
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored sol–gel immobilized lipase preparates for the enzymatic kinetic resolution of heteroaromatic alcohols in batch and continuous flow systems†
Mădălina Elena Moisă a,
Cristina Georgiana Spelmezana,
Cristina Paulb,
Judith Hajnal Bartha-Vária,
László Csaba Benczea,
Florin Dan Irimiea,
Csaba Paizsa,
Fráncisc Péterb and
Monica Ioana Toşa*a
a,
Cristina Georgiana Spelmezana,
Cristina Paulb,
Judith Hajnal Bartha-Vária,
László Csaba Benczea,
Florin Dan Irimiea,
Csaba Paizsa,
Fráncisc Péterb and
Monica Ioana Toşa*a
aBiocatalysis and Biotransformation Research Center, Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University of Cluj-Napoca, Arany János 11, RO-400028, Cluj-Napoca, Romania. E-mail: mtosa@chem.ubbcluj.ro
bBiocatalysis Group, Faculty of Industrial Chemistry and Environmental Engineering, University Politehnica Timişoara, C. Telbisz 6, RO-300001, Timişoara, Romania
First published on 16th November 2017
Abstract
Tailored immobilized lipases from Candida antarctica B and Pseudomonas fluorescens, with improved thermal and operational stability, were prepared through fine tuning of the structure of the sol–gel matrix, using various binary or ternary precursor mixtures for the EKR of various chiral heteroaromatic secondary alcohols with benzofuran, benzo[b]thiophen, phenothiazine and 2-phenylthiazol moieties. The operational stability in batch process was studied for five selected systems by performing reuse experiments, using the conversion, enantiomeric excesses and enantiomeric ratio as parameters, demonstrating the dependence of the sol–gel lipase preparate performance on the structure of both biocatalyst and substrate. The resolution of the benzofuranic substrates with the best performing biocatalysts was studied in continuous-flow mode, using the productivity as a criterion. The specific reaction rates under continuous-flow operation (rflow) were higher than those obtained in batch mode (rbatch) in both cases, sustaining its usefulness for further process development.
Introduction
With the aims of stabilizing enzymes and improving enzymatic activity and selectivity, the development of effective immobilization methods has become crucial for the synthetic applications of lipases. Moreover, the processes performed with immobilized catalysts offer as advantages continuous production, the absence of the biocatalyst in the product stream and the possibility to reuse the biocatalyst. In this way, the synthetic and production processes could be more selective and are for certain more proficient economically and environmentally.Accordingly, immobilization methods such as binding to a carrier, cross-linking, and encapsulation in an organic or inorganic polymeric matrix have been intensively studied.1–8 While the adsorption on solid supports (such as Celite, a diatomaceous earth) is a simple, inexpensive and easy to perform method, suitable for applications in organic solvents, the enzyme leaching from the support is generally the most important disadvantage of this procedure.9 In the covalent immobilization, the protein is multipoint attached to a carrier by covalent bonds, directly or through a spacer. One of the benefits of this method is that enzyme leaching is not possible, but the activity could be significantly modified, decreasing in most of the cases.
For enzymes which are easily deactivated (for instance by covalent immobilization), encapsulation could be the most useful method. In the case of sol–gel encapsulation inert glasses with improved catalytic properties (porosity, thermal and mechanical stability, and tunable hydrophobicity) are prepared. The synthesis can be performed by acid- or base-catalyzed hydrolysis of the organic silane precursors, followed by condensation of the resulted intermediates in the presence of an enzyme.10–12 When some protective additives (crown ether, isopropyl alcohol, etc) were used in the sol–gel immobilization process, for certain substrates a supplementary increase of the lipase activity in esterification and enzymatic kinetic resolution (EKR) processes was already reported.13–15
Lipases are most frequently used for the synthesis of chiral intermediates by kinetic resolution in an immobilized form.16,17
Using continuous-flow reactors, the performances of enzyme-catalyzed reactions are generally significantly improved, including the reduction of reaction time, as compared to the classical batch method. The packed-bed reactor (a column containing the immobilized biocatalyst which is flushed with the reaction mixture using a pump, allowing flow and residence time control) is usually the first and simplest choice.18–21
In the present research, several immobilized lipase preparates (lipases from Candida antarctica B and from Pseudomonas fluorescens immobilized by sol–gel entrapment, adsorption on Celite or their combination) were tested in transesterification reactions of various heteroaromatic secondary alcohols, useful chiral building blocks for the pharmaceutical industry.22–25 Previous studies15,26–28 already stated the versatility of the sol–gel immobilization method and the possibility to fine tune the composition of the binary or ternary precursors mixture to achieve high activity and selectivity, but these studies were carried out on model secondary alcohols (1-phenylethanol or 2-alkanols). The present study demonstrates for the first time the efficiency of the tailored binary and ternary sol–gel immobilized lipase preparates for the EKR of various heteroaromatic alcohols. The recycling capacity of the best performing biocatalysts was also investigated and two continuous-flow procedures were developed by testing the most important reaction parameters: temperature, substrate concentration, and flow rate.
Experimental
Materials and methods
Thin layer chromatography (TLC) was performed on Merck Kieselgel 60F254 sheets. Spots were visualized by treatment with 5% ethanolic phosphomolybdic acid solution followed by heating. Preparative chromatographic separations were performed using column chromatography on Merck Kieselgel 60 (63–200 μm).1H-NMR and 13C-NMR were recorded in CDCl3 at 25 °C using a Bruker spectrometer operating at 300 or 600 MHz and 75 or 151 MHz, respectively.
Chiral HPLC (Agilent 1200 Series instrument) and GC (Agilent 77890A instrument) were employed for the quantitative analysis of the enzymatic kinetic resolutions, by periodically taking samples (50 μL) and analyzing them by chiral chromatography: GC (compounds rac-2,3a,b) or HPLC (compounds rac-2,3c–i). The type of chiral columns used for the enantiomeric separations, separation conditions and retention times for each individual enantiomer are presented in Table 1S.† The absolute configuration of products was assigned according to previously reported data, based on the retention time of a reference enantiomerically enriched compound.29–33
The enantiomeric excesses of the substrate eeS and product eeP were determined from peak areas of GC or HPLC chromatograms. The enantiomeric ratio (E) was calculated with the known equation E = ln[(1 − c)(1 − eeS)]/ln[(1 − c) (1 + eeS)], using the calculated conversion c = eeS/(eeS + eeP).34
Lipase B from Candida antarctica was purchased as solid lyophilized powder from C-Lecta (Leipzig, Germany, product code 20606-1). Lipase AK (beige-brown powder, from Pseudomonas fluorescens) and lipase A from Candida antarctica were bought from Sigma-Aldrich (Germany, product code 534730 and 65986 respectively). Novozyme 435 (lipase from Candida antarctica B, immobilized on acrylic resin) was also purchased from Sigma Aldrich.
The silane precursors, methyl- (MeTMOS), vinyl- (VTMOS), octyl- (OcTMOS), and phenyl-trimethoxysilane (PhTMOS) were purchased from Merck (Germany), excepting tetra-methoxysilane (TMOS), a product of Fluka. The other materials and additives used for immobilization, tris-(hydroxymethyl)-aminoethane, isopropyl alcohol, Celite 545, acetone and hydrochloric acid were products of Merck, while sodium fluoride and the ionic liquid 1-octyl-3-methyl-imidazolium tetrafluoroborate [Omim]BF4 were purchased from Fluka.
Vinyl acetate and the solvents used for enzymatic reactions were of the highest grade from Sigma-Aldrich or Merck and stored over molecular sieves (4 Å). Solvents employed in chemical synthesis were used as purchased (methanol > 99.8%) or dried over molecular sieve (dichloromethane) or sodium (diethyl ether). Other reagents were purchased from Sigma-Aldrich or Merck.
A Heidolph Vibramax 110 shaker equipped with incubator module (Heidolph, Germany) was used for all batch experiments.
Continuous-flow experiments were performed in a stainless steel column (50 × 2.1 mm) using the thermostat and the pump modules of an Agilent 1200 Series HPLC instrument.
Chemical synthesis
The synthesis of the investigated racemic substrates and products was first performed using known methods29–33 from the corresponding prochiral heteroaryl-methyl-ketones (1a–c,f) by chemical reduction with sodium borohydride or from the carbaldehydes (1d,e,g–i) by Grignard reaction. Racemic ethanols rac-2a–i were next transformed with acetyl chloride (1.2 equiv.) and a catalytic amount of 1% DMAP/pyridine in dry CH2Cl2 into the corresponding racemic acetates rac-3a–i (Scheme 1a). The obtained NMR data for rac-2,3a–i are in agreement with those already reported.29–33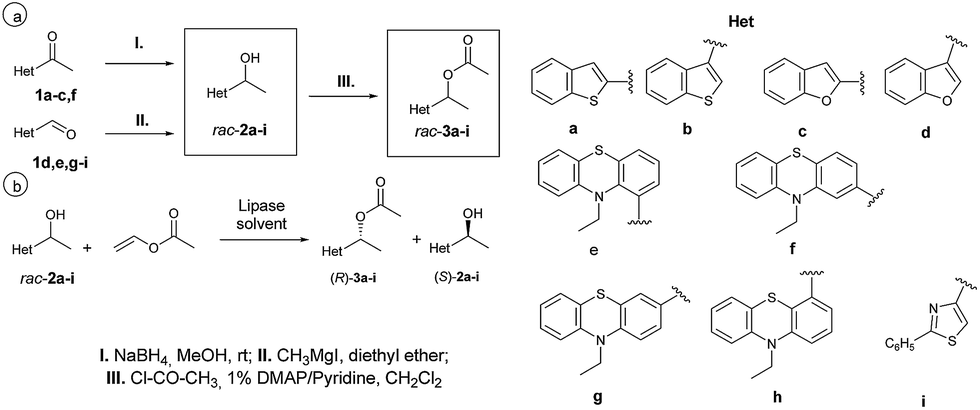 | ||
| Scheme 1 (a) Chemical synthesis of racemic substrates; (b) EKR mediated by lipases. | ||
Immobilization of lipases
The immobilization protocols for the sol–gel entrapment of lipases B from Candida antarctica and Pseudomonas fluorescens (Amano-AK) were accomplished as previously described (Table 1), with NaF as catalyst for simultaneous hydrolysis and polycondensation (Method 1)27 or using HCl for the formation of a prepolymer (Method 2).35 The combined method of sol–gel entrapment and adsorption was also carried out as described elsewhere.26–28 Method 3 was simple adsorption on Celite 545 (see the ESI† for detailed description of all immobilization methods). The protein loading values, calculated for 1 g biocatalyst, were 17.4 mg for sol–gel entrapment and adsorption on Celite and 10.7 mg for the entrapment combined with adsorption, respectively. They were determined as described previously.27 The immobilization method and the silane precursors used for every preparate selected for the present study are described in Table 1.| Preparate codea | Immobilization method | Silane precursorsb (molar ratio) |
|---|---|---|
| a Lipases source: CaL-B-lipase B from Candida antarctica, AK-lipase from Pseudomonas fluorescens.b VTMOS (vinyltrimethoxysilane), PhTMOS (phenyltrimethoxysilane), MeTMOS (methyltrimethoxy-silane), OcTMOS (octyltrimethoxysilane) and TMOS (tetramethoxysilane). | ||
| CaL-B 1 BS | Method 1 | VTMOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TMOS (4:1) :TMOS (4:1) |
| CaL-B 1 BSC | Method 1 + adsorption on Celite 545 | VTMOS:TMOS (4:1) |
| CaL-B 1 TS | Method 1 | PhTMOS:VTMOS:TMOS (1.6:0.4:1) |
| CaL-B 1 TSC | Method 1 + adsorption on Celite 545 | PhTMOS:VTMOS:TMOS (1.6:0.4:1) |
| CaL-B 1 TS* | Method 1 | PhTMOS:MeTMOS:TMOS (1.6:0.4:1) |
| CaL-B 2 TS | Method 2 | PhTMOS:MeTMOS:TMOS (1.6:0.4:1) |
| CaL-B 2 TSC | Method 2 + adsorption on Celite 545 | PhTMOS:MeTMOS:TMOS (1.6:0.4:1) |
| CaL-B Celite | Method 3 (adsorption on Celite 545) | — |
| AK 1 BS | Method 1 | OcTMOS:TMOS (1:1) |
| AK 1 BSC | Method 1 + adsorption on Celite 545 | OcTMOS:TMOS (1:1) |
| AK 1 TS | Method 1 | PhTMOS:VTMOS:TMOS (1.6:0.4:1) |
| AK 1 TSC | Method 1 + adsorption on Celite 545 | PhTMOS:VTMOS:TMOS (1.6:0.4:1) |
| AK 2 TSC | Method 2 + adsorption on Celite 545 | PhTMOS:MeTMOS:TMOS (1.6:0.4:1) |
Enzymatic O-transesterifications
All experiments were performed in triplicate and the average values were further used.Results and discussion
Chemical synthesis of racemic heteroaryl-ethanols rac-2a–i and their corresponding acetates rac-3a–i
Racemic heteroaryl-ethanols rac-2a–c,f were obtained starting from the corresponding prochiral heteroaryl-methyl-ketones (1a–c,f) by chemical reduction with sodium borohydride, while in the case of rac-2d,e,g–i the Grignard reaction of carbaldehydes 1d,e,g–i as starting materials was used (Scheme 1a). Racemic ethanols rac-2a–i were used next as substrates in lipase-catalyzed transesterification reactions (Scheme 1b) and also for the synthesis of the corresponding racemic acetates rac-3a–i (see ESI,† Section 2), used for establishing the chromatographic separation methods (see ESI,† Section 3).Enzymatic kinetic resolution of racemic ethanols rac-2a–i through O-acylation with vinyl acetate and lipase preparates in organic solvents
Efficient enzymatic procedures for the synthesis of enantiomerically enriched (R)- and (S)-ethanols through enzymatic kinetic resolution of the racemic ethanols or their esters (generally acetates) with commercially available lipases have been already described.29–33An optimal immobilization method can improve the biocatalyst efficiency and the operational stability, allowing the process operation in continuous flow reactors, which can provide increased productivity. Lipase encapsulation in sol–gel matrices leads to enzyme preparates with high mechanical and thermal stability and also high porosity. Moreover the surface properties of the sol–gel matrix can be tailored for an optimal interaction of enzymes with particular substrates by choosing the appropriate silane precursors and synthesis method.
The immobilizations were accomplished by adsorption on Celite 545, sol–gel entrapment in mesoporous silica materials obtained from binary (BS) or ternary silane (TS) precursor mixtures, and sol–gel entrapment combined with adsorption on Celite 545, as described in the experimental part (Table 1). Two sol–gel entrapment methods were employed, one that uses NaF as catalyst for simultaneous hydrolysis and polycondensation (Method 1) and another one that uses a sol prepolymer obtained with HCl (Method 2).
The items used in the present work were selected from numerous preparates, according to their activities on model substrates (rac-2-hexanol and rac-1-phenylethanol, data not shown), with the aim to explore several immobilization techniques based on entrapment and adsorption, as well as support matrices with different composition and morphology. In order to be able to compare the catalytic activities of the tested lipase preparates, the same enzyme content was used in all experiments. Vinyl acetate was employed as solvent and as irreversible acyl donor as the liberated vinyl alcohol undergoes spontaneous tautomerization to acetaldehyde, shifting the reaction equilibrium towards product formation.36
In order to find the possible applications of the tested lipase preparates in the chemoenzymatic synthesis of heteroaromatic compounds, first the selected substrates rac-2a–i (40 mM) were acylated in neat vinyl acetate in the presence of immobilized biocatalysts (see ESI, Tables 2S–9S†). Furthermore since the nature of the solvent could significantly influence the selectivity and activity of the immobilized enzyme, the enzymatic acylation of racemic ethanols (10 mM) with vinyl acetate (3 equiv.) in presence of the previously selected optimal biocatalysts was investigated in several solvents with different polarities, such as n-hexane, toluene, diisopropyl ether, methyl-tert-butyl ether, dichloromethane and acetonitrile (see ESI, Tables 10S–18S†). The reactions were evaluated using the conversion c, enantiomeric excesses (of the untransformed alcohol eeS and of the formed ester eeP), and enantiomeric ratio E as parameters.
As a preliminary general conclusion of the screening, all lipase preparates demonstrated high enantiomer selectivity towards the racemic substrates in neat vinyl acetate, except some L-AK preparates (AK 1 TSC and AK 2 TSC in the acylation reaction of rac-2b, E = 25–58 see Table 3S,† entries 14 and 15 in ESI†), while the conversion was influenced by the structure of substrate, as well as by the immobilization method. A more detailed study of the influence of different immobilization conditions was not the subject of this work.
The optimal lipase preparates are summatively presented in Table 2. Some particular information obtained for each substrate is discussed.
| Entry | Substrate rac-2a–i | Optimal biocatalyst | Reaction time (h) | eeSb,c (%) | eePc (%) | cb,d (%) |
|---|---|---|---|---|---|---|
| a E ≫ 200.b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of GC or HPLC chromatograms.d Calculated using the formula c = [eeS/(eeS + eeP)].e No product was detected even after 6 days. | ||||||
| 1 | rac-2a | All CaL-B preparates | 9.5 | >99 | >99 | 50 |
| 2 | rac-2b | CaL-B 1 TS* | 132 | 82 (±1.7) | >99 | 45 (±0.5) |
| CaL-B Celite | 83 (±2.1) | >99 | 46 (±0.5) | |||
| 3 | rac-2c | AK 1 BS | 136 | 94 (±1.) | >99 | 49 (±0.5) |
| 4 | rac-2d | CaL-B 1 TS* | 136 | 91 (±0.9) | >99 | 48 (±0) |
| CaL-B Celite | 24 | 96 (±2.8) | >99 | 49 (±0.9) | ||
| 5 | rac-2ee | All preparates | 144 | <1 | <1 | <1 |
| 6 | rac-2g | AK 1 BS | 11 | 89 (±1.4) | >99 | 47 (±0.5) |
| 7 | rac-2h | CaL-B Celite | 178 | 52 (±1.2) | >99 | 34 (±0.5) |
| 8 | rac-2i | CaL-B 2 TSC | 40 | 75 (±0.5) | >99 | 43 (±0) |
The first tested substrate, rac-1-(benzo[b]thiophen-2-yl)ethan-1-ol rac-2a, was quantitatively resolved (c ≅ 50%) with all tested CaL-B preparates and almost all L-AK preparates in neat vinyl acetate (Table 2S in ESI†). In the given reaction conditions, this substrate was easily acylated regardless to the immobilization method, but it is noteworthy that the catalytic efficiency of the immobilized preparates was higher (c = 45–50%, E ≫ 200) compared to the reaction catalyzed by native CaL-B, with only 16% conversion after the same reaction time (9.5 hours, as presented in Table 2S, entry 1 in ESI†). Since almost all immobilized preparates studied in the acylation reaction of rac-2a gave very good results, for the solvent screening one candidate from each of the two forms of lipases (CaL-B and AK) was selected (CaL-B 1 TSC and AK 1 TSC). The best results were obtained with CaL-B 1 TSC in n-hexane (c ≅ 50% after 1 h, see Table 10S, entry 1 in ESI†), therefore we tested the other CaL-B preparates in the optimal solvent, n-hexane as well. The results (Table 17S in ESI†) indicated that almost all biocatalysts afforded maximum conversion of the substrate in 1 h, except for CaL-B 1 BS (c = 42%, Table 17S, entry 1 in ESI†) and CaL-B 2 TSC (c = 24%, Table 17S, entry 6 in ESI†).
The most efficient biocatalysts (see Table 2) for the acetylation of the racemic alcohols rac-2b,d,i in neat vinyl acetate based on reaction conversion, proved to be the CaL-B preparates obtained by sol–gel entrapment in ternary silane precursor systems with or without deposition on Celite (CaL-B 1 TS* and CaL-B 2 TSC, with 43–48% conversion), depending on the substrate structure, but the adsorbed CaL-B lipase was similarly good with 46–49% conversion. Although the entrapped preparates have the overall advantage of less enzyme leaking, in almost anhydrous organic solvents the adsorbed enzyme could also exhibit high stability, as it was recently demonstrated for Burkholderia cepacia lipase.37
Lipase from Pseudomonas fluorescens (Amano AK), already reported as efficient,29 demonstrated excellent selectivity for the EKR of rac-2c in neat vinyl acetate (entry 3 in Table 2); the optimal biocatalyst was obtained by sol–gel immobilization in a binary silane precursor system (AK 1 BS).
The most appropriate solvent for the transesterification reactions of rac-2a–d,i proved to be n-hexane in all cases (E ≫ 200 at c = 46–50% after 1–28 hours, depending on the substrate, see Table 3).
| Entry | Substrate rac-2a–i | Biocatalyst | Reaction time (h) | eeSb,c (%) | eePc (%) | cb,d (%) |
|---|---|---|---|---|---|---|
| a E ≫ 200.b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of GC or HPLC chromatograms.d Calculated using the formula c = [eeS/(eeS + eeP)]. | ||||||
| 1 | rac-2a | CaL-B 1 BSC | 1 | >99 | >99 | 50 |
| CaL-B Celite | >99 | >99 | 50 | |||
| 2 | rac-2b | CaL-B 1 TS* | 5 | 92 (±1.6) | >99 | 48 (±0.5) |
| CaL-B Celite | >99 | >99 | 50 | |||
| 3 | rac-2c | AK 1 BS | 21 | 86 (±1.2) | >99 | 46 (±0.5) |
| 4 | rac-2d | CaL-B 1 TS* | 19 | 94 (±2.1) | >99 | 49 (±0.5) |
| CaL-B Celite | 2 | 92 (±2.5) | >99 | 48 (±0.8) | ||
| 5 | rac-2f | AK 1 BS | 8 | >99 | >99 | 50 |
| 6 | rac-2g | AK 1 BS | 9 | >99 | >99 | 50 |
| 7 | rac-2h | CaL-B Celite | 27 | 99 (±0.5) | >99 | 50 (±0) |
| 8 | rac-2i | CaL-B 2 TSC | 28 | 95 (±2.2) | >99 | 49 (±0.8) |
In an effort to find a general enzymatic route for the synthesis of enantiomerically enriched phenothiazinic-ethanols, an enzyme and solvent screening was next performed on the model substrate rac-2g and the best reaction conditions (lipase preparate and solvent) were applied to the other phenothiazinic substrates. Accordingly, the enzymatic kinetic resolution of rac-2g with CaL-B and L-AK immobilized preparates in neat vinyl acetate was firstly investigated. AK 1 BS was the only appropriate enzyme preparate in neat vinyl acetate (47% conversion after 11 h, entry 14 in Table 7S in ESI†). When performing the resolution of rac-2g with this biocatalyst and vinyl acetate (3 equiv.) in the selected solvents, the maximum conversion was obtained in n-hexane (34% after 2 h and 50% after 9 h, entries 1 and 4 in Table 14S of ESI†). These reaction conditions were next applied to the other phenothiazinic substrates rac-2e,f,h, leading to very good results in case of rac-2f (50% c and E ≫ 200 after 8 h, entry 2 in Table 18S of ESI†), while for rac-2e and rac-2h further investigations were performed (Tables 6S–8S in ESI†).
In the case of rac-1-(10-ethyl-10H-phenothiazin-1-yl)ethan-1-ol, rac-2e, a sterically hindered secondary alcohol, even sol–gel preparates containing lipase A from C. antarctica, previously reported as an efficient biocatalyst32 were inactive (the acetate was not detected even after 6 days, see Table 6S in ESI†).
The resolution of rac-1-(10-ethyl-10H-phenothiazin-4-yl)ethan-1-ol, rac-2h with CaL-B immobilized preparates in neat vinyl acetate occurred very slowly, but in a selective manner (Table 8S in ESI†). The best results were obtained with CaL-B on Celite 545 (c = 34% after 178 h, E ≫ 200, entry 7, Table 2). Based on the solvent screening, performed in the same manner, n-hexane was also selected as optimal solvent since the reaction rate significantly increased and the maximum 50% conversion was obtained after only 27 h (entry 7 in Table 15S of ESI†). As already reported,31,32 during the enzymatic reactions and particularly during the work-up of the reaction mixture, the instability of the phenothiazinic compounds (ethanol and acetate) was observed, mainly for rac-2h.
It is noticeable that the best performing biocatalysts have shown comparable biocatalytic power with the extensively used Novozyme 435 in the transesterification reactions of rac-2a,b,d,g,i in neat vinyl acetate (Tables 2S, 3S, 5S, 7S and 9S†). Even though the acylation reaction of rac-2d in neat vinyl acetate occurred faster with Novozyme 435 (c = 50% after 16 h, entry 9, Table 5S†) than with any of the sol–gel CaL-B preparates (c = 48% after 136 h with CaL-B 1 TS*, entry 10, Table 5S†), the enantiomeric excess of the product was smaller (eeP = 97% versus eeP > 99%). In terms of conversion Novozyme 435 proved to be more efficient in the acylation reaction of rac-2h in neat vinyl acetate (c = 47% after 42 h, entry 6, Table 8S†), in comparison to all the tested sol–gel preparates (c = 14% after 42 h with CaL-B 1 TS*, entry 3, Table 8S†). It must be taken into consideration that Novozyme 435 is fragile under stirring in batch mode, often resulting in the breakage of the particles and loss of enzyme activity. Consequently, this biocatalyst might not be a suitable candidate for repeated use. Biocatalysts obtained through sol–gel entrapment, on the other hand, do not have this disadvantage and have proved in our previous work27,35,38 and in other studies11,15,39 remarkable recycling stability.
Operational stability of the immobilized lipase preparates at repeated use in batch processes
Long-term catalytic efficiency is a key requirement for any industrial biocatalyst and immobilization should have positive impact on the operational stability of the enzyme. The sol–gel entrapped lipases demonstrated excellent reuse efficiency in the acylation reaction of 2-octanol, showing the significant potential of this technique to provide highly efficient solid-phase biocatalysts for prolonged exploitation.27However, the reuse capability of the biocatalyst must be tested for every individual application, since it depends on the structure of the substrate, the composition, and characteristics of the reaction system, as well as on the operational parameters.
For this reason, the reusability of the optimal immobilized lipase preparates was studied in the enantioselective acylations of rac-2a,c,d,g,i with vinyl acetate, in n-hexane (Fig. 1–4, Tables 19S–24S in ESI†).
 | ||
| Fig. 1 Multiple use efficiency of the selected biocatalysts for the EKR of rac-1-(benzo[b]thiophen-2-yl)ethan-1-ol rac-2a (10 mM) with vinyl acetate (3 equiv.) in n-hexane at 25 °C with: (A) CaL-B 1 BSC and (B) Cal-B on Celite 545 (after 40 min reaction time). Error bars represent standard deviations from average. | ||
 | ||
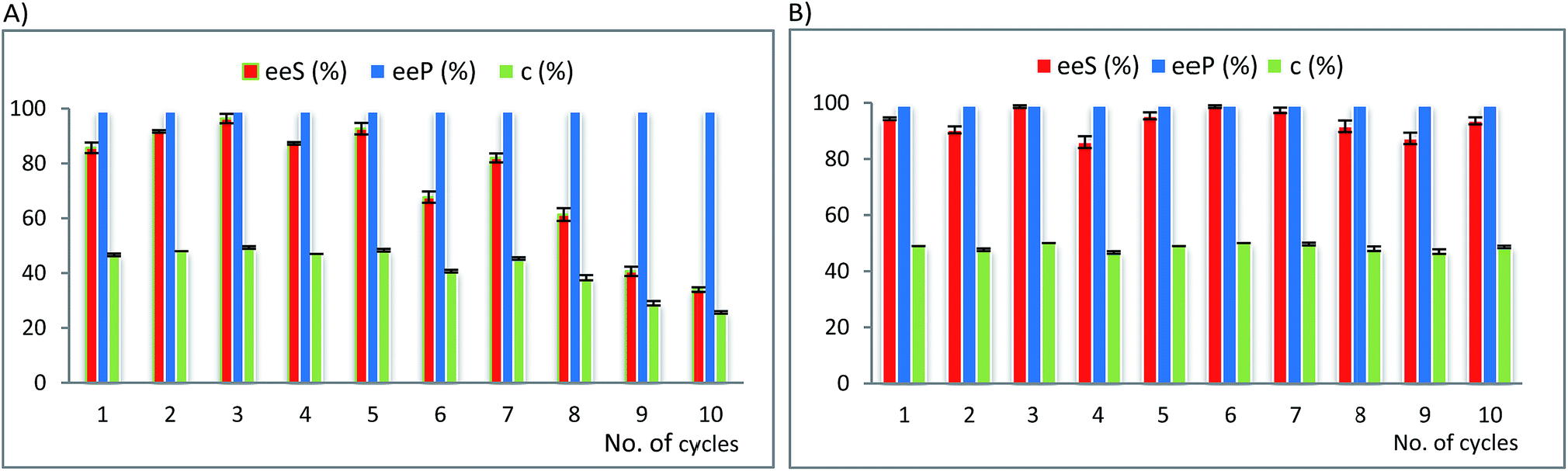
| Fig. 2 Multiple use efficiency of AK 1 BS as selected biocatalyst for the EKR of: (A) rac-1-(benzofuran-2-yl)ethan-1-ol rac-2c (10 mM) with vinyl acetate (3 equiv.) in n-hexane at 25 °C (after 20 hours reaction time) and (B) rac-1-(10-ethyl-10H-phenothiazin-3-yl)ethan-1-ol rac-2g (10 mM) with vinyl acetate (3 equiv.) in n-hexane at 25 °C (after 8 hours reaction time). Error bars represent standard deviations from average. | ||
 | ||
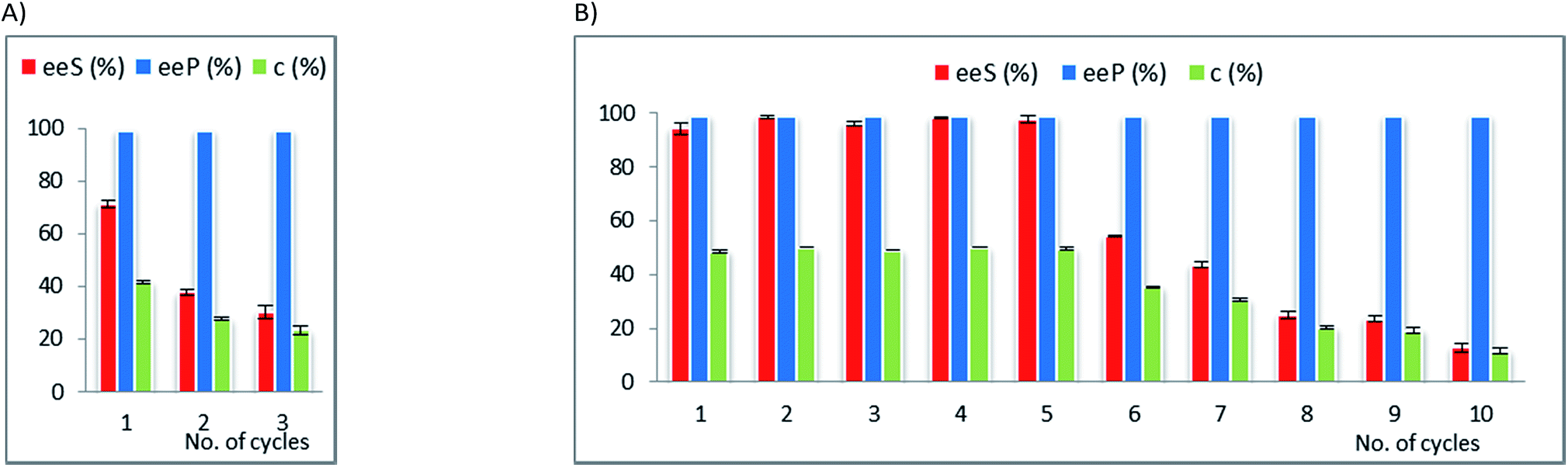
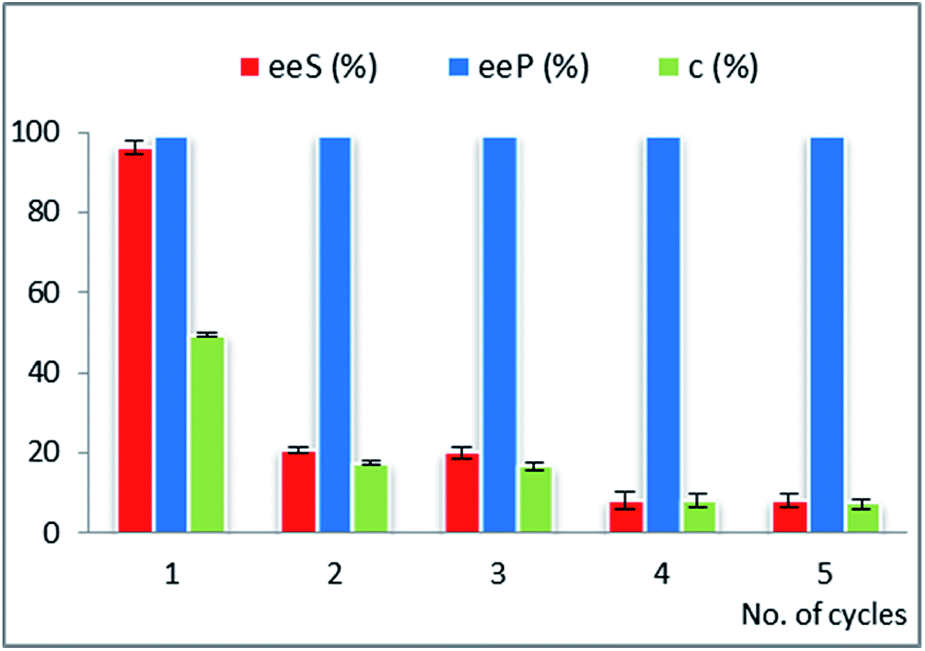
| Fig. 3 Multiple use efficiency of the selected biocatalyst CaL-B 1 TS* for the EKR of rac-1-(benzofuran-3-yl)ethan-1-ol rac-2d (10 mM) with vinyl acetate (3 equiv.) in n-hexane at 25 °C (after 20 hours reaction time). Error bars represent standard deviations from average. | ||
 | ||
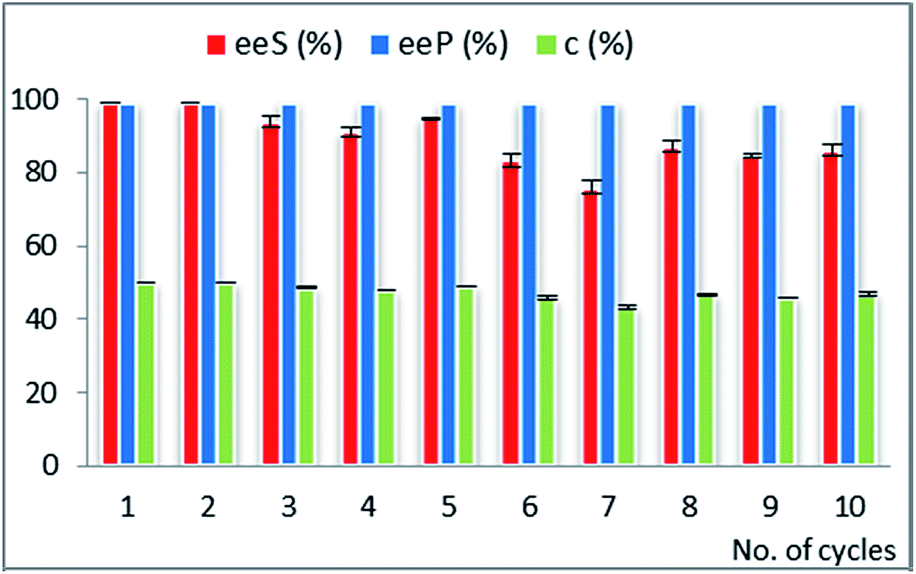
| Fig. 4 Multiple use efficiency of the selected biocatalyst CaL-B 2 TSC for the EKR of rac-1-(2-phenyl-thiazol-4-yl)-ethanol rac-2i (10 mM) with vinyl acetate (3 equiv.) in n-hexane at 25 °C (after 28 hours reaction time). Error bars represent standard deviations from average. | ||
Each reaction was repeated with the same enzyme preparate up to 10 times and has been allowed to proceed to ∼50% conversion, or as long as the preparate remained active and enantioselective, before the catalyst was subjected to the next cycle. Between cycles the catalyst was just washed with anhydrous n-hexane three times and thereafter immediately reused.
As presented in Fig. 1A and B, the immobilized lipases were efficient biocatalysts for the EKR of the benzothiophenic derivative rac-2a in ten consecutive reaction cycles (Tables 19S and 20S†). The adsorbed CaL-B was even more effective than the binary sol–gel preparate containing vinyl pending groups, but by extending the reaction time to more than one hour the entrapped biocatalyst was also able to give almost complete conversion of the substrate with high enantiomer purity.
For the EKR of rac-2c the most active immobilized form of AK was obtained by immobilization of this lipase in a binary sol–gel material as well, but containing octyl groups instead of vinyl, to supply the organic functionality. The recycling results (Fig. 2A) show a significant decrease of the conversion in only three reaction cycles (from 42 to 23% c, Table 21S in the ESI†), while the enantiopurity of the product remained high. The same biocatalyst was also subjected to recyclability studies in the acylation reaction of rac-2g. In this case the conversion started to drop only after 5 acylation cycles (Fig. 2B, Table 22S in ESI†). A rigorous comparative evaluation of this enzyme preparate performance is not possible, since the time needed to reach the maximum conversion strongly depends on the substrate structure (20 h for rac-2c and 8 h for rac-2g). These results confirm that the recycling capacity of a biocatalyst depends on the structure of the substrate.
Although rac-2d is a positional isomer of rac-2c, it was demonstrated to be a substrate with different behaviour in the EKR process. In this case, CaL-B lipase was the most efficient biocatalyst (not Amano AK, as for rac-2c) and the best immobilized enzyme showed high efficiency throughout multiple use (Fig. 3, Table 23S in ESI†).
The ternary sol–gel matrix was obtained from tetramethoxysilane as building block of the inorganic structure, with phenyl-trimethoxysilane and methyl-trimethoxysilane (at 4:1 relative molar ratio), supplying the pending organic functional groups of the hybrid material. The presence of relatively high phenyl group concentration was beneficial for the long-term catalytic efficiency of this biocatalyst, which did not show notable decline of conversion and enantioselectivity throughout 10 recycles. Although it is difficult to correlate the structure of the sol–gel matrix and the catalytic efficiency, the presence of bulky and mid-polar phenyl groups could have a specific influence on the structural organization and hydrophobicity of the formed silica network, resulting in improved performance, as it was also observed in our previous studies.15,40
The operational stability of a biocatalyst obtained by the immobilization method involving the formation of a sol prepolymer in acid catalysis (Method 2) was investigated for the EKR of the phenyl-thiazolyl derivative rac-2i.
As results from Fig. 4, this immobilized CaL-B lipase was not suitable for repeated use, although in the screening study demonstrated comparable catalytic efficiency and enantioselectivity with the best performing preparates obtained with the method using NaF as catalyst (Method 1). The most important drawback was the quick decline of the catalytic performance, as shown by the low conversion values (falling from 49 to 17% c in the second cycle, see Table 24S in ESI†). The stability of this type of sol–gel matrix is probably not appropriate for the employed reaction conditions.
Lipase-mediated acylation of racemic heteroaromatic ethanols in continuous-flow packed-bed microreactors
The best performing immobilized lipase preparates were also tested in continuous-flow experiments using packed-bed microreactors. From all CaL-B preparates, CaL-B 1 TS* was selected and from AK preparates, lipase AK 1 BS was selected. The two benzofuranyl-ethanols rac-2c,d were chosen as substrates (Fig. 5).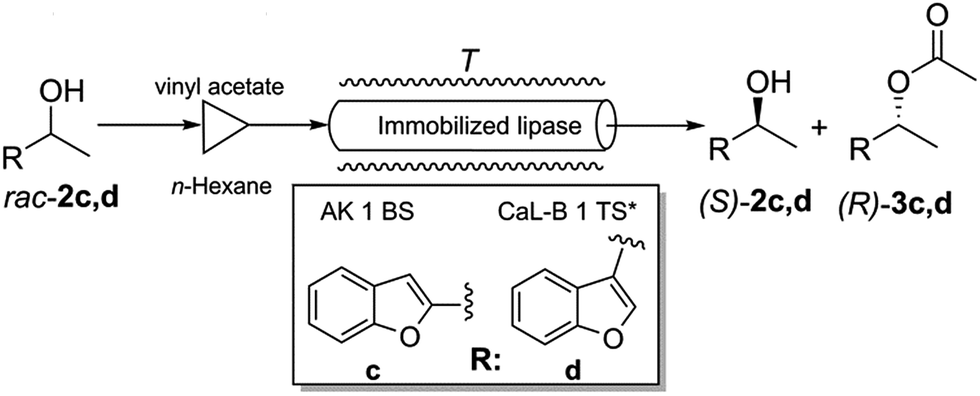 | ||
| Fig. 5 Continuous-flow lipase mediated kinetic resolution of racemic benzofuranyl-ethanols rac-2c,d. | ||
According to preliminary results of experiments performed in continuous-flow system, under identical reaction conditions, an increase in the amount of vinyl acetate for the acylation reaction of rac-2c (catalysed by AK 1 BS, in n-hexane, at 65 °C and 0.5 mL min−1, substrate concentration 10 mM) did not have a major impact on the conversion (20% c with 0.75 equiv. of vinyl acetate and 22% c with 3 equiv. of vinyl acetate), while for the acylation reaction of rac-2d (catalysed by CaL-B 1 TS*, in n-hexane, at 35 °C and 0.5 mL min−1, substrate concentration 10 mM) rising the amount of acyl donor led to a significant increase in conversion (8% c with 0.75 equiv. of vinyl acetate and 23% c with 3 equiv. of vinyl acetate). For this reason, in all further experiments involving rac-2c 0.75 equiv. of vinyl acetate were used and for rac-2d, 3 equiv. of the same acylating agent were employed.
Firstly, the continuous-flow acylation reaction was investigated at different temperatures (25–65 °C) using a 10 mM substrate solution at 0.5 mL min−1 flow rate (Fig. 6A). For both lipase preparates, an increase in conversion was observed by increasing the temperature, the maximum value being reached at 65 °C. Further increase of the reaction temperature was limited by the boiling point of the employed solvent. In the case of CaL-B 1 TS*-mediated acylation of rac-2d, almost a linear increase in conversion with respect to temperature was observed, while for AK 1 BS-catalyzed acylation almost a 10% increase in conversion was seen from 45 to 50 °C.
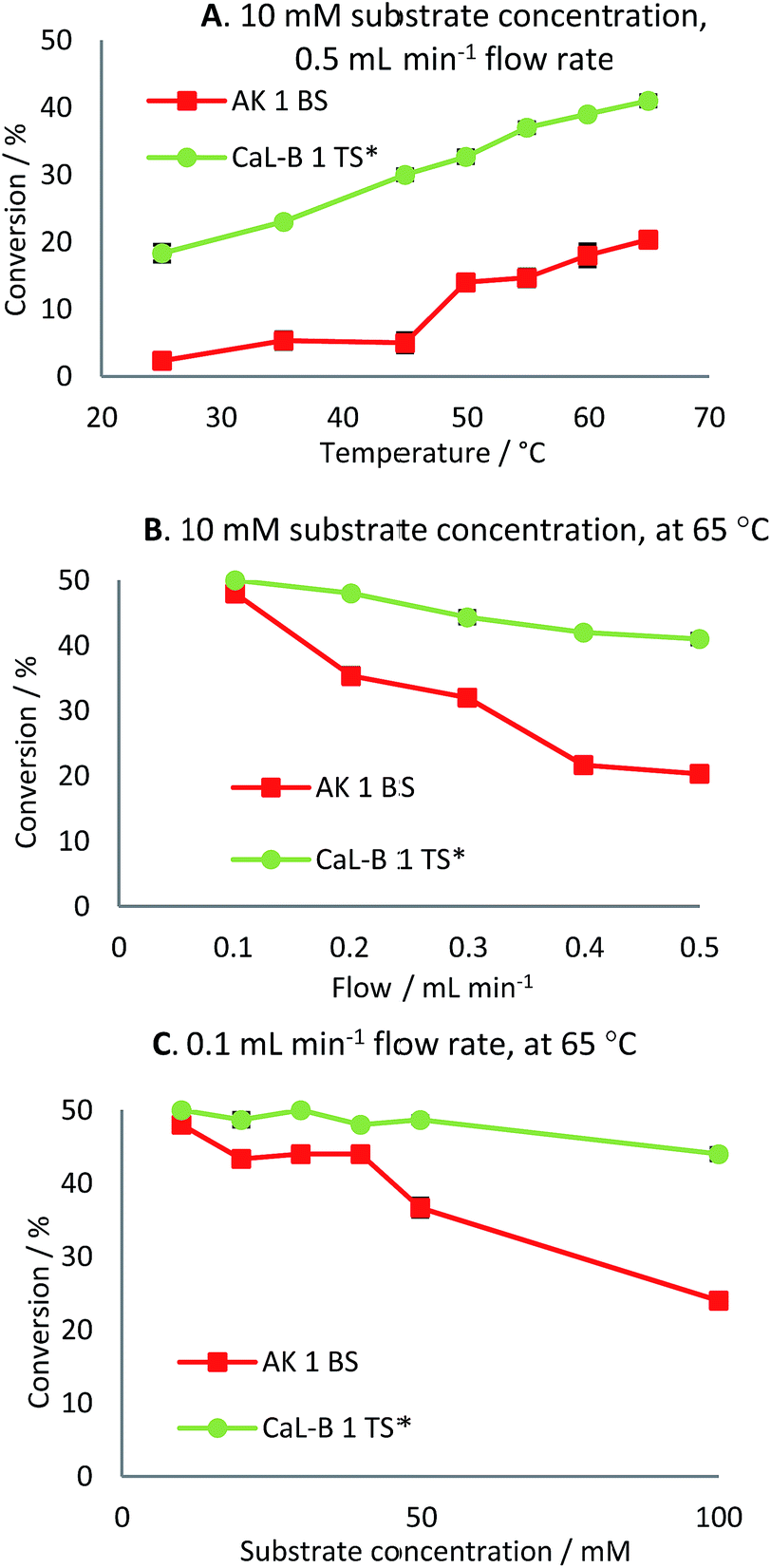 | ||
Fig. 6 Effect of temperature (A), flow rate (B) and substrate concentration (C) on the continuous-flow enzymatic acylation of rac-2c with AK 1 BS ( ) and rac-2d with CaL-B 1 TS*( ) and rac-2d with CaL-B 1 TS*( ) with vinyl acetate (0.75 and 3 equiv., respectively) in n-hexane. ) with vinyl acetate (0.75 and 3 equiv., respectively) in n-hexane. | ||
This experiment revealed that the immobilized lipase B from C. antarctica is more efficient towards rac-2d than is the immobilized lipase from Pseudomonas fluorescens towards rac-2c, as a two times higher conversion was reached at 65 °C (41% vs. 20%). The continuous-flow reactions proceeded with excellent enantioselectivities (E ≫ 200) throughout the investigated temperature domain (25–65 °C).
With the aim to improve the conversion of the enantioselective acylation of the investigated racemic ethanols rac-2c,d, the flow rate was gradually decreased from 0.5 to 0.1 mL min−1, maintaining the temperature at 65 °C, which led to almost complete conversion of the substrates (48% and 50%, Fig. 6B). However it must be taken into consideration that even though the conversion increases, the productivity of the continuous-flow system can decrease at lower flow rates. This however depends on the magnitude of conversion increase and flow rate decrease, since the rate of product formation is not a linear function on conversion.
Furthermore, in an effort to enhance the productivity of the continuous-flow packed-bed microreactor, different substrate concentrations were investigated at 65 °C at 0.1 (Fig. 6C) and 0.5 mL min−1 flow rate (Tables 4 and 5).
| Entry | Flow rate [mL min−1] | Residence timea [min] | Substrate concentration [mM] | Temperature [°C] | eeSb,c (%) | Productivityd [μmol min−1 g−1] |
|---|---|---|---|---|---|---|
a Calculated with the formula 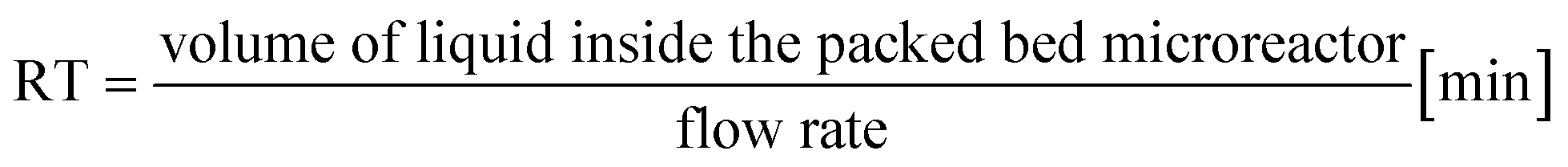 .b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of HPLC chromatograms.d Calculated using the average values of the conversions with the formula .b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of HPLC chromatograms.d Calculated using the average values of the conversions with the formula 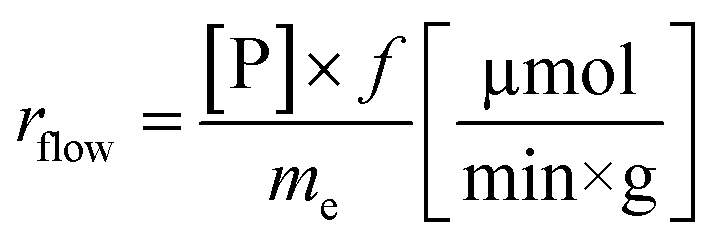 . . |
||||||
| 1 | 0.5 | 0.17 | 10 | 25 | 2 (±0.5) | 0.9 |
| 2 | 0.5 | 0.17 | 10 | 35 | 5 (±1.2) | 2.3 |
| 3 | 0.5 | 0.17 | 10 | 45 | 5 (±1.4) | 2.3 |
| 4 | 0.5 | 0.17 | 10 | 50 | 17 (±0.5) | 7.0 |
| 5 | 0.5 | 0.17 | 10 | 55 | 17 (±1.7) | 7.0 |
| 6 | 0.5 | 0.17 | 10 | 60 | 22 (±2.4) | 8.4 |
| 7 | 0.5 | 0.17 | 10 | 65 | 25 (±0.5) | 9.3 |
| 8 | 0.4 | 0.21 | 10 | 65 | 27 (±1.2) | 8.2 |
| 9 | 0.3 | 0.28 | 10 | 65 | 47 (±1.6) | 9.0 |
| 10 | 0.2 | 0.42 | 10 | 65 | 54 (±2.4) | 6.5 |
| 11 | 0.1 | 0.85 | 10 | 65 | 91 (±3.1) | 4.5 |
| 12 | 0.5 | 0.17 | 20 | 65 | 21 (±1.7) | 15.9 |
| 13 | 0.1 | 0.85 | 20 | 65 | 76 (±1.2) | 8.0 |
| 14 | 0.5 | 0.17 | 30 | 65 | 19 (±2.1) | 22.4 |
| 15 | 0.1 | 0.85 | 30 | 65 | 78 (±1.7) | 12.3 |
| 16 | 0.5 | 0.17 | 40 | 65 | 15 (±1.7) | 26.2 |
| 17 | 0.1 | 0.85 | 40 | 65 | 78 (±2.1) | 16.4 |
| 18 | 0.5 | 0.17 | 50 | 65 | 16 (±0.5) | 32.7 |
| 19 | 0.1 | 0.85 | 50 | 65 | 57 (±2.5) | 17.3 |
| 20 | 0.5 | 0.17 | 100 | 65 | 9 (±1.2) | 37.4 |
| 21 | 0.1 | 0.85 | 100 | 65 | 31 (±0.5) | 22.4 |
| Entry | Flow rate [mL min−1] | Residence timea [min] | Substrate concentration [mM] | Temperature [°C] | eeSb,c (%) | Productivityd [μmol min−1 g−1] |
|---|---|---|---|---|---|---|
a Calculated with the formula 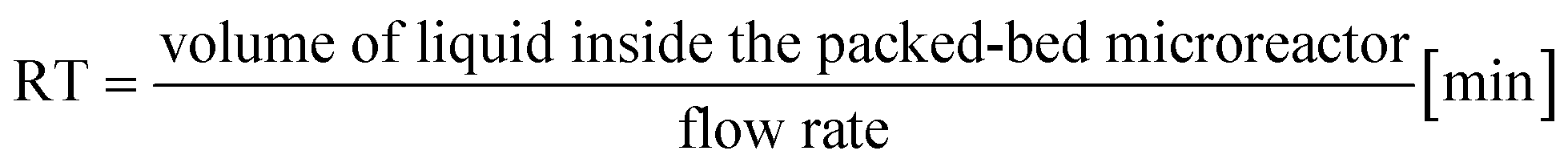 .b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of HPLC chromatograms.d Calculated using the average values of the conversions with the formula .b Experiments were performed in triplicate and standard deviations from average values are given in brackets.c Determined from peak areas of HPLC chromatograms.d Calculated using the average values of the conversions with the formula 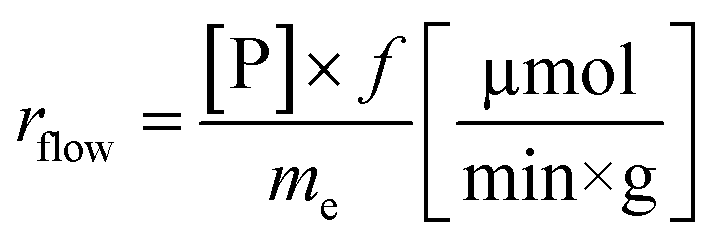 . . |
||||||
| 1 | 0.5 | 0.25 | 10 | 25 | 22 (±2.1) | 17.7 |
| 2 | 0.5 | 0.25 | 10 | 35 | 30 (±0.5) | 22.6 |
| 3 | 0.5 | 0.25 | 10 | 45 | 42 (±2.1) | 29.5 |
| 4 | 0.5 | 0.25 | 10 | 50 | 49 (±2.4) | 32.5 |
| 5 | 0.5 | 0.25 | 10 | 55 | 58 (±2.1) | 36.4 |
| 6 | 0.5 | 0.25 | 10 | 60 | 63 (±0.5) | 38.4 |
| 7 | 0.5 | 0.25 | 10 | 65 | 69 (±1.7) | 40.4 |
| 8 | 0.4 | 0.31 | 10 | 65 | 72 (±0.8) | 33.1 |
| 9 | 0.3 | 0.42 | 10 | 65 | 78 (±2.4) | 26.0 |
| 10 | 0.2 | 0.62 | 10 | 65 | 91 (±0.9) | 18.9 |
| 11 | 0.1 | 1.25 | 10 | 65 | >99 | 9.8 |
| 12 | 0.5 | 0.25 | 20 | 65 | 72 (±1.2) | 82.7 |
| 13 | 0.1 | 1.25 | 20 | 65 | 95 (±2.5) | 19.3 |
| 14 | 0.5 | 0.25 | 30 | 65 | 49 (±2.2) | 97.4 |
| 15 | 0.1 | 1.25 | 30 | 65 | >99 | 29.5 |
| 16 | 0.5 | 0.25 | 40 | 65 | 30 (±0.8) | 90.6 |
| 17 | 0.1 | 1.25 | 40 | 65 | 91 (±0.5) | 37.8 |
| 18 | 0.5 | 0.25 | 50 | 65 | 47 (±2.5) | 157.5 |
| 19 | 0.1 | 1.25 | 50 | 65 | 95 (±2.4) | 48.2 |
| 20 | 0.5 | 0.25 | 100 | 65 | 14 (±1.7) | 118.1 |
| 21 | 0.1 | 1.25 | 100 | 65 | 78 (±1.7) | 86.6 |
It was noted that for CaL-B 1 TS*-mediated transesterification of rac-2d, there was no significant decline in conversion in the range of 10–50 mM substrate concentration at 0.1 mL min−1.
Even when a 100 mM substrate concentration was employed, the conversion was still high (44%), proving the efficiency of this biocatalyst. In the case of the immobilized preparate AK 1 BS, the conversion of the acylation reaction of rac-2c decreased as the substrate concentration increased and at 100 mM the conversion was half of the initial value. At higher substrate concentrations (10–100 mM) the enantiomeric excesses of the products were maximum (>99%), affording excellent enantioselectivities of the continuous-flow acylation reactions.
The maximum productivity achieved by lipase AK 1 BS was 37.4 μmol of product min−1 g−1 at 65 °C, 0.5 mL min−1 flow rate and 100 mM substrate concentration (Table 4, entry 20), while the maximum productivity of CaL-B 1 TS* was 157.5 μmol of product min−1 g−1 at 65 °C, 0.5 mL min−1 flow rate and 50 mM substrate concentration (Table 5, entry 18).
The long term operational stability of the two selected biocatalysts was also studied using a 10 mM substrate concentration and 0.1 mL min−1 flow rate. The obtained results showed that the immobilized preparate AK 1 BS maintained 81% of its initial activity at 65 °C after 44 h of continuous operation (data not shown).
At 55 °C the enzyme exhibited remarkable stability as almost 95% of the initial activity was kept after 100 h. The other biocatalyst, CaL-B 1 TS* proved to be even more stable, the results showing practically no loss of activity after 100 h at 65 °C (Fig. 7). The enantioselectivities of both lipase-mediated continuous-flow reactions were not affected during long term operation of the biocatalysts.
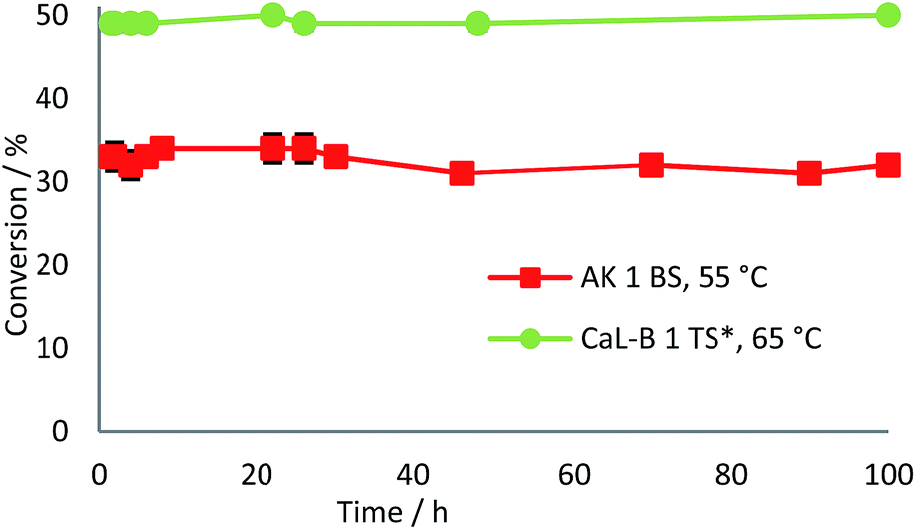 | ||
| Fig. 7 Long term operational stability of immobilized preparates AK 1 BS and CaL-B 1 TS* in the continuous-flow acylation reactions of rac-2c,d (10 mM) with vinyl acetate (0.75 and 3 equiv., respectively) in n-hexane at 55 °C and 65 °C, respectively and 0.1 mL min−1 flow rate. Error bars represent standard deviations from average. | ||
Another goal was to compare the productivities of the continuous-flow systems with those obtained in batch mode by calculating the specific reaction rates (eqn (1) and (2)):
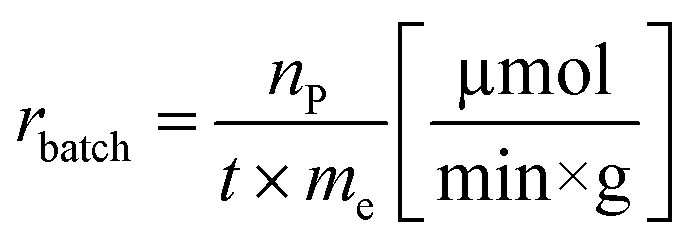 | (1) |
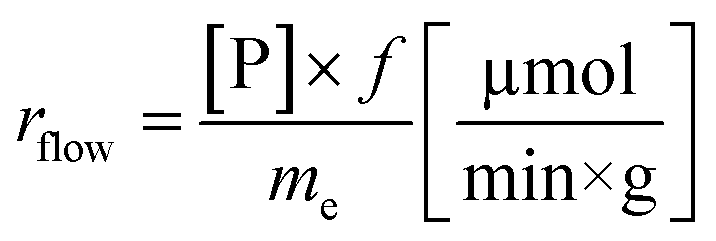 | (2) |
The specific reaction rate of a batch reaction (rbatch) can be calculated from the amount of product formed (np, μmol), the reaction time (t, min), and the mass of the applied enzyme (me, g). Similarly the specific reaction rate of a continuous-flow reaction (rflow) can be determined from the concentration of the product ([P], μmol mL−1), the flow rate (f, mL min−1), and the mass of the applied enzyme (me, g).
A rigorous comparison between these two can be made at the same degree of conversions, using identical experimental conditions (substrate and acyl donor concentrations, solvent, and temperature).41
In the case of AK 1 BS-catalyzed batch and continuous-flow reactions a significant difference in the specific reaction rates values was observed, this biocatalyst affording a 4 times higher productivity under continuous-flow operation (rbatch = 1.1 μmol min−1 g−1, rflow = 4.5 μmol min−1 g−1, at 48% conversion, 65 °C, 10 mM substrate concentration, 0.75 equiv. of vinyl acetate and 0.1 mL min−1 flow rate). On the other hand, CaL-B 1 TS* proved to be efficient in both systems, nevertheless, the specific reaction rate obtained in continuous-flow mode was almost 1.5 times higher than the one obtained in batch mode (rbatch = 6.7 μmol min−1 g−1, rflow = 9.8 μmol min−1 g−1, at 50% conversion, 65 °C, 10 mM substrate concentration, 3 equiv. of vinyl acetate and 0.1 mL min−1 flow rate).
Conclusion
The possibility to obtain tailored immobilized biocatalysts for the EKR of some heteroaromatic secondary alcohols through entrapement in various sol–gel matrix, starting from binary or ternary precursor mixtures, was demonstrated. Using the conversion c, enantiomeric excesses eeP and eeS and enantiomeric ratio E in order to characterize the enzymatic process efficiency, the optimal conditions (enzyme preparates and solvent) were found for each tested substrate.Moreover the operational stability in batch process was studied for five selected systems (consisting of one substrate and one biocatalyst). Despite the well-known advantages of sol–gel immobilization (enhanced thermal and long-term operational stability), the reusability of biocatalysts depends on the analyzed system, being high (more than 10 cycles in the case of rac-2a,d), average (5 cycles for rac-2g) or modest (approx. 55% enzymatic activity recovered after three cycles for rac-2c, 35% enzymatic activity recovered after two cycles for rac-2i).
Furthermore for the resolution of the racemic benzofuranyl-ethanols (rac-2c,d) the best performing biocatalysts were studied in continuous-flow mode at different temperatures, flow rates and substrate concentrations, using the productivity as criterion. The calculated specific reaction rates under continuous-flow operation (rflow) were in both cases higher than those obtained in batch mode (rbatch), 4.5 and 1.1 μmol min−1 g−1 for AK 1 BS preparate and 9.8 and 6.7 μmol min−1 g−1 for CaL-B 1 TS* preparate, respectively, demonstrating the advantages of this operating system. Moreover, these biocatalysts showed very good stability under long term continuous operation at high temperatures (65 °C). In conclusion the use of tailored sol–gel immobilized biocatalysts in packed-bed reactors under continuous flow regime represents a good strategy to perform lipase catalyzed kinetic resolutions of heteroaromatic ethanols and offers a good background for process development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant of the Romanian National Authority for Scientific Research, CNDI–UEFISCDI, project number PN-II-PT-PCCA-2013-4-0734.Notes and references
- J. M. Guisán, Immobilization of enzymes and cells, in Methods in biotechnology, Humana Press, New York, 2006, vol. 22 Search PubMed.
- L. Cao, Carrier-bound immobilized enzymes: principles, applications and design, Wiley-VCH, Weinheim, 2005 Search PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223 RSC.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 3, 8 Search PubMed.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33(5), 435 CrossRef CAS PubMed.
- M. L. E. Gutarra, L. S. M. Miranda and R. O. M. A. de Souza, Enzyme Immobilization for Organic Synthesis, in Organic Synthesis Using Biocatalysis, ed. A. Goswami and J. D. Stewart, Academic Press, Amsterdam, 2016, ch. 4, pp. 99–126 Search PubMed.
- V. V. Pace, J. V. Sinisterra and A. R. Alcántara, Curr. Org. Chem., 2010, 14, 2384 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Angew. Chem., Int. Ed. Engl., 1995, 34, 301 CrossRef CAS.
- M. T. Reetz, A. Zonta and J. Simpelkamp, Biotechnol. Bioeng., 1996, 49, 527 CrossRef CAS PubMed.
- E. Magner, Chem. Soc. Rev., 2013, 42, 6213 RSC.
- M. T. Reetz, P. Tielmann, W. Wiesenhofer, W. Konen and A. Zonta, Adv. Synth. Catal., 2003, 345, 717 CrossRef CAS.
- P. Tielmann, H. Kierkels, A. Zonta, A. Ilie and M. T. Reetz, Nanoscale, 2014, 6, 6220 RSC.
- D. Weiser, F. Nagy, G. Bánóczi, M. Oláh, A. Farkas, A. Szilágyi, K. László, A. Gellért, G. Marosi, S. Kemény and L. Poppe, Green Chem., 2017, 19(16), 3927 RSC.
- P. Dwivedee, J. Bhaumik, S. K. Rai, J. K. Laha and U. C. Banerjee, Bioresour. Technol., 2017, 239, 464 CrossRef PubMed.
- P. Adlercreutz, Chem. Soc. Rev., 2013, 42, 6406 RSC.
- V. M. M. Silva, J. Bassut, I. Ivaldo Jr, S. P. de Souza, M. L. G. Estrada, L. S. M. Miranda and R. O. M. A. de Souza, RSC Adv., 2015, 5, 102409 RSC.
- I. Eş, J. D. G. Vieira and A. C. Amaral, Appl. Microbiol. Biotechnol., 2015, 99(5), 2065 CrossRef PubMed.
- I. Itabaiana Jr, L. S. M. Miranda and R. O. M. A. de Souza, J. Mol. Catal. B: Enzym., 2013, 85–86, 1 CrossRef.
- M. P. Kamble and G. D. Yadav, Ind. Eng. Chem. Res., 2017, 56(7), 1750 CrossRef CAS.
- A. Radadiya and A. Shah, Eur. J. Med. Chem., 2015, 97, 356 CrossRef CAS PubMed.
- S. G. Dastidar, J. E. Kristiansen, J. Molnar and L. Amaral, Antibiotics, 2013, 2, 58 CrossRef CAS PubMed.
- J. Konstantinović, M. Videnović, J. Srbljanović, O. Djurković-Djaković, K. Bogojević, R. Sciotti and B. Šolaja, Molecules, 2017, 22, 343 CrossRef PubMed.
- M. T. Chhabria, S. Patel, P. Modi and P. S. Brahmkshatriya, Curr. Top. Med. Chem., 2016, 16(26), 2841 CrossRef CAS PubMed.
- A. Cimporescu, A. Todea, V. Badea, C. Paul and F. Péter, Process Biochem., 2016, 51, 2076 CrossRef CAS.
- A. Ursoiu, C. Paul, T. Kurtán and F. Péter, Molecules, 2012, 17, 13045 CrossRef CAS PubMed.
- A. Tomin, D. Weiser, G. Hellner, Z. Bata, L. Corici and F. Péter, Process Biochem., 2011, 46, 52 CrossRef CAS.
- C. Paizs, M. I. Toşa, V. Bódai, G. Szakács, I. Kmecz, B. Simándi, C. Majdik, L. Novák, F. D. Irimie and L. Poppe, Tetrahedron: Asymmetry, 2003, 14, 1943 CrossRef CAS.
- M. I. Toşa, S. Pilbák, P. Moldovan, C. Paizs, G. Szatzker, G. Szakács, L. Novák, F. D. Irimie and L. Poppe, Tetrahedron: Asymmetry, 2008, 19, 1844 CrossRef.
- J. Brem, M. I. Toşa, C. Paizs, A. Munceanu, D. Matković-Čalogović and F. D. Irimie, Tetrahedron: Asymmetry, 2010, 21, 1993 CrossRef CAS.
- J. Brem, S. Pilbák, C. Paizs, G. Bánoczi, F. D. Irimie, M. I. Toşa and L. Poppe, Tetrahedron: Asymmetry, 2011, 22, 916 CrossRef CAS.
- D. Hapău, J. Brem, M. Moisă, M. I. Toşa, F. D. Irimie and V. Zaharia, J. Mol. Catal. B: Enzym., 2013, 94, 88 CrossRef.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294 CrossRef CAS.
- M. Ungurean, C. Paul and F. Péter, Bioprocess Biosyst. Eng., 2013, 36, 1327 CrossRef CAS PubMed.
- M. Paravidino and U. Hanefeld, Green Chem., 2011, 13, 2651 RSC.
- E. Abaházi, Z. Boros and L. Poppe, Molecules, 2014, 19, 9818 CrossRef PubMed.
- J. Brem, M. C. Turcu, C. Paizs, K. Lundell, M. I. Toşa, F. D. Irimie and L. T. Kanerva, Process Biochem., 2012, 47, 119 CrossRef CAS.
- P. Tielmann, H. Kierkels, A. Zonta, A. Ilie and M. T. Reetz, Nanoscale, 2014, 6, 6220 RSC.
- C. Paul, P. Borza, A. Marcu, G. Rusu, M. Bîrdeanu, S. Marc Zarcula and F. Peter, Nanomater. Nanotechnol., 2016, 6, 3, DOI:10.5772/62194.
- C. Csajági, G. Szatzker, E. R. Tőke, L. Ürge, F. Darvas and L. Poppe, Tetrahedron: Asymmetry, 2008, 19, 237 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for lipases immobilization, chemical synthesis of racemic compounds, chromatographic chiral separation conditions, lipase and solvents screening results, recyclability studies results. See DOI: 10.1039/c7ra10157k |
| This journal is © The Royal Society of Chemistry 2017 |