction mixture.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21531001, 21402020 and U1608224).Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- O. Altintas and U. Tunca, Chem.–Asian J., 2011, 6, 2584–2591 CrossRef CAS PubMed.
- O. Altintas, U. Tunca and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1146–1155 RSC.
- O. Altintas, A. P. Vogt, C. Barner-Kowollik and U. Tunca, Polym. Chem., 2012, 3, 34–45 RSC.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- L. Y. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- C. L. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–30 CrossRef CAS.
- L. Ackermann, H. K. Potukuchi, D. Landsberg and R. Vicente, Org. Lett., 2008, 10, 3081–3084 CrossRef CAS PubMed.
- A. L. Schöffler, A. Makarem, F. Rominger and B. F. Straub, Beilstein J. Org. Chem., 2016, 12, 1566–1572 CrossRef PubMed.
- S. J. Gu, J. H. Du, J. J. Huang, H. Xia, L. Yang and W. L. Xu, Beilstein J. Org. Chem., 2016, 12, 863–873 CrossRef CAS PubMed.
- K. Namitharan, M. Kumarraja and K. Pitchumani, Chem.–Eur. J., 2009, 15, 2755–2758 CrossRef CAS PubMed.
- K. Yamaguchi, T. Oishi, T. Katayama and N. Mizuno, Chem.–Eur. J., 2009, 15, 10464–10472 CrossRef CAS PubMed.
- B. W. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- E. Bigagli, C. Luceri, S. Bernardini, A. Dei and P. Dolara, Chem.-Biol. Interact., 2010, 188, 214–219 CrossRef CAS PubMed.
- C. Testa, M. Scrima, M. Grimaldi, A. M. D'Ursi, M. L. Dirain, N. L. Germain, A. Singh, C. H. Luevano, M. Chorev, P. Rovero and A. M. Papini, J. Med. Chem., 2014, 57, 9424–9434 CrossRef CAS PubMed.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. R. Batten, N. R. Champness, X. M. Chen, J. Garcia-artinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- H. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- C. Wang, Z. G. Xie, K. E. de Kafft and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639–12641 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- C. Wang, D. Liu and W. B. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- B. J. Adzima, Y. H. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- X. L. Wang, J. Luan, F. F. Sui, H. Y. Lin, G. C. Liu and C. Xu, Cryst. Growth Des., 2013, 13, 3561–3576 CAS.
- J. F. Wang, C. Li, Q. X. Zhou, W. B. Wang, Y. J. Hou, B. W. Zhang and X. S. Wang, Dalton Trans., 2016, 45, 5439–5443 RSC.
- Q. Fu, K. Xie, S. Tan, J. M. Ren, Q. Zhao, P. A. Webley and G. G. Qiao, Chem. Commun., 2016, 52, 12226–12229 RSC.
- Z. H. Xu, L. L. Han, G. L. Zhuang, J. Bai and D. Sun, Inorg. Chem., 2015, 54, 4737–4743 CrossRef CAS PubMed.
- I. Luz, F. X. Llabrés i Xamena and A. Corma, J. Catal., 2010, 276, 134–140 CrossRef CAS.
- P. Li, S. Regati, H. C. Huang, H. D. Arman, J. C.-G. Zhao and B. L. Chen, Inorg. Chem. Front., 2015, 2, 42–46 RSC.
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2016, 52, 19–78 CrossRef CAS.
- Data for Cu–TCTA: C39H32CuN3O8S3, Mr = 830.39, monoclinic, space group P2(1)/c, a = 16.213(13) Å, b = 19.359(14) Å, c = 12.323(9) Å, β = 90.991(12)°, V = 3867(5) Å3, Z = 4, of 6803 reflections, 1553 unique (Rint = 0.1454). Final R1 [with I > 2σ(I)] = 0.0802, wR2 (all data) = 0.2434 and S = 0.985. CCDC no. 1536834.
- K. Idzik, J. Sołoducho, M. Łapkowski and S. Golba, Electrochim. Acta, 2008, 53, 5665–5669 CrossRef CAS.
- P. Y. Wu, J. Wang, Y. M. Li, C. He, Z. Xie and C. Y. Duan, Adv. Funct. Mater., 2011, 21, 2788–2794 CrossRef CAS.
- P. Y. Wu, J. Wang, C. He, X. L. Zhang, Y. T. Wang, T. Liu and C. Y. Duan, Adv. Funct. Mater., 2012, 22, 1698–1703 CrossRef CAS.
- C. Livage, N. Guillou, A. Castiglione, J. Marrot, M. Frigoli and F. Millange, Microporous Mesoporous Mater., 2012, 157, 37–41 CrossRef CAS.
- H. K. Chae, J. Kim, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2003, 42, 3907–3909 CrossRef CAS PubMed.
- E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6374–6381 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- A. C. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592–5629 CrossRef CAS PubMed.
- A. L. Spek, PLATON: A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed.
- B. D. McCarthy, E. R. Hontz, S. R. Yost, T. V. Voorhis and M. Dinca, J. Phys. Chem. Lett., 2013, 4, 453–458 CrossRef CAS PubMed.
- T. P. Liu, L. H. Huo, Z. P. Deng, H. Zhao and S. Gao, RSC Adv., 2014, 4, 40693–40710 RSC.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, VCH, New York, 1993 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945–6947 CrossRef CAS.
- J. S. Wang, F. Z. Jin, H. C. Ma, X. B. Li, M. Y. Liu, J. L. Kan, G. J. Chen and Y.-B. Dong, Inorg. Chem., 2016, 55, 6685–6691 CrossRef CAS PubMed.
- A. Chakraborty, S. Bhattacharyya, A. Hazra, A. C. Ghosh and T. K. Maji, Chem. Commun., 2016, 52, 2831–2834 RSC.
- M. Zhao, Q. Lu, Q. Ma and H. Zhang, Small Methods, 2017, 1, 1600030 CrossRef.
- Z. Zhou, L. Yang, Y. F. Wang, C. He, T. Liu and C. Y. Duan, RSC Adv., 2016, 6, 108010–108016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data, as well as additional tables and figures. CCDC 1536834. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10207k |
| This journal is © The Royal Society of Chemistry 2017 |