
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonium based stabilizers effectively counteract urea-induced denaturation in a small protein: insights from molecular dynamics simulations†
Soham Sarkar,
Soumadwip Ghosh and
Rajarshi Chakrabarti *
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai – 400076, India. E-mail: rajarshi@chem.iitb.ac.in; Fax: +91-22-2576-7152; Tel: +91-22-2576-7192
First published on 15th November 2017
Abstract
Room temperature ionic liquids (IL) and deep eutectic solvents (DES) are known to aid the conformational stability and activity of proteins and enzymes in aqueous solutions. They are popular alternatives to harsh organic protecting osmolytes offsetting the thermal/chemical denaturation of proteins due to their greener attributes and low costs. In this paper, using extensive molecular dynamics (MD) simulations we show that a small helix–rich protein, chicken villin headpiece subdomain (HP-36), is rendered stable by an IL, triethylammonium acetate (TEAA) in aqueous solutions containing very high concentrations of urea (8 M). For comparison, the protein is also simulated in a deep eutectic mixture composed of 4 M choline chloride (ChCl) and 8 M urea under identical simulation conditions. Our simulations clearly show that a sharp increase in the relative proportion of TEAA (from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:2 with respect to the denaturant urea) does not have a pronounced structure-enhancing effect on the protein. In contrast, for ChCl to function as an efficient protein stabilizer against the harsh action of urea, deep eutectic conditions are mandatory. To the best of our knowledge, this is the first study investigating the comparative aspects of the microscopic properties of a protein in an IL and a DES having similar ammonium-based structural frameworks.
:5 to 1:2 with respect to the denaturant urea) does not have a pronounced structure-enhancing effect on the protein. In contrast, for ChCl to function as an efficient protein stabilizer against the harsh action of urea, deep eutectic conditions are mandatory. To the best of our knowledge, this is the first study investigating the comparative aspects of the microscopic properties of a protein in an IL and a DES having similar ammonium-based structural frameworks.
1. Introduction
The native structures of most cellular proteins are sensitive towards changes in environmental conditions and significant perturbations in thermodynamic conditions may induce structural changes, often leading to a partial or complete loss of their activities. Protecting osmolytes are small organic molecules which favor the folded ensemble of the proteins and they belong to various chemical classes including amino acids,1,2 betaine3 and sugars.4 A particularly important example of such stabilizers is trimethylamine-N-oxide (TMAO)5 which is found in large concentrations in shark tissues (containing large amounts of urea) and is instrumental in folding denatured proteins into their native-like states. The human kidney also contains several protecting osmolytes such as glycine-betaine, inositol, sorbitol and taurine which function by counteracting the effect of urea on vital biological activities.6 In general, protecting osmolytes remain excluded from the vicinity of protein backbones and side chains and thus force proteins to adopt a folded conformation with a minimum possible exposed surface area.7,8 A universal model for osmolyte-induced protein stabilization and denaturation was proposed by Street and colleagues.9 Along similar lines of research, Record et al. has shown that the transfer free energy (Δgtr) for the protecting osmolyte, glycine-betaine is proportional to the polar surface area of the protein in aqueous medium.10 In a recent MD study, Ganguly et al., have demonstrated the crucial role of TMAO–water interactions causing self – aggregation of urea and preferentially excluding both the denaturant and the protecting osmolytes from the vicinity of a short peptide backbone in an aqueous mixture containing urea and TMAO (in 2:1 molar ratio).11 The same group has also examined the TMAO–water and TMAO–urea interactions in the hydrophobic association between two neopentane molecules which may be helpful in understanding the functions of cellular proteins in connection with their folding dynamics.12 The influence of co-solvent interactions between TMAO molecules in determining their preferential interaction with protein has also been studied in details by Canchi and Garcia.13 The osmotic model was preferred over the well – known Kast model for TMAO since the former one seemed to capture the preferential exclusion of the stabilizer from a β-hairpin peptide in the co-presence of urea better than the other representation by Borgohain and Paul.14 Using neutron scattering experiments TMAO was earlier shown to interact directly with urea and offset its action on protein by Meersman et al.15 On the contrary, both experimental16 as well as computational17 studies have suggested that the enhancement of water-structuring by TMAO is responsible for the preservation of the protein in its native state, not only from chemical denaturation but also from pressure induced structural collapse.18 A very recent study by Dias and co-workers shows a competing mechanism of TMAO on Trp cage mini proteins, which stabilizes charge–charge interactions, induces collapse of the backbone and causes swelling of peptide with greater degree of hydrophobic residue.19 Among other osmolytes, trehalose mediated counteraction of the effect of urea on a small peptide has been investigated thoroughly by Paul and Paul using extensive MD simulations.20 Trehalose has also been shown to preserve the structure and function of the biologically relevant enzyme, α-chymotrypsin against strong chemical denaturants such as urea21 and guanidium chloride.22 Kumar and Kishore have proposed a synergism between urea and glycine-betaine in an aqueous mixture which can be extrapolated to gain crucial insights into the stabilization of a protein in such a mixture.23 Contrary to popular beliefs, urea has been shown to preferentially bind to the protein side chains, not to the backbone24 in a mixed osmolyte system containing urea and glycine-betaine by Guinn et al.25 Priyakumar and co-workers have also recently looked into the urea-aromatic ring stacking interaction in Trp-cage miniprotein using both micro-second scale molecular dynamics simulation as well as some quantum chemical calculations.26
In the field of biotechnology and biocatalysis, the choice of a proper solvent for the substrate enzyme is imperative which ensures the stability and the activity of the biomacromolecules undergoing enzyme–catalysis reactions. This is also valid for in vitro chemical experiments involving proteins. Safer and non-hazardous solvents are to be employed in chemical reactions as a key principle in green chemistry.27,28 There has always been an urge to replace harsh organic reagents by eco-friendly and biodegradable ones. Keeping this in mind, a variety of non-hazardous solvents like supercritical fluids, ionic liquids been introduced as alternatives for toxic organic solvents in chemical and biological reactions for a few decades.29–31 Among these, ILs have shown an incredible potency as a greener solvent due to its high stability in liquid state close to room temperature, non-flammability and high conductivity.32,33 Its remarkable physical properties along with its biocompatible nature immediately caught the attention of various research groups that ultimately lead to its wide-spread applications in diverse research fields, including the preservation of enzyme/protein activity34,35 and the crystallization of proteins.36 Similarly, Biswas and colleagues have shown the facile chemical transformation of starch and zein proteins solvated in room temperature ionic liquids.37 Summers and flowers have shown that hen egg white lysozyme (HEWL) could be stabilized effectively by ethylammonium nitrate against thermal denaturation.38 HEWL has also been shown to be preserved in its folded state by ethylphosphonium-based ILs under ambient conditions using dynamic light scattering, turbidimetry and spectroscopic techniques.39 In spite of its miscibility in water, immidazolium-based ILs have been found to denature the protein according to the same study.38 Mann et al. have also shown that the stability of HEWL in six different ethylalkyl formate salts against thermal unfolding using near UV circular dichroism (CD) studies.40 Among other important biomolecules, the critical importance of alkylammonium formates and nitrates in the stabilization of cytochrome c has also been explored rigorously using both spectroscopic techniques41 as well as MD simulations.42 Alkylimidazolium acetates have also been used as a pre-treatment solvent for extracting lignin from wood by Lee et al.43 Alkylammonium based ionic liquids, on the other hand, have been shown to exhibit a strong positive effect on the conservation of haemoglobin structure by Attri et al.44 In an important work, Attri et al. have shown that triethylammonium acetate (TEAA), a protic ionic liquid can counteract the deleterious effects of urea on the biologically relevant enzyme α-chymotrypsin using circular dichroism (CD) and NMR techniques.45 They have shown that TEAA can effectively offset the harsh action of urea even when its relative concentration is as low as 1:5 with respect to the denaturant urea. Ammonium-based ILs were found to be better refolding enhancers over the immidazolium-based ones for single chain antibody fragments46 and enzymes like Horseradish peroxidise (HRP)47 and lipase.48
In spite of several promising applications, the preparation of some of the ILs is not straightforward due to the difficulties in cation–anion exchange during chemical transformations. In addition, immidazolium-based ILs are known to have negative impacts on the native structure of some proteins and hence can't be used for protein specific assays.49,50 A new generation of solvents, named deep eutectic solvents (DES) can be an attractive alternative to majority of the ILs because of the ease of preparations and low costs. DESs are composed of an ammonium salt and a halide donor or a hydrogen bond donor compound like urea, organic acids or amines.51 The formation of a DES usually involves the mixing of two solids in appropriate molar ratio, followed often by heating slightly, to form a mixture having a much lesser melting point than either of the precursors.52 One of the most well-known DES is formed by mixing solid choline chloride (ChCl) and urea in 1:2 molar ratio (trade name: reline), giving rise to a liquid having a melting point of ∼12 °C.53 Hammond et al. has recently shown that Reline has a complex radially layered structure formed due to multiple hydrogen bonding networks between the OH functionality of choline, urea and chloride ion using both neutron scattering experiments as well as atomistic modelling.54 Reline has been found to be highly conducting which implies that the ionic species are almost fully dissociated in the liquid and they possess very high ionic mobility.55 The structure and factors stabilizing a urea:ChCl mixture have been studied at atomistic level using MD simulations, FTIR56 and time-resolved fluorescence57 measurements. Similar DESs formed between urea/thiourea and various other organic counterparts such as cresol, oxalic acid, glycerol and fructose have been studied experimentally as well.58
Like room temperature ionic liquids, urea and glycerol-based DESs have been shown to assist the thermal renaturation of lysozyme using intrinsic fluorescence and CD spectroscopy by Esquembre et al.59 Ammonium-based DESs were used for extracting the protein bovine serum albumin (BSA) where the microstructure of the protein remained unaffected by the ions or the hydrogen bond donors constituting the DESs.60 They have also been identified as viable co-solvents for enzyme catalyzed biotransformations61–63 in spite of the presence of urea, which might denature the enzyme. In an important computational work, Monhemi and co-workers have demonstrated the preservation of the native structure of the enzyme, Candida Antarctica lipase B in a deep eutectic mixture containing urea and ChCl in 2:1 molar ratio.64 They have argued that the denaturant urea, due to the formation of hydrogen bonds with choline and chloride ions, experiences low diffusivity and hence is unable to reach the protein domains.64
The merits of ILs and DESs having similar structural frameworks as extraction solvents in natural product synthesis are often compared due to their chemical resemblance.65 However, a comparative analysis of the extent by which two liquids help a protein attenuate the deleterious effects of a chemical denaturant urea has not been studied in details so far to the best of our knowledge. In this paper, using atomistic MD simulations we study the dynamics of a helix-rich protein, chicken villin headpiece sub domain (HP-36) in an aqueous mixture containing urea/TEAA or urea/ChCl at various concentration of the stabilizers. We have emphasized on the concentration-dependent nature of the said counteraction of the denaturing ability of urea for both the liquids. For this, we have performed two simulations for each of the two species, TEAA and ChCl (at 1.6 M and 4 M concentrations respectively) in aqueous solutions containing 8 M urea. Visual representations of the protein and the chemical structure of TEAA and ChCl have been depicted in Fig. 1. Specific information on the derived force field parameters for the TEAA and ChCl (Tables S1–S5†) and a brief overview of each of the simulated systems under various conditions (Table S6†) have been presented in the ESI† Section. We have calculated preferential binding interaction parameters and transfer free energies of various co-solvents with respect to the solvent water in order to elucidate their binding affinity for the protein surface at each concentration of the stabilizers. Our study sheds light on the mechanism by which two ammonium-based species protect a model protein from the stressful effects of urea. The counteracting ability of the two has been found to be concentration dependent. This might act as a guiding tool for both qualitative as well as quantitative selection of an environmentally benign stabilizer for in vitro experiments involving a protein under ambient conditions.
 | ||
| Fig. 1 (a) The initial helical structure of the chicken villin headpiece (HP-36) protein, the chemical structures of (b) triethylammonium acetate and (c) choline chloride. | ||
2. Simulation details
To account for the counteracting ability of ILs and DESs on urea induced denaturation of a small protein, we perform extensive classical MD simulation using GROMACS molecular dynamics package (version 4.5.6)66 along with OPLS-AA force field.67 Five mixed osmolyte systems along with a control simulation of the protein in pure SPC/E water model68 (system designation PW) are considered in this study. The topological parameters for ammonium-based stabilizers are obtained using OPLS parameters following our previous work.69 8 M urea concentration is kept constant for observing the denaturation of the small protein in the binary mixture (system designation PUW), as well as in the other ternary systems, with varying concentration of the protecting osmolytes for triggering its counteraction. For addressing TEAA and ChCl mediated stabilization of the protein, first we take 1.6 M of TEAA and ChCl each and 8 M urea (1:5 molar ratio) in two different systems which are designated as PUILW (5:1) and PUChClW (5:1) respectively. It is to be noted that ChCl under this concentration ratio doesn't form deep eutectic mixture with urea. One of the remaining two systems is constructed in such a way that 1:2 molar ratio of the ChCl with urea (4 M ChCl and 8 M urea) is maintained for attaining the deep eutectic condition. This system is designated as PUChClW (2:1). In order to be consistent with the concentration ratio of DES, we also increase the concentration of TEAA to 4 M in presence of 8 M urea, the system being designated as PUILW (2:1).
The initial configuration of HP-36 is collected from Protein Data Bank (PDB entry 1VII).70 In order to get rid of technical problems during simulation, C-terminus (Met-1) of the protein is amidated and N-terminus (Phe-36) is acetylated71 using the mole fracture utility in VMD (Version 1.9.2).72 Simulations are performed at 330 K, in order to accelerate both the unfolding as well as the counteraction process. At this temperature the protein doesn't undergo thermal denaturation since the melting point of HP-36 is 343 K.73 However, addition of 8 M urea at an elevated temperature of 330 K, greatly enhances the unfolding rate of this small protein. The choice of maintaining higher temperature for expediting the structural unfolding process resembles other studies64,74 dealing with the complete conformational denaturation of native proteins. This allows us to account for several properties within the time scale of 500 ns. We prefer SPC/E water model over SPC and TIP3P due to the better reproduction of bulk dynamics and structure by the former model.75 The Duffy model,76,77 consistent with the OPLS-AA force field68 has been adopted for urea. The OPLS force field parameters for urea employed by various eminent groups for investigating a broad spectrum of research interest have been listed below. We, however, would like to mention that we are completely aware of the limitations of the methods adopted in this study as computational modelling of urea molecule is quite sensitive and challenging at best. Lack of reliable experimental data on similar systems could not confirm the accuracy of the force field under consideration. Thus, the molecular level insights obtained from this study depend significantly on the used force field and it might vary substantially when treated with different force field parameters without altering its broad outcomes. Some of the works involving the OPLS model for representing urea includes the comparative study78 between Kuharsky and Rossky (KR)79 model and the Tsai, Gerstein and Levitt (TGL)80 model. The OPLS model for urea reproduces the experimental density for concentrated urea/water system better compared to the other ones. In analyzing denaturation of Aβ16–22 oligomers, Klimov et al. using OPLS urea model regarding the solvation of the monomers and the enhancement of their β contents.81 Similar kind of force field response was noted by Xiu et al. who showed the permeability of highly concentrated urea through a small single walled carbon nanotube (SWCNT).82 The hydrophobic association of methane molecules in presence of urea and glycine betaine (GB) studied by Dixit et al. had OPLS derived parameters for urea in it.83 The synergistic behaviour shown by glycine betaine–urea mixture on a small model peptide by Kumar and Kishore84 indicates exclusion of GB and decrease in interaction of the peptide surface with the urea molecules that leads to the structural stability of the peptide. The OPLS model of urea has been adopted in this study.84 The above force field representation for urea shows fair agreement with experimentally obtained thermodynamic parameters like density, free enthalpy of urea hydration, mixing enthalpy, and urea diffusion.85 A combination of TIP4P water model along with the OPLS force field parameters including planar and non-planar model for urea was preferred by Bertran et al.86 in an extensive computational study based on Monte Carlo simulations. The focus of the authors was to account for the structuring of water and the aggregation between urea molecules on increasing the concentration of the latter from 5 M to 8 M.86 In another study involving water–urea mixture by Grubmuller and co-workers, parameters from OPLS in modelling urea were used to investigate three distinct pair of urea conformations with special reference to translational and orientational populations.87 The same research group used identical pair of model and methods for exploring the molecular level interaction between urea and amino acids which in turn is indicative of the urea mediated protein denaturation.88 The Duffy model representing urea was also adopted by Paul et al. to probe the structural and energetics of solutions containing water, urea and trimethylamine-N-oxide (TMAO).89 They have found that the TMAO–water and TMAO–urea interactions through hydrogen bonding play a crucial role in counteracting the urea induced denaturation.89 Vegt and co-workers used two nonpolarizable urea force field; OPLS and KBFF in combination with three nonpolarizable water force field namely, SPC, SPC/E and TIP4P to frame the significant perturbation of urea and subsequent formation of a “glue” bridge in between the hydrophobic (neopentane) pairs.90 Duffy parameters were found to be useful in representing urea while the kinetic steps of its dissolution and crystal growth were investigated.91 Shimizu et al. obtained comparable potential of mean force values for methane in comparison to the solubility data of small hydrocarbons in presence of urea.92 In this paper, the aggregation of methane was shown to be driven by the increase in free energy and urea was computationally designed according to OPLS parameters.92 MD simulations with reference interaction site model (RISM) integral equation theory were adopted to study the solvation structure and thermodynamics of the transfer process from water to a water–urea mixture in presence of the cations and anions together along with an uncharged species of the same size. Urea molecule containing the parameters from OPLS force field was used.93 The choice of OPLS parameters of urea is based on the idea that it would efficiently exhibit the denaturation of the helix rich protein, at least qualitatively. This is particularly true when the action of urea on the protein and its counteraction by an additive are the main focus of the work and is not meant for capturing microscopic properties qualitatively especially in the absence of reliable experimental data on related systems. TEAA and choline cation has also been modelled using the OPLS-AA derived parameters. Keeping all these issues in mind, we hope the force field descriptions for urea and TEAA, choline chloride as the denaturant and the protecting osmolytes respectively can predict the correct dynamics of the small protein in such mixtures. The geometry optimized structures of urea, TEAA, ChCl molecules are obtained using Gaussian 09 software package94 in combination with density functional theory (DFT) at the B3LYP/6-311+G* level for calculating the OPLS charges.94,95 We have used similar geometry optimization and consequent calculation of partial charges on TEAA and reline. An OPLS-AA force field compatible automatic topology construction program MKTOP96 is used to evaluate the correct partial charges assigned for each of its atoms for TEAA (Tables S1 and S2, ESI†) and reline (Table S3, ESI†). The bonded interaction parameter of TEAA and reline are listed in Tables S4 and S5 (ESI†). A cubic box of volume 141.05 nm3 is built for all systems. 85 ions of each type, namely triethylammonium and acetate (PUILW (5:1) & PUILW (2:1)) and choline (PUChClW (5:1) & PUChClW (2:1)) along with chlorine (after offsetting the charge of the system) are added to the box to maintain the experimentally observed density of 1 M for both TEAA and choline chloride in water. Considering the ionic liquid systems (PUILW (5:1) & PUILW (2:1)) minimizing the energy of the box, the molecules are further equilibrated for another 100 ps under NVT ensemble at 293 K. A 2 ns run is carried out in the NPT ensemble to maintain the system pressure at 1 bar. Then the system is simulated for 10 ns with the OPLS-AA65 force field to obtain an equilibrated TEAA. The calculated density of 1 M TEAA in water at 1 bar and 293 K is found to be 1006.580 (±0.095) kg m−3 which is in fair agreement with the experimental value, 1002 kg m−3.97 Similar procedure is followed for choline chloride. However, to be consistent with the experimentally obtained value the temperature of the system is maintained at 313 K. The calculated density of the system comes to be 1090.990 (±0.067) kg m−3, which is close enough with the experimental value 1190 kg dm−3.98 These equilibrated choline chloride and TEAA are used as co-solvents in the systems. We calculated the osmotic pressure of the ionic liquid and choline chloride
systems according to the statistical mechanical theory of osmotic pressure, suggested by Mayer and McMillan.99 The deviation from ideality can be measured from osmotic coefficient (φ) which is expressed as
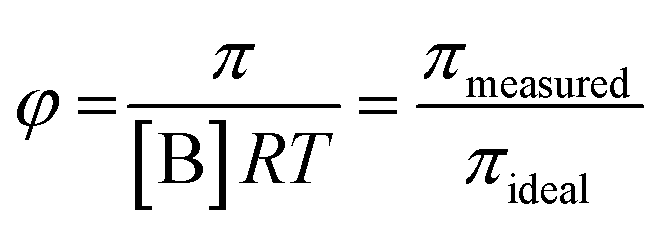 | (1) |
| πmeasured ≈ RT{[B] + B2[B]2 + …} | (2) |
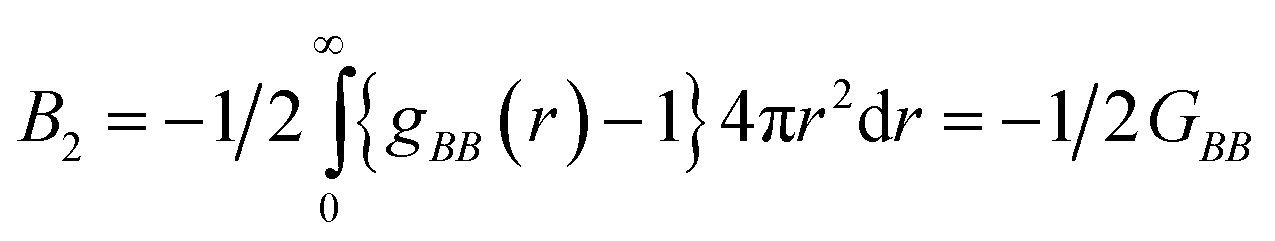 | (3) |
:5 molar ratio of the stabilizers with respect to urea existing already in the system. In order to perform the simulations at higher concentration of stabilizers, each system is packed with total 340 ions of each type. For obtaining solvent relaxed proper initial structures, a 5000-steps energy minimization is performed for each system using steepest decent method.101 Subsequently, a 200 ps equilibration at NVT ensemble is performed to heat each system from 0 K to 330 K to avoid void formation in a box followed by a 5 ns equilibration at isothermal-isobaric (NPT) ensemble to attain a steady pressure of 1 bar. This period of equilibration is found to be sufficient enough for the convergence of properties like pressure, temperature, cell volume and density for each system. Temperature is kept constant by applying the V – rescale thermostat.102 Parrinello–Rahman barostat103 with a pressure relaxation time of 2 ps is used for the attainment of desired pressure for all simulations. Production runs for 500 ns with a time step of 2 fs is performed and the trajectory is saved at a frequency of 2 ps for analysis. Short range Lennard–Jones interactions are calculated using the minimum image convention.104 For estimating nonbonding interactions including electrostatic as well as van der Waals forces, a spherical cut-off distance 1 nm is chosen. Periodic boundary conditions have been used in all three directions for removing edge effects. SHAKE algorithm105 is applied to constrain bonds involving hydrogen atom of the water molecules. Long range electrostatic interactions are calculated using the particle mesh Ewald (PME) method.106 Specific species of interest are labelled as energy groups prior to beginning each MD run. VMD 1.9. 2 (ref. 72) is used to visualize the trajectories and capture snapshots of the protein. Simulations are repeated twice for estimating statistical uncertainties and testing the convergence of the results obtained. Root-mean-square deviation (RMSD), and radius of gyration (Rg) of Cα atoms and solvent accessible surface area (SASA) of protein backbone for all six systems are calculated and plotted together for comparison. The preferential binding interactions and the subsequent transfer free energy of various co-solvents to the protein backbone from the solvent water for different systems are evaluated to shed light on the mechanism by which the stabilizers compensate the action of urea on the protein. A hydrogen bond is considered between a donor and an acceptor if the hydrogen bond length is less than or equal to 0.35 nm and donor–hydrogen–acceptor angle is less than or equal to 30° as followed by other studies.107,108 Average numbers of hydrogen bonds are calculated using the above geometric criteria.109 Three dimensional spatial density distribution functions (SDF) of urea/TEAA and urea/ChCl around the time – averaged HP-36 are calculated by employing the g_spatial utility of GROMACS 4.5.6.66 The secondary structure of HP-36 is calculated under various simulation conditions using DSSP program.110
3. Results and discussions
3.1. Initial signatures of the counteraction of urea-induced denaturation of the small protein HP-36
Snapshots in Fig. 2 highlight the structural changes of the small protein during simulations under various conditions. It gets denatured in urea very rapidly (2nd row, Fig. 2) and the denatured state persists till the end of 500 ns. The native structure of the protein seems to start getting preserved from the next row onwards. It turns out that the extent of stabilizer – induced counteraction increases quite significantly with increasing concentration of ChCl from its non-deep eutectic condition (4th row, Fig. 2) to the corresponding deep eutectic one (6th row, Fig. 2). On the contrary, the qualitative change in TEAA – mediated counteraction of urea is not that much profound while moving from low concentration (3rd row, Fig. 2) of the protecting osmolyte to its higher concentration (5th row, Fig. 2). | ||
| Fig. 2 Snapshots for small protein HP-36 representing systems PW (1st row), PUW (2nd row), and PUILW (5:1) (3rd row). PUChClW (5:1) (4th row), PUILW (2:1) (5th row), PUChClW (2:1) (6th row) at different instances of the simulations, 100, 200, 300, 400, 500 ns snapshots respectively from left to right (a–e). These snapshots are created using VMD 1.9. 2 (ref. 72) solvent molecules and ionic species are ignored for clarity of visualization. | ||
It follows from Fig. 3a that the average RMSD for the Cα – carbon atoms of HP-36 does not go beyond 0.2–0.25 nm in pure water indicating its stability in native conformation (black line, Fig. 3a). However, in the binary protein/urea mixture under aqueous condition, the RMSD abruptly shoots up to around 0.75 nm beyond 20 ns and further up to 1 nm around 200 ns of the simulation indicating progressive urea – assisted denaturation of the protein (red line, Fig. 3a). It is interesting to note that in the urea/TEAA mixture, the calculated RMSD for PUILW (5:1) (blue line, Fig. 3a) and PUILW (2:1) (green line, Fig. 3a) evolves quite similarly to that in presence of neat water (black line, Fig. 3a) with time. In the mixture containing urea and ChCl, different trends are observed for two systems. An increase in RMSD approximately around 0.6 nm from 250 ns is observed for PUChClW (5:1) (pink line, Fig. 3a), while for PUChClW (2:1) (orange line, Fig. 3a), the RMSD value (∼0.1 nm) seems to be slightly lower than that of the urea/TEAA systems. As simulation progresses, variation of the radius of gyration (Rg) of the Cα – carbon atoms of HP-36 (Fig. 3b) with time exhibits a pattern which is similar to the time evolution of the RMSD discussed earlier for the six systems under consideration. Comparable average Rg values are observed for the systems PW (black line, Fig. 3b), PUILW (5:1) (blue line, Fig. 3b), PUILW (2:1) (green line, Fig. 3b), and PUChClW (2:1) (orange line, Fig. 3b) throughout the simulations. The Rg value for the small protein keeps on fluctuating heavily beyond 170 ns of the simulation (red line, Fig. 3b) suggesting the complete denaturation of HP-36. Another system, PUChClW (5:1) shows lesser extent of counteraction against urea assisted unfolding of the small protein (pink curve, Fig. 3b) for which the average Rg value is above the one estimated for pure water throughout the simulation. The time evolution of both RMSD and Rg shed light on the urea-induced protein unfolding and its subsequent counteraction by ammonium-based stabilizers. Solvent accessible surface area (SASA), is another useful parameter that probes the exposure of the protein to its surrounding solvent molecules and thus accounting for the folded and unfolded state. It is obvious that the extent of exposure to solvent would be proportional to the amount of denaturation caused in the native structure of protein. Backbone atoms of HP-36 in six different systems are considered for the time evolution of SASA (Fig. 3c). Throughout 500 ns time frame of the simulation, protein in pure water (PW) (black line, Fig. 3c), in urea/TEAA ternary mixture both PUILW (5:1) (blue line, Fig. 3c) and PUILW (2:1) (green line, Fig. 3c) and in urea/ChCl mixture (PUChClW (2:1) (orange line, Fig. 3c) exhibit almost identical SASAs with an average value of roughly 21 nm2 implying the reasonable preservation of the native structure of the small protein under these conditions. The protein–urea binary (system PUW (red line, Fig. 3c)) mixture shows higher value of SASA throughout simulation time scale than the other systems, most prominently beyond 150 ns. The system PUChClW (5:1) shows an intermediate change in the SASA value with time indicating a relatively lower extent of counteraction by the protecting osmolytes against urea induced denaturation (pink line, Fig. 3c) under this condition.
 | ||
| Fig. 3 Time evolution of (a) RMSD, (b) radius of gyration (Rg) of the Cα carbon atoms of HP-36 and (c) solvent accessible surface area (SASA) of the backbone atoms of HP-36 for six systems containing pure water (PW), binary aqueous urea (PUW), ternary aqueous urea/TEAA mixture at 5:1 molar ratio (PUILW (5:1)), ternary aqueous urea/choline chloride mixture at 5:1 molar ratio (PUChClW (5:1)), ternary aqueous urea/TEAA mixture at 2:1 molar ratio (PUILW (2:1)) and ternary aqueous urea/choline chloride mixture at 2:1 molar ratio (PUChClW (2:1)) respectively. | ||
Analysis of the secondary structure of the protein for six different systems under consideration (Fig. 4a–f) highlights the changes in its helical properties. It appears from Fig. 4b that the protein undergoes significant helix–coil transitions during the 500 ns simulation. The helices get entirely distorted in the absence of the protecting osmolytes (Fig. 4b). Fig. 4a shows the expected retention of the initial helical structure of HP-36 in pure water. The secondary structure at lower TEAA concentration (5:1) shows comparatively larger number of turns and bends than those at relatively higher proportion of the ionic liquid (2:1). This underlines the slight improvement of the native structure of the protein on moving from 5:1 to 2:1 urea/TEAA mixtures. ChCl, on the other hand, in its non deep eutectic condition with urea (PUChClW (5:1)) is not that much potent in resisting the protein unfolding caused by urea. This is justified from the time evolution of the secondary structure obtained for the system PUChClW (5:1) (Fig. 4d) which shows the presence of a larger number bends and turns as time progress. A considerable loss in the helicity of residues 10–15 can be observed beyond 200 ns for this combination of the denaturant and the stabilizer. Under the deep eutectic condition, however, ChCl (system PUChClW (2:1)) provides better resistance towards urea-induced chemical denaturation of HP-36. The secondary structure under deep eutectic condition (Fig. 4f) shows stability of the constituent alpha helices almost throughout the simulated trajectory. Residues 30–34 undergo visibly substantial conformational changes as compared to the same in presence of the IL in 1:2 molar ratios with respect to urea. As a consequence, the stability of the protein seems to be relatively lesser in the former case. The deep eutectic mixture involving choline chloride and urea, nevertheless turns out to be a more benign choice of solvation medium for the protein over the non deep eutectic one (PUChClW (5:1)) and the one containing IL in a much less proportion with respect to urea (PUILW (5:1)).
 | ||
| Fig. 4 Changes of the secondary structures of the native protein in six different systems designated as (a) PW, (b) PUW, (c) PUILW (5:1), (d) PUChClW (5:1), (e) PUILW (2:1) and (f) PUChClW (2:1) during the simulation. | ||
In this study of urea induced denaturation of protein structure, the folding–unfolding equilibrium of the small model protein can be solely defined by the presence and absence of the α-helices in the protein structure. Three distinct α-helices are present in the native structure of the HP-36 which is evident from three pillars in Fig. 5. We have used the g_helix utility to account for the percentage of total trajectory existing over the residues containing the α-helices. This tool calculates α-helix property of a protein structure. We choose the total protein structure for this analysis. Hence the residues that do not contain the α-helices exhibit sharp dips in between the pillars like patterns. For PW system α-helices exist maximum time throughout the trajectory, which is roughly ∼90% for all three α-helices. A sharp decrease is observed over the time trajectory to 30% for the first and third α-helices and 20% for the second α-helix on moving to the PUW system. PUILW (2:1) and PUChClW (2:1) show comparable persistance of the α-helices, which is in fair correlation with the previous plots containing the time evolution of RMSD, Rg and SASA. They exhibit almost similar extent of counteraction against urea denaturation. The α-helices get preserved for more fraction of total simulation time which is around 70% of the total time the helix content of the protein exist. Furthermore, the PUChClW (2:1) exhibits somewhat better extent of preservation towards the native structure of the small protein. Choline chloride in its non deep eutectic state (PUChClW (5:1)) shows decreased counteracting efficiency on urea mediated denaturation of the protein structure than that of TEAA under same concentration ratio (PUILW (5:1)) with respect to the denaturant urea. Fig. 5 indicates that TEAA at its lower concentration ratio (PUILW (5:1)) shows a comparable counteraction against the urea induced denaturation as three α-helices is sustained for almost 70% of the total simulation time. This gets reduced for the big third α-helix to 30%. PUChClW (5:1) can retain the 2nd α-helix for 70% of the total simulation time but for the 1st and 3rd helices it is around 50% & 60% of the total time trajectory.
 | ||
| Fig. 5 Variation of percentage of simulation time with residue number of HP-36 for six systems containing pure water (PW), binary aqueous urea (PUW), ternary aqueous urea/TEAA mixture at 5:1 molar ratio (PUILW (5:1)), ternary aqueous urea/choline chloride mixture at 5:1 molar ratio (PUChClW (5:1)), ternary aqueous urea/TEAA mixture at 2:1 molar ratio (PUILW (2:1)) and ternary aqueous urea/choline chloride mixture at 2:1 molar ratio (PUChClW (2:1)) respectively. | ||
Another way to quantify the α-helix content in the system is to calculate the time evolution of the number of residues containing the α-helices. We use the do_dssp programme of GROMACS for performing this analysis. In the protein–water (PW) system, α-helices reside over ∼24 residues over the trajectory, which is the highest among the six systems. For PUW system, starting from a value of 15 residues at the initial, the number of residues started gradually decreasing with time. After 175 ns it can be seen that only 4–5 residues of the total protein structure contain the α-helices, which is a prominent signature of urea-induced denaturation on the native protein structure in the protein–urea–water aqueous binary mixture. PUILW (2:1) and PUChClW (2:1) systems exhibit the α-helices containing roughly 21 & 20 residues of the protein structure respectively while for the system PUILW (2:1) it fluctuates slightly in between 250 and 450 ns of the simulation time. Thus, PUChClW (2:1) is much more instrumental in preserving the α-helices of the small protein structure. TEAA in its lower concentration ratio with respect to the denaturant urea (PUILW (5:1)) exhibit approximately 15 residues with α-helices over the time. This is even lower in the case of PUChClW (5:1) system where the number of residues containing alpha-helical motifs get reduced from 15 to 10 as the simulation progresses. The outcome of Fig. 6 is consistent with the insights obtained from the time evolution of RMSD, Rg and SASA where PUILW (2:1) and PUChClW (2:1) show comparable degree of protection towards the urea mediated denaturation but the PUChClW (2:1) brings forth further consistency as a counteracting agent against the harsh actions of urea over the protein throughout the simulated trajectory.
 | ||
| Fig. 6 Time evolution of number of residues containing α-helices of HP-36 for six systems containing pure water (PW), binary aqueous urea (PUW), ternary aqueous urea/TEAA mixture at 5:1 molar ratio (PUILW (5:1)), ternary aqueous urea/choline chloride mixture at 5:1 molar ratio (PUChClW (5:1)), ternary aqueous urea/TEAA mixture at 2:1 molar ratio (PUILW (2:1)) and ternary aqueous urea/choline chloride mixture at 2:1 molar ratio (PUChClW (2:1)) respectively. | ||
3.2. Preferential interaction coefficient
Preferential interaction coefficient, (ΓXP) accounts for the effects of solvent on protein thermodynamics.111 The preferential interaction coefficient of a protein in a mixed solvent is not only a thermodynamic quantity for solvent effects on protein thermodynamics, but it provides a measure of the excess number of co-solute molecules, X present in the immediate surroundings of a protein, P with respect to the solvent water. The positive value of ΓXP indicates the preferential interaction and its negative value indicates the preferential exclusion of a co-solute from a protein surface. Recently, different groups have studied theoretically for further refinement of the expressions for ΓXP based on statistical thermodynamics.112–116 However, the essence of ΓXP lies in the average local solvent composition in the immediate vicinity of the protein. Protein solvation characteristics determine solvent composition at the local regime, thus relating ΓXP to the protein solvation with solvent affecting the protein thermodynamics.
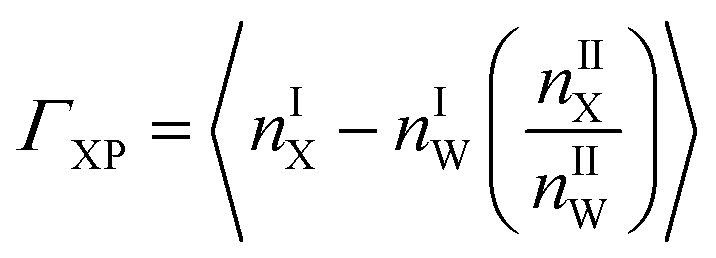 | (4) |
Solvent molecules in the local domain of protein contribute significantly to ΓXP (eqn (4)). Hence, the local solvent composition demands extensive characterization to obtain a molecular level insight of ΓXP. Where, nX and nW are the number of a particular co-solute (urea or the ionic species belonging to TEAA or ChCl) and water molecules, respectively. I and II represent the local and bulk environments of the protein, respectively. The cut-off distance of 0.45 nm is used for counting the number of various co-solutes which is in accordance with the metadynamics study on β-hairpin stability by Saladino et al.117 This specific cut-off of 0.45 nm for the boundary between the small protein and bulk domains, shows an agreement with the suggested osmophobic effect.16,118
The ΓXP value of a co-solute is related to the Gibbs free energy of transfer of a protein from aqueous solution to its co-solute environment.119 The value of ΓXP can be calculated by using the following expression120 In this study, Gibbs free energy of transfer per peptide bond (Δμtr) of the protein backbone from water to per unit molar concentration of urea/mixed osmolyte solutions was calculated using the following equation121,122
 | (5) |
Considering different co-solutes in the system, the significant contribution in ΓXP came from the backbone, which is in fair agreement with previous results.9 While validating the obtained value of transfer free energy with the experimental ones, difference comes due to the variable extent of the exposed protein backbone but it doesn't have any direct effect on preferential binding/preferential exclusion characteristics of the co-solutes under consideration.90 The values of preferential interaction coefficient are calculated from 4.5 Å distance of the protein backbone for different species of the all the systems, which are presented in Table 1. Urea preferentially interacts with protein backbone strongly in the absence of the protecting osmolytes (PUW). The large positive ΓPX value for this system is indicative of high concentration of urea in the vicinity of protein backbone. In presence of IL, however, at lower concentration (PUILW (5:1)) the preferential interaction coefficient of urea with protein backbone is found to be decreased to 0.526, which shows around 81% of reduction of the binding of urea with protein backbone when compared with the ΓPX value with PUW system. It is important to note here that increasing the relative proportion of TEAA does not enhance the preferential binding affinity of urea for protein to a large extent. Non Deep Eutectic combination of choline chloride with urea (PUChClW (5:1)) shows a much larger ΓPX value for urea which accounts for roughly 42% reduction of the binding interaction of urea with protein as compared to that in the absence of ChCl in the system. While going from non-DES condition to DES condition of ChCl (PUChClW (2:1)), ΓPX value for urea is decreased almost by 56%. This justifies the preservation of the native structure of the protein under deep eutectic condition. All the ΓPX values obtained for urea with protein backbone are positive and they indicate preferential interaction, though the values are reduced prominently in the presence of protecting osmolytes. Triethyl-ammonium cation and acetate anion of TEAA are found to be preferentially excluded from protein backbone at both the IL concentrations. At lower concentration of TEAA of 1.6 M (PUILW (5:1)) both of the species exhibit ΓPX values without significant changes in preferential exclusions of these ionic species even though the concentration of TEAA is increased to 4 M (PUILW (2:1)). A similar trend of preferential exclusion of ionic species from protein backbone is encountered for the ions constituting choline chloride.
| System | X | ΓPX | Δμtr |
|---|---|---|---|
| PUW | Urea | 2.780 | −6.51 |
| PUILW (5:1) |
Urea | 0.526 | −1.23 |
| PUILW (2:1) |
Urea | 0.578 | −1.35 |
| PUChClW (5:1) |
Urea | 1.599 | −3.74 |
| PUChClW (2:1) |
Urea | 0.702 | −1.64 |
| PUILW (5:1) |
TEA | −0.673 | 7.88 |
| PUILW (2:1) |
TEA | −0.500 | 2.34 |
| PUILW (5:1) |
ACE | −0.026 | 0.304 |
| PUILW (2:1) |
ACE | −0.019 | 0.089 |
| PUChClW (5:1) |
Choline | −1.320 | 15.456 |
| PUChClW (5:1) |
Cl− | −0.069 | 0.808 |
| PUChClW (2:1) |
Choline | −2.2099 | 10.35 |
| PUChClW (2:1) |
Cl− | −0.0678 | 0.318 |
The positive magnitude of preferential interaction coefficient (ΓPX) produces negative value of transfer Gibbs free energy (Δμtr) and vice versa. All Δμtr values of urea with protein backbone considered here turns out to be negative. It depicts the feasible transfer of the co-solutes (urea) to the protein backbone. This, however, decreases in the co-presence of stabilizers similar to the preferential interaction coefficients which are consistent other studies.123,124 On the other hand, the transfer of the protecting osmolytes from water to the protein has been found to be energetically unfavourable.
3.3. Coordination number
Urea-promoted protein denaturation and the alteration in the protein folding-unfolding dynamics can be probed by calculating the coordination number of particular species of interest surrounding the protein at a specified cut-off of 5 Å in various systems. Coordination number can be expressed by the following equation
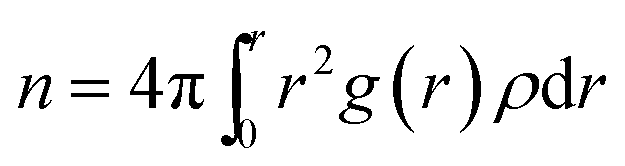 | (6) |
| Reference group | Group | PW | PUW | PUILW (5:1) |
PUILW (2:1) |
PUChClW (5:1) |
PUChClW (2:1) |
|---|---|---|---|---|---|---|---|
| Protein | Water | 16.025 | 10.149 | 5.576 | 5.972 | 7.365 | 3.929 |
| Protein | Urea | — | 13.149 | 7.822 | 6.165 | 11.916 | 7.841 |
| Protein | TEA | — | — | 3.177 | 5.162 | — | — |
| Protein | ACE | — | — | 2.190 | 2.922 | — | — |
| Protein | Choline | — | — | — | — | 1.915 | 5.560 |
| Protein | Cl | — | — | — | — | 0.144 | 0.366 |
It is evident from Table 2 that the numbers of water surrounding the protein in neat water (PW) is quite high. It indicates the higher solubility of the small protein in water. Roughly 36% decrease in co-ordination number of water around the protein in protein–urea binary system is observed, which accounts for the presence of urea molecules within the first solvation shell of the protein. In the ternary aqueous mixtures lesser number of water molecules is observed in the vicinity of the protein. The urea content is diminished by almost 40% and 50% on adding TEAA in 1:5 and 1:2 molar ratios (row 3, columns 5 and 6 in Table 2) with respect to urea. This hints at the sequestering of the denaturant urea by an ionic liquid under ambient conditions and is consistent with the preferential interaction coefficients calculated earlier. The overall increase in TEAA concentration causes a mere 10% decrease in the urea content around the protein. On the other hand, ChCl seems to reduce the accumulation of urea around the protein in a more concentration-dependent manner. For 5:1 urea/ChCl mixture, a small decrease (∼9.3%) in the number of urea surrounding the protein is observed which is in direct contrast to the reduction by 40% for 2:1 urea/ChCl mixture. With increase in the relative concentration of the TEAA in the ternary mixture, coordination number of both cations and anions surrounding the protein increases inevitably. Similar insights can be drawn from the systems containing ChCl as well. It seems that the population of cationic species is larger than the anions near the protein in mixed osmolytes. In the essence of the above calculations, we speculate that urea gets almost saturated within the solvation sphere formed by TEAA ions and as a consequence, an increase in its relative proportion does not have a profound impact on the removal of urea from the surface of the protein. Thus the stabilization of the protein in its native conformation does not get significantly improved (see Section III.D). On the contrary, ChCl-mediated counteraction of the denaturant urea appears to be largely concentration-dependent and the attainment of deep eutectic condition seems to be absolutely imperative. The saturation in the number of urea in TEAA containing systems can further be validated by counting the number of denaturant molecules within a predefined cut-off from the corresponding stabilizing ions as presented in Table 3.
| Reference group | Group | Systems | |||||
|---|---|---|---|---|---|---|---|
| PW | PUW | PUILW (5:1) |
PUILW (2:1) |
PUChClW (5:1) |
PUChClW (2:1) |
||
| TEA | Urea | — | — | 18.856 | 18.462 | — | — |
| ACE | Urea | — | — | 18.922 | 18.096 | — | — |
| Choline | Urea | — | — | — | — | 18.753 | 15.889 |
| Cl− | Urea | — | — | — | — | 17.553 | 14.189 |
It immediately follows from Table 3 that the number of the denaturant urea molecules in the vicinity of TEAA ions remains virtually unaltered despite the change in its concentration for two systems (row 3 and 4, column 5 and 6, Table 3). This indicates that the masking of urea afforded by the ionic liquid does not get enhanced on increasing its concentration. Thus, the aggregate formed due to the networking between urea and TEEA ions can't accommodate more urea molecules on going from the system PUILW (5:1) to PUILW (2:1). As a consequence of this saturation of urea around TEAA, the refolding of the protein does not experience a very prominent improvement (as seen previously in Fig. 3a–c). Choline chloride, on the other hand, exhibits subtle differences in its urea-masking efficiency at two different concentrations. It can be seen that the aggregate formed between urea and ChCl in 2:1 molar ratios respectively involves a relatively smaller number of urea molecules as compared to that representing the 5:1 combination. As a result, the protein differs vastly in the extent of its stabilizations attained under deep eutectic/non-deep eutectic conditions.
3.4. Hydrogen bond calculation
Hydrogen bonding plays a crucial role in determining the stability of protein. Jaffrey had classified hydrogen bonding to be electrostatic in nature.120 Due to the charge neutrality of urea, the electrostatic interaction between urea and the protein is categorized as the H-bond interaction. The average numbers of H-bonds between various combinations of species have been calculated based on certain geometric criteria, discussed earlier (Section II, simulation details). These values are given in Table 4.| Combinations | Systems | |||||
|---|---|---|---|---|---|---|
| PW | PUW | PUILW (5:1) |
PUILW (2:1) |
PUChClW (5:1) |
PUChClW (2:1) |
|
| Intra-protein | 23.592 | 14.339 | 19.949 | 21.592 | 16.722 | 21.383 |
| Protein–urea | — | 46.615 | 35.662 | 22.094 | 43.779 | 31.942 |
| Protein–TEA | — | — | 1.598 | 2.388 | — | — |
| Protein–ACE | — | — | 14.221 | 17.520 | — | — |
| Protein–choline | — | — | — | — | 1.588 | 4.060 |
| Protein–chloride | — | — | — | — | 0 | 0 |
Intra-protein hydrogen bonds provide useful insights into the stability of the folded state of the protein in a specific system. It is quite obvious that a greater number of hydrogen bonds are formed between different residues due to the proximities of the amino acids with each other maintained in a folded state. The average number of hydrogen bonds in neat water (PW) experiences a sharp decrease by ∼40% in the co-presence of urea but in the absence of any protecting osmolytes (system PUW). This shifts the equilibrium in favour of the unfolded ensemble of the protein. Presence of ionic liquid TEAA increases the number of hydrogen bonding within the protein, indicating its stabilization in the folded state. The intra-protein H-bonds only increase by 8% on increasing the relative proportion of TEAA in the ternary mixtures containing urea and IL. The same trend associated with ChCl is somewhat different. There is an enhancement in the number of average intra-protein hydrogen bonds by 30% on going from non-deep eutectic condition (PUChClW (5:1)) to the corresponding deep eutectic condition (PUChClW (2:1)). The reasonable persistence of these hydrogen bonds speaks in favour of the stabilization of the protein in its native state as shown in Fig. 2–4. The extents by which direct protein – urea interactions occur via hydrogen bonding can be illustrated from Table 4. In absence of protecting osmolytes the obtained number of H-bonds between HP-36 and urea comes out to be 46.615, which first decreases in ternary mixtures containing TEAA and ChCl and keeps on decreasing on increasing their concentrations further. Around 30% decrease in the number of protein–urea H-bonds with respect to the ones in the binary aqueous urea mixture can be observed on going from 5:1 to 2:1 PUILW systems. In a similar fashion, the average numbers of H-bonds for protein–urea combinations get diminished significantly for aqueous protein/urea systems containing ChCl in varying amounts. The very low values of average number of hydrogen bonds between the cations of both the species with the protein correlate well with the preferential exclusion of the ammonium-based stabilizers from protein surface separately.
3.5. Spatial density distribution functions (SDFs)
Three dimensional spatial density distribution function (SDF) provides a time-averaged insight into the dynamics of the protein in different systems containing osmolytes. The snapshots obtained for the protein structure in Fig. 2 represent particular time frames in which the protein structure is frozen. Advantage of SDFs lies in the fact, that it takes into account the entire trajectory of the system in a single snapshot which sheds light on the aggregation of the osmolytes (denaturing or stabilizing or both) around a time-averaged protein. The ‘Isosurface’ representation is used to compute and draw a surface within a volumetric data field, on a 3 dimensional surface associated with the points of a single scalar value. The isovalue keeps control over the value for which the isosurface will be computed. The isovalue 7 is selected on the basis of trial and error in order to retain the best visual clarity of the time averaged density of urea (yellow in Fig. 7), TEAA (red in Fig. 8), choline chloride (green in Fig. 8) and has been adopted consistently for all the SDF plots below. | ||
| Fig. 7 Three dimensional spatial density distribution function (SDF) of urea (yellow isosphere) around the time-averaged small protein HP-36 (licorice representation, violet colour) for systems (a) PUW, (b) PUILW (5:1), (c) PUILW (2:1), (d) PUChClW (5:1) and (e) PUChClW (2:1). Isovalue ∼7 has been maintained in all the cases for the best display. Snapshots are rendered using VMD 1.9.2.70 Solvent molecules are ignored for clarity. | ||
 | ||
| Fig. 8 Three dimensional spatial density distribution function (SDF) of protecting osmolytes around the time-averaged small protein HP-36 (violet colour, licorice representation) and urea (bond type, licorice representation) for systems (a) PUILW (5:1), (b) PUILW (2:1), (c) PUChClW (5:1) and (d) PUChClW (2:1). TEAA is represented as red isosphere and choline chloride is represented as green isosphere. Isovalue of around 7 has been maintained in all the cases for the best display. Snapshots are rendered using VMD 1.9.2.70 Solvent molecules are ignored for clarity. | ||
It seems from Fig. 7b–e that the small protein shows unwillingness to stay in the vicinity of urea in the co-presence of the protecting osmolytes for most of the systems under consideration. On the contrary, in the protein/urea binary mixture the unfolded protein structure is almost embedded within the density of urea (Fig. 7a). For the system PUChClW (5:1), the protein, however, still has a considerable amount of urea in its immediate vicinity (Fig. 7d). In the systems PUILW (5:1) (Fig. 7b) and PUILW (2:1) (Fig. 7c) the protein structure is out of the urea density zone and there is no such significant difference in the time average snapshots obtained for two systems. It depicts that increasing the concentration of the ionic liquid (TEAA) does not account for further betterment in counteraction property. Deep eutectic condition of choline chloride (PUChClW (2:1)) (Fig. 7e) shows the protein structure is out of urea density zone, utters profound increase in counteraction property on urea denaturation of protein structure.
It is revealed Fig. 8a–d that in the ternary mixture containing urea and protecting osmolytes, the denaturant forms aggregates with the stabilizers. In Fig. 8c, at lower ChCl concentration (PUChClW (5:1)) it can be seen that the protein structure is still within the urea–ChCl domain implying the inefficient counteraction of urea-induced denaturation at this simulation condition. The other snapshots representing Fig. 8a, b and d for PUILW (5:1), PUILW (2:1) and PUChClW (2:1) respectively demonstrate that the protecting osmolytes scavenge the urea away from the protein structure effectively. The folded state of the protein manifests the better preservation of the native structure under these conditions. TEAA sequesters urea from the surrounding of the protein roughly to the same extent for high concentration (PUILW (2:1)) (Fig. 8b) in comparison to that for low concentration of the ionic liquid (PUILW (5:1)) (Fig. 8a). Fig. 8d shows that ChCl under deep eutectic condition pulls more urea towards itself keeping the native structure of the protein relatively intact as compared to the non-deep eutectic condition.
4. Conclusions
In this paper, using extensive molecular dynamic simulation we show how a small protein can attain reasonable conformational stability in mixed osmolyte systems containing high amounts of urea. Ammonium-based stabilizers have been found to protect the protein from the harsh action of the denaturant urea under aqueous conditions. Initial assessments, such as the time evolution of RMSD, radius of gyration and solvent accessible surface area of the protein reveal the stability of the protein in the two systems containing TEAA/urea and the one under deep eutectic condition in connection with ChCl/urea mixture. The stability of the protein under these conditions is almost identical to the one in neat water (Fig. 3a–c). From these figures, it is also apparent that the presence of urea alone forces the protein to undergo spontaneous unfolding. Secondary structure analyses (Fig. 4a–f) also exhibit a similar trend of protein stability in various systems, most notably, the partial unfolding of the protein under non-deep eutectic condition (system PUChClW (5:1)).
In order to gain molecular level insights into the counteraction of the urea-induced denaturation we calculate the preferential binding interactions between the ionic components of the stabilizers and the protein backbone. Addition of protecting osmolytes causes progressive depletion in the binding affinity of urea for protein with the increment in ChCl concentration. On the other hand, the presence of TEAA in varying amount in the mixed osmolyte systems does not guarantee a significant change in the binding affinity of urea for protein. In addition to the depletion in urea concentration, it is also seen that all the stabilizer ions are preferentially excluded from the protein surface for all the mixed osmolyte systems. From SDF calculations, it can be seen that urea forms stable aggregates with TEAA/ChCl under all the conditions and the protein is forced to remain solubilised in water which accounts for its stability against the urea-induced denaturation. The favourable urea/stabilizer interactions and the proteins preference to stay in water agree well with the synergistic behaviour of urea–glycine-betaine mixture as proposed by Kumar and Kishore.21,89
It is apparent from the analyses throughout this study that the stability of the protein does not get significantly improved on increasing the TEAA content in the co-presence of urea while the attainment of deep eutectic condition brings forth better stability to the protein as compared to the non-deep eutectic one. This may be rationalized on the basis of the coordination numbers of urea within a specified distance from various ionic species under consideration. It follows from Table 3 that the number of urea molecules around both the ions constituting TEAA remains virtually unaltered on increasing the TEAA concentration from 1:5 to 1:2 with respect to urea. In case of ChCl, however, these numbers change appreciably which attributes to the different conformations adopted by the protein in urea/ChCl mixtures. The average numbers of H-bonds between various species of interest also shed light on the variation of the conformational flexibility of the protein as a function of the type/concentration of the stabilizer in mixed osmolyte systems. We would like to mention here that the force filed description for various species in this study is merely a choice since the in-built OPLS urea model is easier to use in combination with GROMACS. This was done in order to obtain qualitative trends in the dynamics of the substrate peptide in aqueous mixtures containing the denaturant and two ammonium based stabilizers. This extensive computational work is neither aimed at establishing absolute/quantitative dynamic parameters nor does it attempt to extrapolate experimental evidences which are sparse anyways for similar systems to the best of our knowledge. Moreover, we are fully aware of the shortcomings of the model used for urea and the fact that a different atomistic force field might alter some of the solvation parameters and free energy values reported here. However, we also strongly opine that the peptide denaturation and its attenuation induced by the stabilizers are sufficiently evident from the 500 ns long trajectories for each system under consideration and this qualitative trend won't change significantly even if an alternative force field is employed. The properness of the equilibration protocols used here is also another valid concern. However, it is well known that the equilibration of a complex system such as ours is very tricky and the appropriateness of such protocols could not be ascertained in absence of suitable experimental evidences. Nevertheless, our study has important bearings towards designing custom-made stabilizers composed of molecular ions for in vitro experiments where the maintenance of the native structure of the substrate protein or an enzyme is of primordial importance.125,126
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
R. C. acknowledges SERB (SB/SI/PC-55/2013) for financial support. S. S. thanks IIT Bombay for a fellowship. S. G. thanks CSIR, Govt. of India and IIT Bombay-IRCC for providing senior research fellowship.References
- A. P. Golovanov, G. M. Hautbergue, S. A. Wilson and L. U. Lian, A Simple Method for Improving Protein Solubility and Long-Term Stability, J. Am. Chem. Soc., 2004, 126, 8933–8939 CrossRef CAS PubMed.
- H. M. Shahjee, K. Banerjee and F. Ahmad, Comparative analysis of naturally occurring L-amino acid osmolytes and their D-isomers on protection of Escherichia coli against environmental stresses, J. Biosci., 2002, 27, 515–520 CrossRef CAS PubMed.
- S. Ortiz-Costa, M. M. Sorenson and M. Sola-Penna, Betaine protects urea-induced denaturation of myosin subfragment-1, FEBS J., 2008, 275, 3388–3396 CrossRef CAS PubMed.
- L. S. Taylor, P. York, A. C. Williams, H. G. Edwards, V. Mehta, G. S. Jackson, I. G. Badcoe and A. R. Clarke, Sucrose reduces the efficiency of protein denaturation by a chaotropic agent, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1253, 39–46 CrossRef.
- H. R. Palmer, J. J. Bedford, J. P. Leader and R. A. Smith, 31P and 1H NMR studies of the effect of the counteracting osmolyte trimethylamine-N-oxide on interactions of urea with ribonuclease A, J. Biol. Chem., 2000, 275, 27708–27711 CAS.
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Living with water stress: evolution of osmolyte systems, Science, 1982, 217, 1214–1222 CAS.
- B. J. Bennion, M. L. DeMarco and V. Daggett, Preventing misfolding of the prion protein by trimethylamine N-oxide, Biochemistry, 2004, 43, 12955–12963 CrossRef CAS PubMed.
- C. Y. Hu, G. C. Lynch, H. Kokubo and B. M. Pettitt, Trimethylamine N-oxide influence on the backbone of proteins: an oligoglycine model, Proteins, 2010, 78, 695–704 CAS.
- T. O. Street, D. W. Bolen and G. D. Rose, A molecular mechanism for osmolyte-induced protein stability, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13997–14002 CrossRef CAS PubMed.
- L. Ma, L. Pegram, M. T. Record and Q. Cui, Preferential interactions between small solutes and the protein backbone: A computational analysis, Biochemistry, 2010, 49, 1954–1962 CrossRef CAS PubMed.
- P. Ganguly, T. Hajari, J. Shea and N. F. A. van der Vegt, Correction to Mutual Exclusion of Urea and Trimethylamine N-Oxide from Amino Acids in Mixed Solvent Environment, J. Phys. Chem. Lett., 2015, 6, 581–585 CrossRef CAS PubMed.
- P. Ganguly, N. F. A. van der Vegt and J. Shea, Hydrophobic association in mixed urea–TMAO solutions, J. Phys. Chem. Lett., 2016, 7, 3052–3059 CrossRef CAS PubMed.
- D. R. Canchi and A. Garcia, Cosolvent effects on protein stability, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- G. Borgohain and S. Paul, Model Dependency of TMAO's Counteracting Effect Against Action of Urea: Kast Model versus Osmotic Model of TMAO, J. Phys. Chem. B, 2016, 120, 2352–2361 CrossRef CAS PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. Koch, Counteraction of Urea by Trimethylamine N-Oxide is Due to Direct Interaction, Biophys. J., 2009, 97, 2559–2566 CrossRef CAS PubMed.
- Q. Zou, B. J. Bennion, V. A. Daggett and K. P. Murphy, The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea, J. Am. Chem. Soc., 2002, 124, 1192–1202 CrossRef CAS PubMed.
- J. Hunger, N. Ottosson, K. Mazur, M. Bonn and H. J. Bakker, Water-mediated interactions between trimethylamine-N-oxide and urea, Phys. Chem. Chem. Phys., 2015, 17, 298–306 RSC.
- S. Shimizu and P. E. Smith, How Osmolytes Counteract Pressure Denaturation on a Molecular Scale, ChemPhysChem, 2017, 18, 2243–2249 CrossRef CAS PubMed.
- Z. Su, F. Mahmoudinobar and C. L. Dias, Effects of Trimethylamine-N-oxide on the Conformation of Peptides and its Implications for Proteins, Phys. Rev. Lett., 2017, 119, 108102 CrossRef PubMed.
- S. Paul and S. Paul, Investigating the Counteracting Effect of Trehalose on Urea Induced Protein Denaturaion Using Molecular Dynamics Simulation, J. Phys. Chem. B, 2015, 119, 10975–10988 CrossRef CAS PubMed.
- N. Zhang, F. Liu, X. Dong and Y. Sun, Molecular Insight into the Counteraction of Trehalose on Urea-Induced Protein Denaturation Using Molecular Dynamics Simulation, J. Phys. Chem. B, 2012, 116, 7040–7047 CrossRef CAS PubMed.
- M. Sola-Penna, A. Ferreira-Pereira, A. dos Passos Lemos and J. R. Meyer-Ferwandes, Carbohydrate Protection of Enzyme Structure and Function against Guanidinium Chloride Treatment Depends on the Nature of Carbohydrate and Enzyme, Eur. J. Biochem., 1997, 248, 24–29 CAS.
- N. Kumar and N. Kishore, Synergistic behavior of glycine betaine-urea mixture: a molecular dynamics study, J. Chem. Phys., 2013, 139, 115104–115113 CrossRef PubMed.
- Q. Zou, S. M. Habermann-Rottinghaus and K. P. Murphy, Urea effects on protein stability: Hydrogen bonding and the hydrophobic effect, Proteins, 1998, 31, 107–115 CrossRef CAS.
- E. J. Guinn, L. M. Pegram, M. W. Capp, M. N. Pollock and M. T. Record, Quantifying why urea is a protein denaturant, whereas glycine betaine is a protein stabilizer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16932–16937 CrossRef CAS PubMed.
- S. Goyal, A. Chattopadhyay, K. Kasavajhala and U. D. Priyakumar, Role of Urea-Aromatic Stacking Interactions in Stabilizing the Aromatic Residues of the Protein in Urea-Induced Denatured State, J. Am. Chem. Soc., 2017, 139, 14931–14946 CrossRef CAS PubMed.
- Y. Dai, van Spronsen., G. Witkamp, R. Verpoorte and Y. H. Choi, Ionic liquids and deep eutectic solvents in natural products research: mixtures of solids as extraction solvents, J. Nat. Prod., 2013, 76, 2162–2173 CrossRef CAS PubMed.
- R. A. Sheldon, Green solvents for sustainable organic synthesis: state of the art, Green Chem., 2005, 7, 267–278 RSC.
- H. R. Hobbs and N. R. Thomas, Biocatalysis in supercritical fluids, in fluorous solvents, and under solvent-free conditions, Chem. Rev., 2007, 107, 2786–2820 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Chemistry. Ionic liquids-solvents of the future?, Science, 2003, 302, 792–793 CrossRef PubMed.
- P. E. Savage, Organic Chemical Reactions in Supercritical Water, Chem. Rev., 1999, 99, 603–622 CrossRef CAS PubMed.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Ionic liquids: a pathway to environmental acceptability, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- S. Zhang, N. Sun, X. He, X. Lu and X. J. Zhang, Physical Properties of Ionic Liquids: Database and Evaluation, J. Phys. Chem. Ref. Data, 2006, 35, 1475 CrossRef CAS.
- T. Vasantha, P. Attri, P. Venkatesu and R. S. Rama Devi, Structural Basis for the Enhanced Stability of Protein Model Compounds and Peptide Backbone Unit in Ammonium Ionic Liquids, J. Phys. Chem. B, 2012, 116, 11968–11978 CrossRef CAS PubMed.
- R. Patel, M. Kumari and A. B. Khan, Recent advances in the applications of ionic liquids in protein stability and activity: a review, Appl. Biochem. Biotechnol., 2014, 172, 3701–3720 CrossRef CAS PubMed.
- D. F. Kennedy, C. J. Drummond, T. S. Peat and J. Newman, Evaluating protic ionic liquids as protein crystallization additives, Cryst. Growth Des., 2011, 11, 1777–1785 CAS.
- A. Biswas, R. L. Shogren, D. G. Stevenson, J. L. Willett and P. K. Bhowmik, Ionic liquids as solvents for biopolymers: Acylation of starch and zein protein, Carbohydr. Polym., 2006, 66, 546–550 CrossRef CAS.
- C. A. Summers and R. A. Flowers II, Protein renaturation by the liquid organic salt ethylammonium nitrate, Protein Sci., 2000, 9, 2001–2008 CrossRef CAS PubMed.
- N. Bryne, L. M. Wang, J. P. Belieres and C. A. Angell, Reversible folding-unfolding, aggregation protection, and multi-year stabilization, in high concentration protein solutions, using ionic liquids, Chem. Commun., 2007, 2714–2716 RSC.
- J. P. Mann, A. McCluskey and R. Atkin, Activity and thermal stability of lysozyme in alkylammonium formate ionic liquids—influence of cation modification, Green Chem., 2009, 11, 785–792 RSC.
- W. Wei and N. D. Danielson, Fluorescence and Circular Dichroism Spectroscopy of Cytochrome c in Alkylammonium Formate Ionic Liquids, Biomacromolecules, 2011, 12, 290–297 CrossRef CAS PubMed.
- M. Jaganathan, C. Ramakrishnan, D. Velmurugan and A. Dhathathreyan, Understanding ethylammonium nitrate stabilized cytochrome c – Molecular dynamics and experimental approach, J. Mol. Struct., 2015, 1081, 334–341 CrossRef CAS.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Ionic liquid-mediated selective extraction of lignin from wood leading to enhanced enzymatic cellulose hydrolysis, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- P. Attri, P. Sarinont, T. Amano, K. Koga, A. E. Cho, E. H. Choi and M. Shiratani, Influence of ionic liquid and ionic salt on protein against the reactive species generated using dielectric barrier discharge plasma, Sci. Rep., 2015, 5, 17781 CrossRef CAS PubMed.
- P. Attri, P. Venkatesu, A. Kumar and N. Byrne, A protic ionic liquid attenuates the deleterious actions of urea on α-chymotrypsin, Phys. Chem. Chem. Phys., 2011, 13, 17023–17026 RSC.
- C. Lange, G. Patil and R. Rudolph, Ionic liquids as refolding additives: N′-alkyl and N′-(ω-hydroxyalkyl) N-methylimidazolium chlorides, Protein Sci., 2005, 14, 2693–2701 CrossRef CAS PubMed.
- D. Das, A. Dasgupta and P. K. Das, Improved activity of horseradish peroxidase (HRP) in ‘specifically designed’ionic liquid, Tetrahedron Lett., 2007, 48, 5635–5639 CrossRef CAS.
- V. Rodrigues, D. Ruivo, A. Rodríguez, F. J. Deive, J. S. M. Esperança, I. M. Marrucho, C. M. Gome and L. P. N. Rebelo, Structural–functional evaluation of ionic liquid libraries for the design of co-solvents in lipase-catalysed reactions, Green Chem., 2014, 16, 4520–4523 RSC.
- K. M. Docherty and C. F. Kulpa Jr, Toxicity and antimicrobial activity of imidazolium and pyridinium ionic liquids, Green Chem., 2005, 7, 185–189 RSC.
- D. Constantinescu, H. Weingartner and C. Herrmann, Protein denaturation by ionic liquids and the Hofmeister series: a case study of aqueous solutions of ribonuclease A, Angew. Chem., Int. Ed., 2007, 46, 8887–8889 CrossRef CAS PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jerome, Deep eutectic solvents: syntheses, properties and applications, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Novel solvent properties of choline chloride/urea mixtures, Chem. Commun., 2003, 70–71 RSC.
- F. S. Mjalli and O. U. Ahmed, Physical properties and intermolecular interaction of eutectic solvents binary mixtures: reline and ethaline, Asia-Pac. J. Chem. Eng., 2016, 11, 549–557 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Liquid structure of the choline chloride-urea deep eutectic solvent (reline) from neutron diffraction and atomistic modelling, Green Chem., 2016, 18, 2736–2744 RSC.
- D. Rengstl, V. Fischer and W. Kunz, Low-melting mixtures based on choline ionic liquids, Phys. Chem. Chem. Phys., 2014, 16, 22815–22822 RSC.
- S. L. Perkins, P. Painter and C. M Colina, Molecular dynamic simulations and vibrational analysis of an ionic liquid analogue, J. Phys. Chem. B, 2013, 117, 10250–10260 CrossRef CAS PubMed.
- A. Das and R. Biswas, Dynamic Solvent Control of a Reaction in Ionic Deep Eutectic Solvents: Time-Resolved Fluorescence Measurements of Reactive and Nonreactive Dynamics in (Choline Chloride + Urea) Melts, J. Phys. Chem. B, 2015, 119, 10102–10113 CrossRef CAS PubMed.
- P. Liu, J. Hao, L. Mo and Z. Zhang, Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions, RSC Adv., 2015, 5, 48675–48704 RSC.
- R. Esquembre, J. M. Sanz, J. G. Wall, F. del Monte, C. R. Mateo and M. L. Ferrer, Thermal unfolding and refolding of lysozyme in deep eutectic solvents and their aqueous dilutions, Phys. Chem. Chem. Phys., 2013, 15, 11248–11256 RSC.
- Q. Zeng, Y. Wang, Y. Huang, X. Ding, J. Chen and K. Xu, Deep eutectic solvents as novel extraction media for protein partitioning, Analyst, 2014, 139, 2565–2573 RSC.
- J. T. Gorke, F. Srienc and R. J. Kazlaukas, Hydrolase-catalyzed biotransformations in deep eutectic solvents, Chem. Commun., 2008, 1235–1237 RSC.
- D. Lindberg, M. de la Fuente Revenga and M. Widersten, Deep eutectic solvents (DESs) are viable cosolvents for enzyme-catalyzed epoxide hydrolysis, J. Biotechnol., 2010, 147, 169–171 CrossRef CAS PubMed.
- H. Zhao, G. A. Baker and S. Holmes, Protease activation in glycerol-based deep eutectic solvents, J. Mol. Catal. B: Enzym., 2011, 72, 163–167 CrossRef CAS PubMed.
- H. Monhemi, M. R. Housaindokht, A. A. Moosavi-Movahedi and M. R. Bozorgmehr, How a protein can remain stable in a solvent with high content of urea: insights from molecular dynamics simulation of Candida antarctica lipase B in urea:choline chloride deep eutectic solvent, Phys. Chem. Chem. Phys., 2014, 16, 14882–14893 RSC.
- F. Pena-Pereira and J. Namiesnik, Ionic liquids and deep eutectic mixtures: sustainable solvents for extraction processes, ChemSusChem, 2014, 7, 1784–1800 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- K. Kahn and T. C. Bruice, Parameterization of OPLS–AA force field for the conformational analysis of macrocyclic polyketides, J. Comput. Chem., 2002, 23, 977–996 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- S. Ghosh, S. Dey, M. Patel and R. Chakrabarti, Can ammonium-based room temperature ionic liquid counteract the urea-induced denaturation of a small peptide?, Phys. Chem. Chem. Phys., 2017, 19, 7772–7787 RSC.
- C. J. McKnight, P. T. Matsudaira and P. S. Kim, NMR structure of the 35-residue villin headpiece subdomain, Nat. Struct. Biol., 1997, 4, 180–184 CrossRef CAS PubMed.
- R. Ghosh, S. Roy and B. Bagchi, Solvent Sensitivity of Protein Unfolding: Dynamical Study of Chicken Villin Headpiece Subdomain in Water–Ethanol Binary Mixture, J. Phys. Chem. B, 2013, 117, 15625–15638 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. Jang, N. Sreerama, V. H. Liao, S. H. Lu, F. Li, S. Shin, R. W. Woody and S. H. Lin, Theoretical investigation of the photoinitiated folding of HP-36, Protein Sci., 2006, 15, 2290–2299 CrossRef CAS PubMed.
- S. Roy and B. Bagchi, Comparative Study of Protein Unfolding in Aqueous Urea and Dimethyl Sulfoxide Solutions: Surface Polarity, Solvent Specificity, and Sequence of Secondary Structure Melting, J. Phys. Chem. B, 2014, 118, 5691–5697 CrossRef CAS PubMed.
- P. K. Yuet and D. Blankschtein, Molecular Dynamics Simulation Study of Water Surfaces: Comparison of Flexible Water Models, J. Phys. Chem. B, 2010, 114, 13786–13795 CrossRef CAS PubMed.
- E. M. Duffy, P. W. Kowalczyk and W. L. Jorgensen, Do denaturants interact with aromatic hydrocarbons in water?, J. Am. Chem. Soc., 1993, 115, 9271–9275 CrossRef CAS.
- E. M. Duffy, D. L. Severance and W. L. Jorgensen, Urea: Potential Functions, logP, and Free Energy of Hydration, Isr. J. Chem., 1993, 33, 323–330 CrossRef CAS.
- S. Sokolic, A. Idrissi and A. Perera, Concentrated Aqueous Urea Solutions: A Molecular Dynamics Study of Different Models, J. Chem. Phys., 2002, 116, 1636–1646 CrossRef.
- R. A. Kuharsky and P. J. Rossky, Molecular Dynamics Study of Solvation in Urea Water Solution, J. Am. Chem. Soc., 1984, 106, 5786–5793 CrossRef.
- J. Tsai, M. Gerstein and M. Levitt, Keeping the Shape but Changing the Charges: A Simulation Study of Urea and its Iso-Steric Analogs, J. Chem. Phys., 1996, 104, 9417–9430 CrossRef CAS.
- D. K. Klimov, J. E. Straub and D. Thirumalai, Aqueous Urea Solution Destabilizes Ab16-22 Oligomers, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14760–14765 CrossRef CAS PubMed.
- P. Xiu, Y. Tu, X. Tian, H. Fang and R. Zhou, Molecular Wire of Urea in Carbon Nanotube: A Molecular Dynamics Study, Nanoscale, 2012, 4, 652–658 RSC.
- M. K. Dixit, A. A. Siddique and B. L. Tembe, Salting-Out of Methane in the Aqueous Solution of Urea and Glycine-Betaine, J. Phys. Chem. B, 2015, 119, 10941–10953 CrossRef CAS PubMed.
- N. Kumar and N. Kishore, Protein Stabilization and Counteraction of the Denaturing Effect of Urea by Glycine Betaine, Biophys. Chem., 2014, 189, 16–24 CrossRef CAS PubMed.
- L. J. Smith, H. J. C. Berendsen and W. F. van Gunsteren, Computer Simulations of Urea–Water Mixtures: A Test of Force Field Parameters for Use in Bimolecular Simulations, J. Phys. Chem. B, 2004, 108, 1065–1071 CrossRef CAS.
- C. A. Bertran, J. J. V. Cirino and L. C. G. Freitas, Theoretical Studies of Concentrated Aqueous Urea Solutions Using Computacional Monte Carlo Simulation, J. Braz. Chem. Soc., 2002, 13, 238–244 CrossRef CAS.
- M. C. Stumpe and H. Grubmuller, Aqueous Urea Solutions: Structure, Energetics, and Urea Aggregation, J. Phys. Chem. B, 2007, 111, 6220–6228 CrossRef CAS PubMed.
- M. C. Stumpe and H. Grubmuller, Interaction of Urea with Amino Acids: Implications for Urea-Induced Protein Denaturation, J. Am. Chem. Soc., 2007, 129, 16126–16131 CrossRef CAS PubMed.
- S. Paul and G. N. Patey, Structure and Interaction in Aqueous Urea–Trimethylamine-N-oxide Solutions, J. Am. Chem. Soc., 2007, 129, 4476–4482 CrossRef CAS PubMed.
- M. E. Lee and N. F. A. van der Vegt, Does Urea Denature Hydrophobic Interactions, J. Am. Chem. Soc., 2006, 128, 4948–4949 CrossRef CAS PubMed.
- S. Piana and J. D. Gale, Understanding the Barriers to Crystal Growth: Dynamical Simulation of the Dissolution and Growth of Urea from Aqueous Solution, J. Am. Chem. Soc., 2005, 127, 1975–1982 CrossRef CAS PubMed.
- M. Ikeguchi, S. Nakamura and K. Shimizu, Molecular Dynamics Study on Hydrophobic Effects in Aqueous Urea Solutions, J. Am. Chem. Soc., 2001, 123, 677–682 CrossRef CAS PubMed.
- T. Yamazaki, A. Kovalenko, V. V. Murashov and G. N. Patey, Ion Solvation in a Water–Urea Mixture, J. Phys. Chem. B, 2010, 114, 613–619 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Z. Xu, H. H. Luo and D. P. Tieleman, Modifying the OPLS-AA force field to improve hydration free energies for several amino acid side chains using new atomic charges and an off-plane charge model for aromatic residues, J. Comput. Chem., 2007, 28, 689–697 CrossRef CAS PubMed.
- A. S. T. Ribeiro, B. A. C. Horta and R. B. de Alencastro, MKTOP: A Program for Automatic Construction of Molecular Topology, J. Braz. Chem. Soc., 2008, 19, 1433–1435 CrossRef CAS.
- Aldrich Catalogue Handbook of Fine Chemicals, Aldrich Chemicals, Milwaukee, WI, 2011 Search PubMed.
- R. B. Leron and M. H. Li, High-pressure density measurements for choline chloride: Urea deep eutectic solvent and its aqueous mixtures at T = (298.15 to 323.15) K and up to 50 MPa, J. Chem. Thermodyn., 2012, 54, 293–301 CrossRef CAS.
- W. G. McMillan and J. E. Mayer, The Statistical Thermodynamics of Multicomponent Systems, J. Chem. Phys., 1945, 13, 276–305 CrossRef CAS.
- D. R. Canchi, P. Jayasimha, D. C. Rau, G. I. Makhatadze and A. E. Garcia, Molecular Mechanism for the Preferential Exclusion of TMAO from Protein Surfaces, J. Phys. Chem. B, 2012, 116, 12095–12104 CrossRef CAS PubMed.
- S. S. Petrova and A. D. Solev'ev, The Origin of the Method of Steepest Descent, Hist. Math., 1997, 24, 361–375 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquid, Oxford University Press, Clarendon, U.K., 1987 Search PubMed.
- H. C. A. Anderson, Rattle: A “Velocity” version of the Shake Algorithm for Molecular Dynamics Calculation, J. Comp. Physiol., 1983, 52, 24–32 Search PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- A. Luzar, Resolving the hydrogen bond dynamics conundrum, J. Chem. Phys., 2000, 113, 10663–10675 CrossRef CAS.
- N. Kumar and N. Kishore, Mechanistic insights into osmolyte action in protein stabilization under harsh conditions: N-methylacetamide in glycine betaine–urea mixture, Chem. Phys., 2014, 443, 133–141 CrossRef CAS.
- M. C. Stumpe and H. Grubmüller, Aqueous Urea Solutions: Structure, Energetics, and Urea Aggregation, J. Phys. Chem. B, 2007, 111, 6220–6228 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features, Biopolymers, 1983, 22, 2576–2637 CrossRef PubMed.
- S. N. Timasheff, Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated, Adv. Protein Chem., 1998, 51, 355–432 CrossRef CAS PubMed.
- S. Shimizu and C. L. Boon, The Kirkwood–Buff theory and the effect of cosolvents on biochemical reactions, J. Chem. Phys., 2004, 121, 9147–9155 CrossRef CAS PubMed.
- P. E. Smith, Cosolvent Interactions with Biomolecules: Relating Computer Simulation Data to Experimental Thermodynamic Data, J. Phys. Chem. B, 2004, 108, 18716–18724 CrossRef CAS.
- J. M. Schurr, D. P. Rangel and S. R. Aragon, A contribution to the theory of preferential interaction coefficients, Biophys. J., 2005, 89, 2258–2276 CrossRef CAS PubMed.
- I. L. Shulgin and E. Ruckenstein, A protein molecule in an aqueous mixed solvent: fluctuation theory outlook, J. Chem. Phys., 2005, 123, 54909 CrossRef PubMed.
- P. E. Smith, Equilibrium Dialysis Data and the Relationships between Preferential Interaction Parameters for Biological Systems in Terms of Kirkwood–Buff Integrals, J. Phys. Chem. B, 2006, 110, 2862–2868 CrossRef CAS PubMed.
- G. Saladino, M. Merenchino, S. Pieraccini, R. Campos-Olivas, M. Sironi and F. L. Gervasio, A Simple Mechanism Underlying the Effect of Protecting Osmolytes on Protein Folding, J. Chem. Theory Comput., 2011, 7, 3846–3852 CrossRef CAS PubMed.
- D. W. Bolen and I. V. J. Baskakov, The osmophobic effect: natural selection of a thermodynamic force in protein folding, Mol. Biol., 2001, 310, 955–963 CrossRef CAS PubMed.
- B. M. Baynes and B. L. Trout, Proteins in Mixed Solvents: A Molecular-Level Perspective, J. Phys. Chem. B, 2003, 107, 14058–14067 CrossRef CAS.
- V. Vagenende, M. S. Yap and B. L. Trout, Molecular Anatomy of Preferential Interaction Coefficients by Elucidating Protein Solvation in Mixed Solvents: Methodology and Application for Lysozyme in Aqueous Glycerol, J. Phys. Chem. B, 2009, 113, 11743–11753 CrossRef CAS PubMed.
- L. Ma, L. Pegram, M. T. Record and Q. Cui, Preferential interactions between small solutes and the protein backbone: A computational analysis, Biochemistry, 2010, 49, 1954–1962 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Additive transfer free energies of the peptide backbone unit that are independent of the model compound and the choice of concentration scale, Biochemistry, 2004, 43, 1329–1332 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Predicting the energetics of osmolyte-induced protein folding/unfolding, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15065–15068 CrossRef CAS PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- P. Atrri and P. Venkatesu, Influence of protic ionic liquids on the structure and stability of succinylated Con A, Int. J. Biol. Macromol., 2012, 51, 119–128 CrossRef PubMed.
- C. L. Louros, A. F. Claudio, C. M. Neves, M. G. Freire, I. M. Marrucho, J. Pauly and J. A. Coutinho, Extraction of Biomolecules Using Phosphonium-Based Ionic Liquids + K3PO4 Aqueous Biphasic Systems, Int. J. Mol. Sci., 2010, 11, 1777–1791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10712a |
| This journal is © The Royal Society of Chemistry 2017 |