
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrones and nucleobase-containing spiro-isoxazolidines derived from isatin and indanone: solvent-free microwave-assisted stereoselective synthesis and theoretical calculations†
Loredana Maiuolo *a,
Pedro Merinob,
Vincenzo Algieria,
Monica Nardiac,
Maria Luisa Di Gioiad,
Beatrice Russoa,
Ignacio Delsoe,
Matteo Antonio Tallaridaa and
Antonio De Nino*a
*a,
Pedro Merinob,
Vincenzo Algieria,
Monica Nardiac,
Maria Luisa Di Gioiad,
Beatrice Russoa,
Ignacio Delsoe,
Matteo Antonio Tallaridaa and
Antonio De Nino*a
aDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Via P. Bucci, cubo 12C, 87036 Rende, CS, Italy. E-mail: maiuolo@unical.it; denino@unical.it
bInstitute of Biocomputation and Physics of Complex Systems (BIFI), Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain
cDipartimento di Agraria, Università Telematica San Raffaele, Via di Val Cannuta, 247, Roma, 00166, Italy
dDipartimento di Farmacia e Scienze della Salute e della Nutrizione, Università della Calabria, Edificio Polifunzionale, 87036 Rende, CS, Italy
eServicio de Resonancia Magnética Nuclear, CEQMA, Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain
First published on 17th October 2017
Abstract
The spiro-oxindoles have found wide application because of their antiviral properties. However, in the literature few examples of synthesis of their precursors, oxindole-nitrones, are reported. In this paper, we initially present a rapid and efficient synthetic approach to ketonitrones by solvent-free microwave-assisted reaction between isatin or indanone derivatives and various hydroxylamines. The synthetic protocol is facile, clean, fast, high-yielding and stereoselective. Then, we explored the possibility to synthesize nucleobase-containing spiro-isoxazolidines with isatin and indanone nuclei by solvent-free MW-assisted 1,3-dipolar cycloaddition, obtaining good results in yields (74–85%), and regio- and diastereoselectivity. Theoretical calculations were done to analyze the difference of reactivity of isatin and indanone derivatives with hydroxylamines.
Introduction
The spiro-oxindole scaffold is the characteristic structural core of numerous alkaloids and unnatural biologically active compounds.1 Inspired by this important moiety, a number of methods to synthesize isatin and oxindole derivatives were realized during the last few years. In fact, the biological importance of compounds containing spiro-carbon at the C-3 position of the indoline or oxindole skeleton has recently been emphasized in the literature.2,3 In particular, spiro-oxindoles and spiro-isoxazolidines possess antiviral activity for various human diseases, inhibiting poxvirus,4 ectromelia,5 rhinovirus,6 HIV-1 (ref. 7) and, more recently, as inhibitors of MDM2/p53 interaction.8,9The 1,3-dipolar cycloaddition between a ketonitrone and a vinylic substrate may be considered a common procedure for the preparation of these compounds.10–12 Although a broad range of methodologies to synthesis of aldonitrones is available today,13–20 in contrast the preparation of ketonitrones is not always accomplished by simple condensation reaction.21–23 A thorough search of the relevant literature yielded only few articles related to synthetic procedures of isatin and oxindole nitrone.24,25 In particular, a methodology regards the preparation of N-substituted isatin nitrones via a multi-step reaction sequence.26
Recently, the synthesis of (Z)-N-aryl oxindole nitrones was performed through a N-arylation of 3-(hydroxyimino) indolin-2-ones with diaryliodonium salts, demonstrating the current interest in having efficient routes towards those sort of nitrones.27 On the contrary, to our knowledge, methodologies of indanone nitrones are not present in literature.
As a result, the development of a simple and convenient method for the synthesis of isatinyl and indanyl nitrones realized from ketones and hydroxylamines through mild conditions could be highly desirable.
In recent years, the use of microwave technology in organic chemistry, allows to prepare organic compounds very fastly, with high purity and better yield respect to other conventional methods.28,29
Recently, we described a green approach to synthesize aldo- and ketonitrones by solvent-free condensation of alkyl- or arylhydroxylamines hydrochlorides with aromatic aldehydes and ketones under microwave irradiation, bypassing the critical results obtained with ketonitrones.30
Then, in present work, we firstly describe a facile synthesis of isatinyl and indanyl nitrones via microwave-assisted reaction between ketones and hydroxylamines under solvent-free conditions. In our approach, we observed very short reaction times and a reduced formation of by-products, isolating the expected products in high yields and excellent stereoselectivity. Successively, we report for the first time a regio- and diastereomeric synthesis of nucleobase-containing spiro-isoxazolidines with an indoline or an indane ring by MW-assisted 1,3-dipolar cycloaddition, obtained in yields between 74–85% and via solvent-free procedure.
Results and discussion
To find the optimal conditions for the synthesis of nitrones, we chose N-methylhydroxylamine hydrochloride 1a and 1-isatin 2a and indanone 3a as ketones. The methodology consists of the co-grinding of the ketone and hydroxylamine in a mortar, followed by transfer of the mixture in a sealed vessel and further mixing by a vortex; finally the mixture is placed in a microwave oven. The optimization study is collected in Table 1.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ketoneb | MW (W) | T (°C) | Time (min) | Product | Yield (%) |
a Reaction conditions: 2.0 eq. of 1a were used unless otherwise indicated.b 1.0 eq. was used.c 1.0 eq. of 1a was used.d 1.0 eq. of NaOAc was added.e 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOH/water was used as a solvent and 2.0 eq. of NaOAc were added. :1 EtOH/water was used as a solvent and 2.0 eq. of NaOAc were added. |
||||||
| 1c | 2a | 200 | 180 | 10 | 4a | 33 |
| 2c | 2a | 400 | 180 | 10 | 4a | 42 |
| 3 | 2a | 400 | 180 | 10 | 4a | 53 |
| 4 | 2a | 400 | 180 | 20 | 4a | 61 |
| 5 | 2a | 600 | 180 | 10 | 4a | 97 |
| 6d | 2a | 600 | 180 | 10 | 4a | 80 |
| 7e | 2a | — | rt | 1440 | 4a | 70 |
| 8 | 3a | 600 | 180 | 10 | 5a | 37 |
| 9 | 3a | 600 | 180 | 15 | 5a | 51 |
| 10 | 3a | 400 | 180 | 10 | 5a | 25 |
| 11 | 3a | 400 | 180 | 30 | 5a | 82 |
| 12d | 3a | — | rt | 1440 | 5a | 78 |
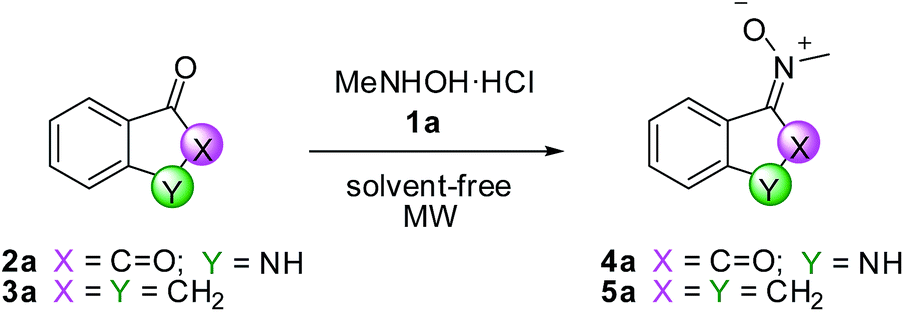
The best conditions for both ketones (entries 5 and 11) were those corresponding to an irradiation of 600 W (T = 180 °C) for 10 min for ketone 2a and an irradiation of 400 W (T = 180 °C) for 30 min for ketone 3a. The corresponding products 4a and 5a were obtained in 97% and 82% chemical yield, respectively. In both cases, nitrones were obtained as single isomers, E-4a and Z-5a respectively, as confirmed by NOESY experiments. When the reaction was carried out in EtOH/H2O as a solvent and in the presence of 2 equivalent of sodium acetate as reported in literature on similar substrates,31 compounds 4a and 5a were obtained in 70% and 78% chemical yield, respectively, in 24 hours (entries 7 and 12). Moreover, the presence of sodium acetate in reaction mixture does not significantly change the trend of the reaction (entry 6) and therefore we chose not to use it.
In an effort to expand the scope of the reaction, we explored coupling of substituted ketones with N-methyl hydroxylamine hydrochloride (1a) N-benzyl hydroxylamine hydrochloride (1b),32 N-phenyl hydroxylamine (1c),33 N-3-chloro-phenyl hydroxylamine (1d) and N-t-butylhydroxylamine hydrochloride (1e) (Table 2).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Hydroxylamine | R | Ketone | X | Y | R1 | Time (min) | Product | Yield (%) |
| a Ketone/phenylhydroxylamine ratio 1:3. |
|||||||||
| 1 | 1b | Bn | 2a | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
NH | H | 12 | 4b | 95 |
| 2 | 1c | Pha | 2a | CO |
NH | H | 10 | 4c | 92 |
| 3 | 1d | 3-Cl-Ph | 2a | CO |
NH | H | 10 | 4d | 87 |
| 4 | 1a | Me | 2b | CO |
NMe | H | 10 | 4e | 90 |
| 5 | 1b | Bn | 2b | CO |
NMe | H | 12 | 4f | 92 |
| 6 | 1c | Pha | 2b | CO |
NMe | H | 10 | 4g | 90 |
| 7 | 1d | 3-Cl-Ph | 2b | CO |
NMe | H | 10 | 4h | 88 |
| 8 | 1a | Me | 2c | CO |
NH | 5-Br | 10 | 4i | 95 |
| 9 | 1b | Bn | 2c | CO |
NH | 5-Br | 10 | 4j | 93 |
| 10 | 1c | Pha | 2c | CO |
NH | 5-Br | 10 | 4k | 92 |
| 11 | 1a | Me | 2d | CO |
NH | 5-NO2 | 10 | 4l | 95 |
| 12 | 1b | Bn | 2d | CO |
NH | 5-NO2 | 13 | 4m | 92 |
| 13 | 1c | Pha | 2d | CO |
NH | 5-NO2 | 10 | 4n | 92 |
| 14 | 1a | Me | 2e | CO |
NH | 5,7-Cl | 25 | 4o | 80 |
| 15 | 1e | t-Bu | 2a | CO |
NH | H | 60 | 4p | — |
| 16 | 1b | Bn | 3a | CH2 | CH2 | H | 30 | 5b | 85 |
| 17 | 1a | Me | 3b | CHMe | CH2 | H | 30 | 5c | 61 |
| 18 | 1a | Me | 3c | CH2 | CHMe | H | 35 | 5d | 78 |
| 19 | 1a | Me | 3d | CHnPr | CH2 | H | 32 | 5e | 20 |
| 20 | 1a | Me | 3e | CH2 | CH2 | 5-Br | 30 | 5f | 88 |
| 21 | 1a | Me | 3f | CH2 | CH2 | 5-F | 28 | 5g | 83 |
| 22 | 1a | Me | 3g | CH2 | CH2 | 4-Br-7-OH | 60 | 5h | 67 |
| 23 | 1b | Bn | 3g | CH2 | CH2 | 4-Br-7-OH | 48 | 5i | 78 |
| 24 | 1e | t-Bu | 3a | CH2 | CH2 | H | 60 | 5j | — |
| 25 | 1a | Me | 3h | CH2 | CH2CH2 | H | 50 | 5k | Traces |
| 26 | 1b | Bn | 3h | CH2 | CH2CH2 | H | 50 | 5l | Traces |
| 27 | 1a | Me | 3i | CH2 | CH2CH2 | 7-F | 45 | 5m | Traces |
| 28 | 1a | Me | 3j | CH2 | CH2CH2 | 6-OMe | 55 | 5n | — |
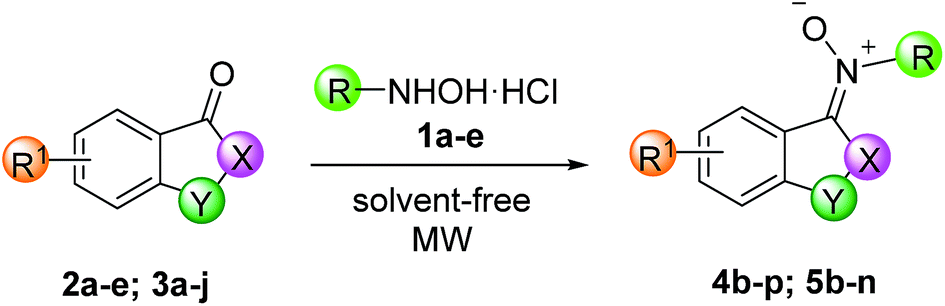
The reaction works well with various isatin ketones 2 and we noticed that the presence of an electron-withdrawing group (Br or NO2) on C-5 of phenyl ring of isatin does not significantly alters their performance (entries 8–13). Methylation of nitrogen does not reduce the reactivity (entries 4–7). Phenylhydroxylamine 1c was used in further excess (ratio ketone/phenylhydroxylamine 1:3) because of its known degradability at temperature higher than 50 °C.34 Attempts to reduce by-products by lowering power of MW (200 or 400 W) or by reducing the maxim temperature were unnecessary. In all cases the E-isomer was the only obtained with the exceptions of compounds 4d and 4h for which small quantities of Z isomers (ratio E/Z: 78:22 and 75:25 for 4d and 4h, respectively) were obtained. In general, ketones 3 present lower reactivity with respect to isatin ketones 2. In all cases, the Z-isomer was the only obtained. When an alkyl group is present on the 2-C or 3-C of the carbon backbone of indanone ring (entries 17 and 18), the yield drops presumably due to a minor electrophilicity of carbonyl group. The propyl group introduces additional steric hindrance, and the yield drops to 20% (entry 19).
The presence of Br or F in C-5 of indanone ring (entries 20 and 21) does not produce significant differences of reactivity respect to unsubstituted indanone. On the other hand, the reaction is very sensitive to the size of the cycloalkyl ring (tetralone versus indanone). Tetralone derivatives showed very little reactivity and longer reaction times than other substrates (entries 25–27) are required for obtaining just traces of products.
Moreover, resonance effect in C-4 or in C-7 of isatine and indanone ring (entries 14, 22 and 23) causes major reaction times and/or minor yields because the electrophilicity of the acceptor carbonyl group is reduced. Unfortunately, the large size of t-butyl group of 1e dramatically influences both reactions of isatin 2a and indanone 3a, not leading to the formation of any product (entries 15 and 24). Attempts to carry out the reactions using the conditions previously applied in entries 7 and 12 of Table 1 confirmed the reaction trend, revealing that these reactions are sensitive to steric effects.
As a follow-up work, some nucleobase-containing spiro-isoxazolidines with an indoline or an indane ring were synthesized by MW-assisted 1,3-dipolar cycloaddition between nitrones as dipole and vinylnucleobases as dipolarophile. It is generally been recognized that the incorporating of different bioactive scaffolds into one molecule is the powerful strategy to construct substrates with structural novelty and biological potential.
Considering that, some spiroindoline–isoxazolidines have shown different degrees of anticancer activity mainly based on the isoxazolidine ring fused at C-3 position of oxindole backbone,35–37 we hypothesized that the simultaneous presence of an spiro carbon, an oxindole-like ring, an isoxazolidine portion and a nucleobase might generate novel molecular entities with amplified anticancer activity as, for example, inhibitors of MDM2/p53 interaction. Moreover, to our knowledge, in literature spiro-isoxazolidines with nucleobases and indoline or indane ring on their backbone have been never synthesized.
As is well known, the typical insertion of a nucleobase on a sugar or isoxazolidine scaffold consists of the method of Vorbrüggen,38 but for a long time now, we have developed a simple method by solvent-free MW-assisted 1,3-dipolar cycloaddition between nitrones as dipole and vinylnucleobases39 as dipolarophile.40
The formation reactions between N-1-vinylthymine 6a and nitrones 4e and 5a, respectively, were taken as examples for optimization of reaction conditions. The target conjugates were prepared following the synthetic procedure similar to ketonitrones previously prepared, consisting of the co-grinding of the ketonitrone and vinylnucleobase in a mortar, followed by transfer of the mixture in an appropriate vessel and further mixing of the solids in a vortex without use of solvents; finally the solid mixture was placed in a microwave oven. The results are summarized in Table 3.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Nitroneb | MW (W) | T (°C) | Solvent | Time (min) | Product | Yield (%) |
| a 1.0 eq. of 6a were used.b 2.0 eq. was used. | |||||||
| 1 | 4e | 650 | 180 | — | 30 | 7a | 59 |
| 2 | 4e | 750 | 180 | — | 20 | 7a | 62 |
| 3 | 4e | 850 | 180 | — | 10 | 7a | 85 |
| 4 | 4e | — | 110 | Toluene | 4320 | 7a | 25 |
| 5 | 5a | 650 | 180 | — | 10 | 8a | 22 |
| 6 | 5a | 650 | 80 | — | 30 | 8a | 34 |
| 7 | 5a | 650 | 125 | — | 30 | 8a | 38 |
| 8 | 5a | 750 | 80 | — | 10 | 8a | 41 |
| 9 | 5a | 750 | 100 | — | 30 | 8a | 61 |
| 10 | 5a | 750 | 125 | — | 30 | 8a | 77 |
| 11 | 5a | — | 110 | Toluene | 4320 | 8a | 20 |
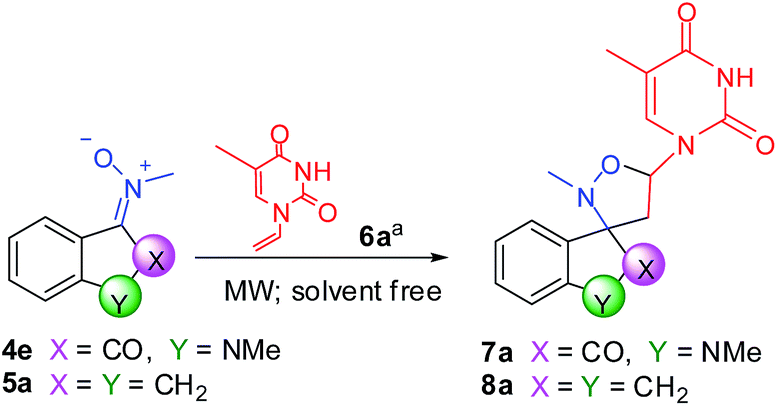
We prevalently changed the MW power that, based upon our experience, turns out to be the key parameter in MW-assisted cycloadditions, by keeping the nitrone/vinylnucleobase ratio to 2:1, respectively. The optimized conditions for the formation of 7a are listed in entry 3 (Table 3). On the contrary, the indanone nucleus obliged us to reduce maintained temperature (T = 125 °C) to avoid the formation of a lot quantity of by-products (entries 5–10, Table 3). Despite everything, reaction times were major respect to isatin derivatives and yields were good (entry 10, Table 3). Finally, we compared our results with those obtained from classical conditions of 1,3-dipolar cycloaddition, using toluene and reflux (entries 4 and 11, Table 3). It is possible to highlight that reaction times had to be extended to three days, obtaining very poor yields and many by-products.
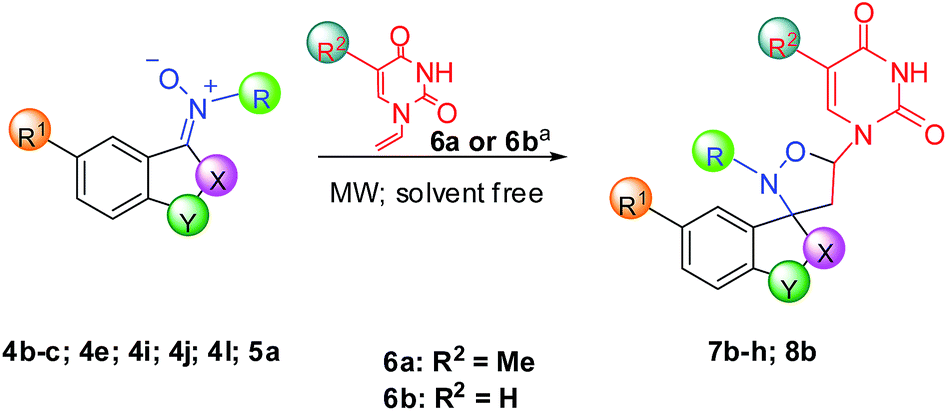
Then, the reaction conditions of 7a and 8a were expanded to the formation of some substituted nucleobase-containing spiro-isoxazolidines, using various our nitrones and some vinylnucleobases (N-1-vinylthymine 6a or N-1-vinyluracil 6b). The results are collected in Table 4. All cycloadducts were isolated in good yields and short reaction times (8–30 minutes). The effect of substituents on indoline ring was also investigated, observing slightly yields in presence of bromo or nitro group as substituents because of formation of by-products (entries 5–7, Table 4). All cycloaddition reactions were highly regioselective and goodly diasteroselective, furnishing only one regioisomer (5-substituted) and prevalent exo-adduct (Table 4), as confirmed through NOESY experiments. In particular, we observed a specific correlation between the proton on C-6 of thymine or uracil and the proton on C-4 of aromatic ring, only possible in the exo approach of cycloaddition.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Nitroneb,c | R | R1 | R2 | Time (min) | 7,8 | Yield (%) | exo/endo ratio |
| a 1.0 eq. of 6a or 6b were used.b 2.0 eq. was used.c X = CO; Y = NH unless otherwise indicated.d Y = NMe.e X = Y = CH2. | ||||||||
| 1 | 4b | Bn | H | H | 12 | 7b | 85 | 83:17 |
| 2 | 4c | Ph | H | H | 8 | 7c | 84 | 73:27 |
| 3 | 4c | Ph | H | Me | 8 | 7d | 85 | 72:28 |
| 4 | 4ed | Me | H | H | 10 | 7e | 84 | 82:18 |
| 5 | 4i | Me | Br | Me | 10 | 7f | 77 | 86:14 |
| 6 | 4j | Bn | Br | Me | 12 | 7g | 75 | 88:12 |
| 7 | 4l | Bn | NO2 | Me | 12 | 7h | 74 | 75:25 |
| 8 | 5ae | Me | H | H | 30 | 8b | 76 | 78:22 |
Theoretical calculations
In order to correlate the reactivity of ketones with hydroxylamines we evaluated the electrophilicity of both reagents by calculating ω reactivity index. The ω index establishes an absolute scale of electrophilicity in the sense that the hierarchy of electrophilicity is built up from the electronic structure of molecules. The electrophilicity hierarchy may be systematically rationalized on the basis of substituent effects. As a result, electron-withdrawing groups lead to electrophilic activation, and electron-releasing groups lead to electrophilic deactivation. Accordingly, by establishing a scale in which electrophiles and nucleophiles are present, couples of reagents (ketones and hydroxylamines in our case) presenting large differences of electrophilicity (i.e. Δω > 2.0) will be the more reactive.Calculations were carried out at b3lyp-d3bj/def2svp level of theory according to the procedure described elsewhere. We considered the experimentally studied ketones 2a–e and 3a–j but also ketones 2f–k, 3k–3p were calculated in order to predict an extended reactivity. The results are illustrated in Fig. 1.
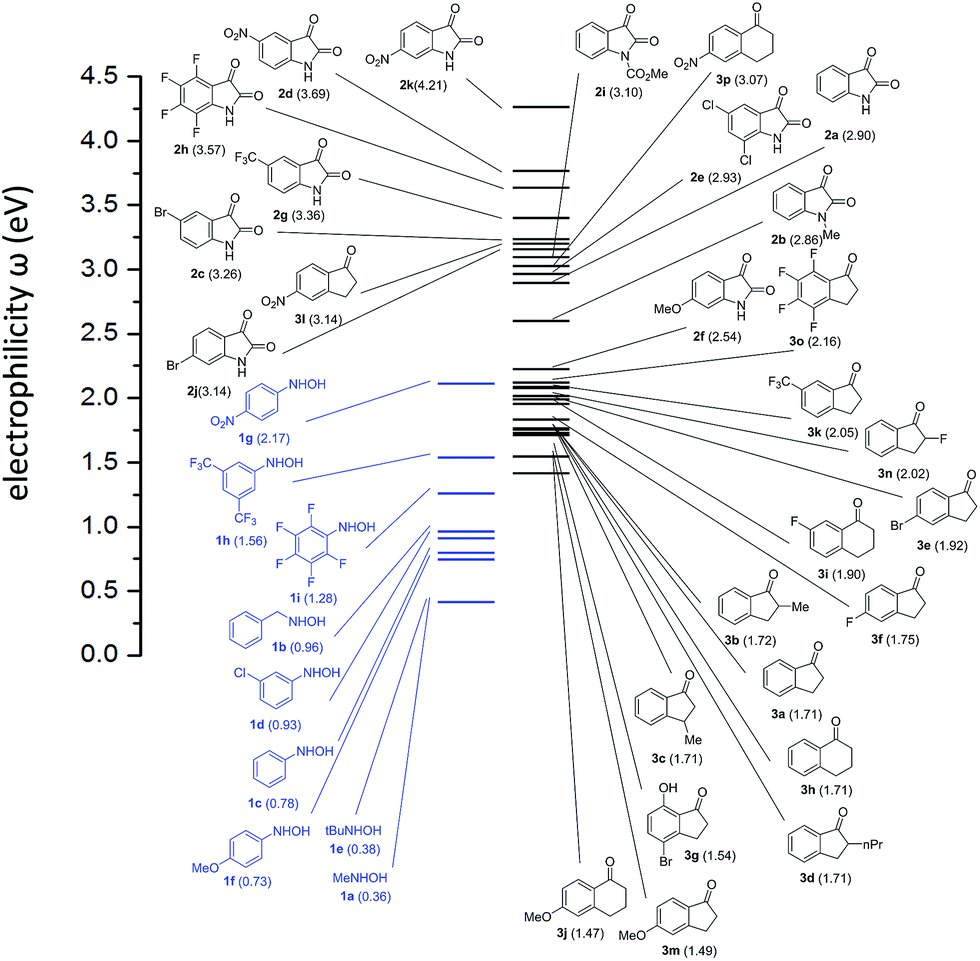 | ||
| Fig. 1 Electrophilicity (ω in eV) scale for ketones and hydroxylamines. | ||
Among the ketones the most electrophilic one is predicted to be 2d, which has been studied experimentally. Among the hydroxylamines the most nucleophilic one resulted 1a, also experimentally studied. Consequently, the reaction between 2d and 1a (Table 2, entry 11) is the most favoured as, indeed, is experimentally demonstrated (high yield and only 10 min of reaction). In general, isatin derivatives are the most reactive with values of Δω > 2 with some exception. The lower reactivity predicted (Δω < 2) for indanyl and tetralonyl derivatives is experimentally confirmed by the longer reaction times required and lower yields obtained. In fact, the absence of product obtained for the reaction between ketone 3j and hydroxylamine 1a correlates with a low difference in electrophilicity (Δω = 1.11).
Experimental
Commercial starting materials were used without further purification. Reactions were monitored by TLC using silica plates 60-F264, commercially available from Merk. 1H and 13C NMR spectra were recorded at 300, 400 and 500 MHz and 75, 100 and 125 MHz, respectively, in CDCl3 and DMSO-d6 using tetramethylsilane (TMS) as internal standard (Bruker ACP 300 MHz, 400 MHz and 500 MHz). Chemical shifts are given in parts per million and coupling constants in Hertz. The stereochemistry were established by NOESY experiments.LC-MS analysis were carried using an Agilent 6540 UHD Accurate - Mass Q-TOF LC-MS (Agilent, Santa Clara, CA) fitted with a electrospray ionisation source (Dual AJS ESI) operating in positive ion mode. Chromatographic separation was achieved using a C18 RP analytical column (Poroshell 120, SB-C18, 50 × 2.1 mm, 2.7 μm) at 30 °C with a elution gradient from 5% to 95% of B over 13 min., a being H2O (0.1% FA) and B CH3CN (0.1% FA). Flow rate was 0.4 mL min−1.
MW-assisted reactions were performed in Synthos 3000 instrument from Anton Paar, equipped with a 4 × 24MG5 rotor and an IR probe as external control of the temperature. 0.3–3 mL glass vials sealed with a dedicated PEEK screw-cup together with a reliable PTFE seal were used for all reactions. In synthesis of all derivatives the setting of temperature was always maintained to 180 °C in each experiment, except for 5a–i where temperature was always maintained to 125 °C.
General procedure for synthesis of nitrones 4a–o and 5a–i
The selected ketone (0.5 g) and appropriate hydroxylamine derivative (2 eq. or 3 eq. for N-phenylhydroxylamine) were grinded in a mortar, placed in apposite vessel and mixed in a vortex. The mixture was transferred to a microwave oven and was irradiated with the opportune power. After the appropriate time the crude oil was recrystallized with ethyl acetate (4a–o) or cyclohexane (5a–i).General procedure for synthesis of nucleobase-containing spiro-isoxazolidines 7a–h and 8a–b
The opportune nitrone (2 eq.) and N-1-vinylthymine 6a or N-1-vinyluracil 6b (0.05 g) were grinded in a mortar, placed in apposite vessel and mixed in a vortex. The mixture was transferred to a microwave oven and was irradiated with the opportune power and temperature (when required). After the appropriate time the crude oil was purified by flash chromatography with hexane:AcOEt 7.75:2.25 v/v for 7c–e or CHCl3/MeOH 9.75:0.25 v/v for all the other crudes.
Conclusions
In summary, we have initially developed an environmentally benign and fast method by microwave irradiation and solvent-free conditions for the synthesis of significant number of substituted isatinyl and indanyl nitrones as important precursors of spiro-isoxazolidines. Then, we applied a MW-assisted 1,3-dipolar cycloaddition to some previously synthesized ketonitrones as dipole and vinylthymine or vinyluracil as dipolarophile, obtaining nucleobase-containing spiro-isoxazolidines with indoline or indane ring in good yields, excellent regioisomery and high diastereoisomery. The importance of this application is surely the direct insertion of a nucleobase on spiroisoxazolidines with indoline or indane scaffold that in our opinion represents a great chemical novelty. Finally, we have also enriched this work, studying by theoretical calculations the effect of various substituents about the reactivity of the carbonyl groups with different hydroxylamines in synthesis of nitrones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Italian Ministry of University and Scientific Research (MIUR) for a doctoral grant and the University of Calabria for financial support. Thanks to the Ministerio de Ciencia e Innovación (MICINN) and FEDER Program (Madrid, Spain, project CTQ2016-76155-R) and the Gobierno de Aragón (Zaragoza, Spain. Bioorganic Chemistry Group. E-10). We acknowledge the Institute of Biocomputation and Physics of Complex Systems (BIFI) at the University of Zaragoza (Spain) for computer time at clusters.Notes and references
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed.
- D. Cheng, Y. Ishihara, C. F. Barbas III and B. Tan, ACS Catal., 2014, 4, 743 CrossRef CAS.
- B. Tan, X. Zeng, W. W. Y. Leong, Z. Shi, C. F. Barbas III and G. Zhong, Chem.– Eur. J., 2012, 18, 63 CrossRef CAS PubMed.
- M. C. Pirrung, S. V. Pansare, K. D. Sarma, K. A. Keith and E. R. Kern, J. Med. Chem., 2005, 48, 3045 CrossRef CAS PubMed.
- D. J. Bauer and P. W. Sadler, Nature, 1961, 190, 1167 CrossRef CAS PubMed.
- J. M. Z. Gladich, J. H. Hunt, D. Jack, R. F. Haff, J. J. Boyle, R. C. Stewart and R. J. Ferlauto, Nature, 1969, 221, 286 CrossRef.
- D. Sriram, T. R. Bal and P. Yogeeswari, J. Pharm. Pharm. Sci., 2005, 8, 565 CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432 CrossRef CAS PubMed.
- S. Wang, Y. Zhao, D. Bernard, A. Aguilar and S. Kumar, Top. Med. Chem., 2012, 8, 57 CAS.
- F. Shi, Z.-L. Tao, S.-W. Luo, S.-J. Tu and L.-Z. Gong, Chem.– Eur J., 2012, 18, 6885 CrossRef CAS PubMed.
- C.-S. Wang, R.-Y. Zhu, J. Zheng, F. Shi and S.-J. Tu, J. Org. Chem., 2015, 80, 512 CrossRef CAS PubMed.
- W. Dai, X.-L. Jiang, Q. Wu, F. Shi and S.-J. Tu, J. Org. Chem., 2015, 80, 5737 CrossRef CAS PubMed.
- C. Chavarría, D. I. Pérez, C. Pérez, J. Morales-García, S. Alonso-Gil, A. Pérez Castillo, C. Gil, J. Souza and W. Porcal, Eur. J. Med. Chem., 2012, 58, 44 CrossRef PubMed.
- V. Gautheron-Chapoulaud, S. U. Pandya, P. Cividino, G. Masson, S. Py and Y. Vallée, Synlett, 2001, 8, 1281 CrossRef.
- G. Masson, S. Py and Y. Valleé, Angew. Chem., Int. Ed., 2002, 41, 1772 CrossRef CAS PubMed.
- H. I. Petkes, E. Gal, L. Gaina, M. Sabou, C. Majdik and L. Silaghi-Dumitrescu, C. R. Chim., 2014, 17, 1050 CrossRef CAS.
- G. Soldaini, F. Cardona and A. Goti, Org. Lett., 2007, 9, 473 CrossRef CAS PubMed.
- S. Franco, F. L. Merchant, P. Merino and T. Tejero, Synth. Commun., 1995, 25, 2275 CrossRef CAS.
- Y. Tomioka, C. Nagahiro, Y. Nomura and H. Maruoka, J. Heterocycl. Chem., 2003, 40, 121 CrossRef CAS.
- R. Y. Suman, P. Kadigachalam, V. R. Doddi and Y. D. Vankar, Tetrahedron Lett., 2009, 50, 5827 CrossRef.
- H. G. Aurich and W. Weiss, Tetrahedron, 1976, 32, 159 CrossRef CAS.
- K. Torssell and O. Zeuthen, Acta Chem. Scand., Ser. A, 1978, 32, 118 CrossRef.
- H. Valizadeh, Heteroat. Chem., 2010, 21, 78 CrossRef CAS.
- H.-B. Yang and S. Min, Org. Biomol. Chem., 2012, 10, 8236 CAS.
- Y.-H. Zhang, M.-Y. Wu and W.-C. Huang, RSC Adv., 2015, 5, 105825 RSC.
- I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 26, 3023 CrossRef PubMed.
- S.-Y. Wu, X.-P. Ma, C. Liang and D.-L. Mo, J. Org. Chem., 2017, 3232 CrossRef CAS PubMed.
- A. Loupy, in Microwave in Organic Synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed.
- J. P. Tieney and P. Lidstrom, in Microwave Assisted Organic Chemistry, Blackwell Publishing, Oxford, 2005 Search PubMed.
- L. Maiuolo, A. De Nino, P. Merino, B. Russo, G. Stabile, M. Nardi, N. D'Agostino and T. Bernardi, Arabian J. Chem., 2016, 9, 25 CrossRef CAS.
- F. M. Murphy and M. J. Meegan, J. Chem. Res., Miniprint, 1996, 301 Search PubMed.
- T. B. Nguyen, A. Martel, R. Dhal and G. Dujardin, Synthesis, 2009, 18, 3174 Search PubMed.
- B. S. Furniss; A. J. Hannaford; P. W. G. Smith and A. R. Tatchell, in Vogel's, Textbook of Practical Organic Chemistry, Pearson Education Limited, 1989 Search PubMed.
- C. S. Marvel and O. Kamm, J. Am. Chem. Soc., 1919, 41, 276 CrossRef CAS.
- J. Khazir, P. P. Singh, D. M. Reddy, I. Hyder, S. Shafi, S. D. Sawant, G. Chashoo, A. Mahajan, M. S. Alam, A. K. Saxena, S. Arvinda, B. D. Gupta and H. M. S. Kumar, Eur. J. Med. Chem., 2013, 63, 279 CrossRef CAS PubMed.
- Raunak, V. Kumar, S. Mukherjee, Poonam, A. K. Prasad, C. E. Olsen, S. J. C. Schäffer, S. K. Sharma, A. C. Watterson, W. Errington and V. S. Parmar, Tetrahedron, 2005, 61, 5687 CrossRef CAS.
- S. Malhotra, S. Balwani, A. Dhawan, B. K. Singh, S. Kumar, R. Thimmulappa, S. Biswal, C. E. Olsen, E. Van der Eycken, A. K. Prasad, B. Ghosh and V. S. Parmar, Med. Chem. Commun., 2011, 2, 743 RSC.
- J. M. Vorbrüggen, K. Kvolikiewicz and B. Bennua, Chem. Ber., 1981, 114, 1234–1255 CrossRef.
- R. Dalpozzo, A. De Nino, L. Maiuolo, A. Procopio, R. Romeo and G. Sindona, Synthesis, 2002, 2, 172–174 Search PubMed.
- O. Bortolini, A. De Nino, T. Eliseo, R. Gavioli, L. Maiuolo, B. Russo and F. Sforza, Bioorg. Med. Chem., 2010, 18, 6970 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09995a |
| This journal is © The Royal Society of Chemistry 2017 |