
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-active copper triangles as an enzymatic molecular flask for light-driven hydrogen production†
Liang Zhao ,
Jianwei Wei,
Feili Zhang,
Cheng He,
Sijia Zheng and
Chunying Duan*
,
Jianwei Wei,
Feili Zhang,
Cheng He,
Sijia Zheng and
Chunying Duan*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: cyduan@dlut.edu.cn
First published on 18th October 2017
Abstract
A positively charged redox-active metal–organic triangle containing three redox-active copper centres was developed to encapsulate anionic organic dyes (fluorescein) through a weak host–guest interaction for photocatalytic hydrogen production. The unique geometry enforces a distorted square planar coordination suitable for proton reduction. Control experiments with a mononuclear copper complex as a reference photocatalyst and inactive ATP as an inhibitor were performed to confirm this enzymatic photocatalytic behaviour.
The development of catalytic synthetic methods inspired by natural enzyme prototypes that react under an ambient atmosphere and use benign solvents and clean energy is a major endeavour in synthetic chemistry.1–4 To match the efficiency and selectivity of enzymatic systems, chemists use small molecules with defined hydrophobic cavities that emulate enzyme active site properties to catalyse specific chemical transformations.5–8 An exciting area in this research includes the incorporation of transition-metal moieties as redox-active centres that mimic highly evolved9 and finely tuned natural photocatalytic systems by catching organic dyes in their pockets.10,11 When positively charged, these pockets have important properties in anion encapsulation.12–14 As the majority of enzyme substrates are anionic, any replication of their catalysed reactivity under mild biological conditions using synthetic hosts represents a useful advance.
Photocatalytic hydrogen production from water represents an important process in future sustainable solar energy conversion. Inspired by photosynthetic complexes in nature, this process has been realized using a homogeneous reaction environment towards the optimal availability of active catalytic sites for solar hydrogen production.15–18 New reaction pathways have emerged for such substrate molecules inside these containers19–21 by enhancing the proximity between the substrate and the catalytic centre and by increasing the effective concentration of the reaction within the confined space.22,23 This novel, well-elucidated reaction strategy has developed for application in light-driven hydrogen production and has indicated that these supramolecular systems are superior to other relevant systems. Accordingly, the construction of host–guest photosynthetic systems should be a promising approach to increase hydrogen production efficiency.24,25
Copper is an earth-abundant and low-cost material that also participates in many biological metabolic processes, for example, photosynthesis and respiration, with various functions.26–29 A CuII complexes with well-defined coordination chemistry and diverse redox chemistry have attracted extensive attention and have been used as catalysts for various transformations.30–32 To our surprise, only a few copper complexes have been used for the electrocatalytic reduction33 and oxidation of water.34,35 Herein, we report a copper triangular metallo-macrocycle Cu–OBP (where H2OBP = 2,2′-(([1,1′-biphenyl]-4,4′-diylbis(azanylylidene))bis(methanylyl idene))diphenol) encapsulating an organic dye molecule for homogeneous light-driven hydrogen production. To the best of our knowledge, this is the first example of a homogeneous copper metal–organic macrocycle for photo-catalytic hydrogen generation from water (Scheme 1).
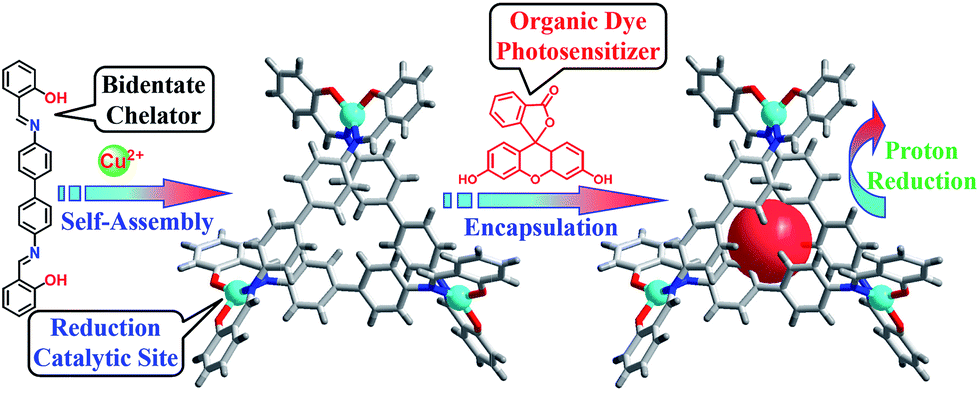 | ||
| Scheme 1 Procedure for the synthesis of the molecular Cu–OBP triangle and construction of the artificial supramolecular system for photocatalytic proton reduction. | ||
The ligand H2OBP and complex Cu–OBP were obtained according to previous literature procedures.36 Single-crystal X-ray structural analysis indicated that two of three ligands were linked by one metal centre during self-assembly, while the third ligand position on the same side as the other ligands constructed a non-C3 symmetry triangular geometry.37–40 Due to the comfortable metal⋯metal distances of approximately 12.2 Å, the three ligands overlap to form a double-layer structure with a height of approximately 9.3 Å. The structural features of the molecular triangle most likely allow the encapsulation of one planar fluorescein (Fl) molecule in the pocket,41 producing an artificial supramolecular system for the light-driven generation of hydrogen from water.42
The solution-phase stability of Cu–OBP was characterized by electrospray ionisation-mass spectrometry (ESI-MS), revealing an intense peak at m/z = 1362.20 (Fig. 1). A simple comparison with the simulation results based on natural isotopic abundances suggests that this peak is properly assigned to the singly charged [HCu3(OBP)3]+ species, indicating the formation and stability of the M3L3 species in solution. When an equimolar amount of Fl was added into the solution of Cu–OBP, a new intense peak at m/z = 1694.28 was observed, which could clearly attributed to a [HCu3(OBP)3(Fl)]+ species. A comparison of the experimentally obtained peak with that obtained via simulation based on natural isotopic abundances confirms the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric host–guest complex species Fl ⊂ Cu–OBP in the solution.
:1 stoichiometric host–guest complex species Fl ⊂ Cu–OBP in the solution.
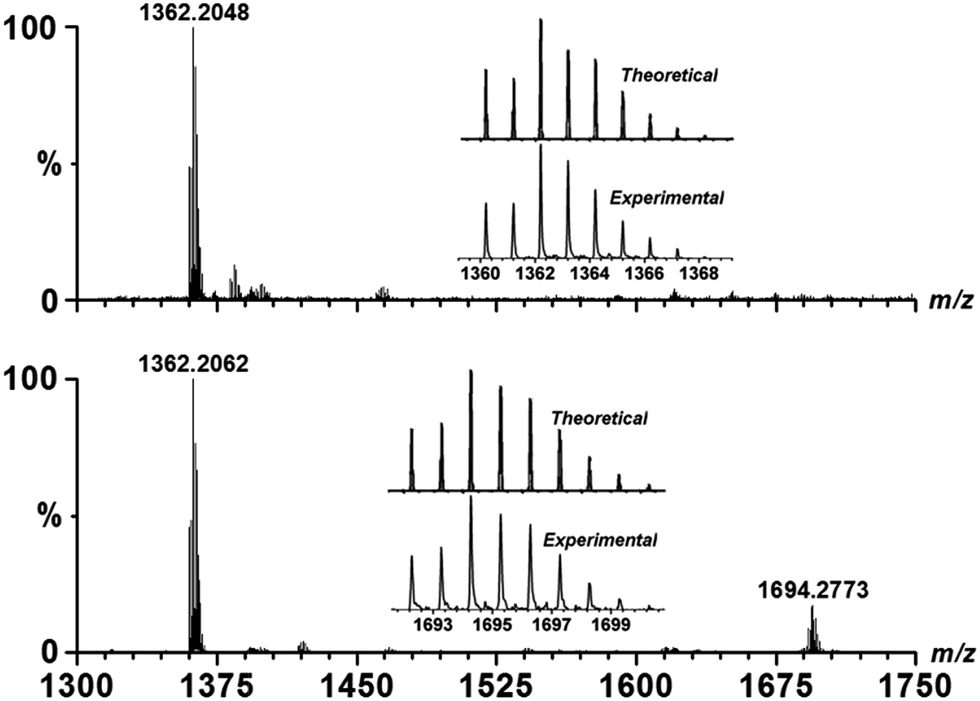 | ||
| Fig. 1 ESI-MS spectra of Cu–OBP in EtOH solution (top) and of Fl in the aforementioned solution (bottom). The insets show the measured and simulated isotopic patterns at m/z = 1362.20 (top) and 1694.28 (bottom). | ||
Isothermal titration microcalorimetry (ITC) was used to ensure the quantitative accuracy of the titration data and provide insight into the thermodynamics for host–guest complexation.43–45 A typical titration curve is shown in Fig. 2a; the enthalpy change (ΔH) and entropy change (TΔS) for Cu–OBP ⊃ Fl are −1.5 and 30.0 kJ mol−1, respectively, with a good “n” value of 1.0 in the curve fitting by computer simulation using an “independent” model.
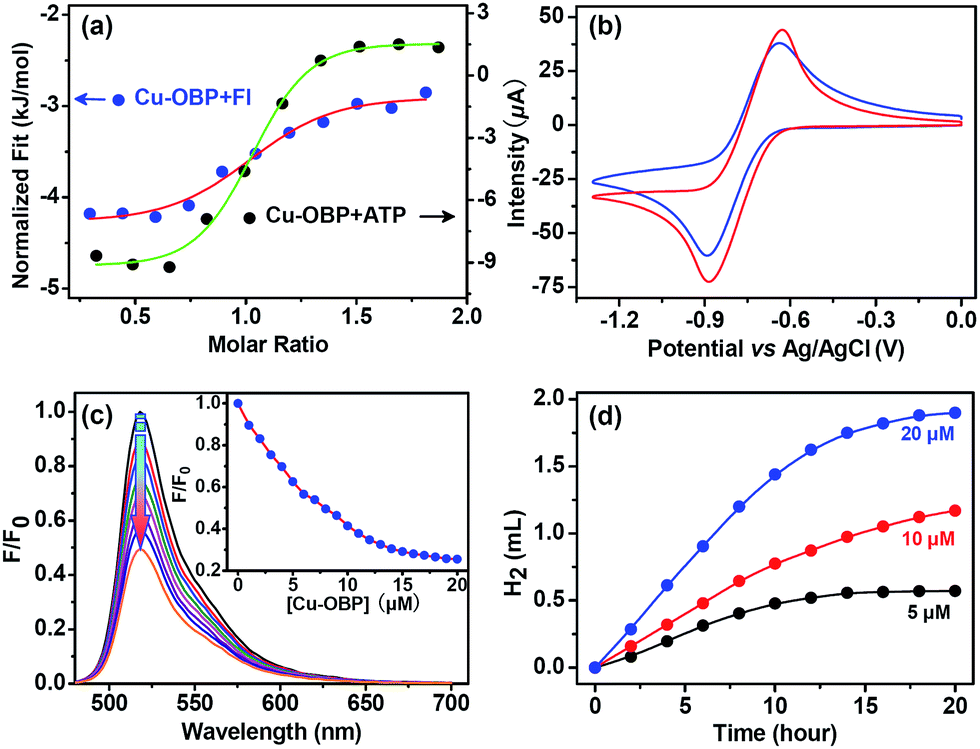 | ||
| Fig. 2 (a) ITC experiments of Cu–OBP upon addition of Fl or ATP in EtOH solution, showing the formation of host–guest complex species. (b) Cyclic voltammograms of 0.1 mM Cu–OBP (blue line) and 0.3 mM Cu–OMP (red line), which is a mononuclear copper complex possessing the same coordination geometry as Cu–OBP, in CHCl3 containing 0.1 M TBAPF6. Scan rate: 100 mV s−1. (c) Set of emission spectra for Fl (10 μM) upon the addition of Cu–OBP. The inset shows the normalized luminescence vs. [Cu–OBP]. (d) Light-driven hydrogen evolution of systems containing Fl (4.0 mM), NEt3 (15% v/v), and Cu–OBP in an EtOH/H2O solution (1:1, pH = 12.5) with the concentration of Cu–OBP fixed at 5.0 μM (black), 10.0 μM (red line), and 20.0 μM (blue line). | ||
Cyclic voltammetry of the molecular Cu–OBP triangle (1.0 mM) recorded in CHCl3 solution shows the coupled CuI/Cu0 reduction process at approximately −0.75 V (vs. Ag/AgCl) (Fig. 2b).33,46 The CuI/Cu0 potential falls well within the range of that of proton reduction in aqueous media, indicating that the reduced Cu–OBP complex is capable of directly reducing protons.46–48 The molecular Cu–OBP triangle is also demonstrated to be an efficient quencher of the Fl excited state. The addition of an equivalent molar ratio of Cu–OBP to an EtOH/H2O solution (1:1 in volume, pH = 12.5, ensuring the same pH condition as that of the reaction mixture mentioned below for photocatalytic hydrogen evolution) containing Fl (10 μM) quenched approximately 50% of the Fl emission intensity (Fig. 2c). The quenching behaviour is attributed to the photoinduced electron transfer (PET) process from the excited state Fl* to the redox catalyst Cu–OBP.49
Irradiation of a solution containing Fl (4.0 mM), Cu–OBP (10.0 μM), and triethylamine (Et3N, 15% in volume) in an EtOH/H2O (1:1 in volume) solution at 25 °C resulted in direct hydrogen generation.25,50 A higher hydrogen production efficiency was achieved at pH = 12.0–13.0. Control experiments revealed that the absence of any of these individual components led to failure to produce hydrogen, thus demonstrating that all three species are essential for hydrogen generation. Of course, the artificial system could not function well in the absence of light. When the concentrations of Fl (4.0 mM) and Et3N (15% in volume) were fixed, the produced hydrogen volume exhibited a linear relationship with the Cu–OBP catalyst concentration in the range of 5.0 μM to 20.0 μM (Fig. 2d). The calculated turnover number (TON) was approximately 1200 moles of hydrogen per mole of catalyst (Fig. 3a). The modified supramolecular system exhibits the highest TON among related homogenous copper/FI systems. This is the first example of a homogeneous copper metal–organic macrocycle for photocatalytic hydrogen generation from water,33,46 providing an opportunity for the development of highly efficient copper-based catalysts for photocatalytic proton reduction.
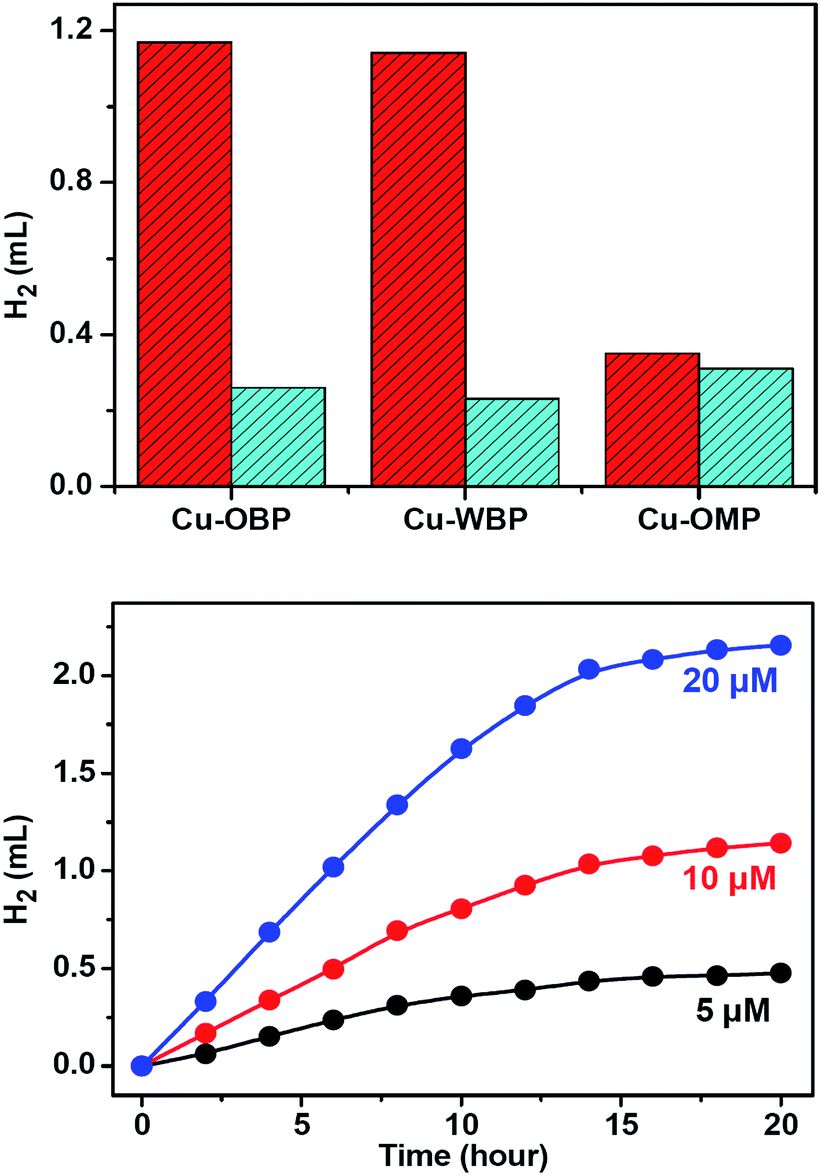 | ||
| Fig. 3 (a) Produced hydrogen volume by systems containing Fl (4.0 mM), Et3N (15% in volume), and redox catalysts (red bars) in EtOH/H2O (1:1 in volume, pH = 12.5): Cu–OBP (10.0 μM), Cu–WBP (10.0 μM, in EtOH/H2O = 2:8, in volume, pH = 12.5), and Cu–OMP (30.0 μM). The cyan bars show the aforementioned systems in the presence of ATP (4.0 mM). (b) Light-driven hydrogen evolution of systems containing Fl (4.0 mM), NEt3 (15% v/v) and Cu–WBP in an EtOH/H2O solution (1:1, pH = 12.5) with the Cu–WBP concentration fixed at 5.0 μM (black), 10.0 μM (red line) and 20.0 μM (blue line). | ||
To further determine whether photocatalytic hydrogen evolution occurred inside the pocket of Cu–OBP through a typical enzymatic fashion or outside of the pocket in a normal homogeneous manner, an important biomolecule without any suitable redox potential for hydrogen production, adenosine triphosphate (ATP), was chosen as an inhibitor for our enzymatic system.9 The inhibition of the photocatalytic reaction was displayed through the addition of this non-reactive species.24 Microcalorimetric titration curve generated for Cu–OBP upon addition of ATP revealed the formation of a host–guest system. The higher ΔH (−11.0 kJ mol−1) and TΔS (22.9 kJ mol−1) compared to those of the Cu–OBP/Fl system suggested that ATP could replace Fl and become encapsulated in the pocket of the macrocycle (Fig. 2a). As expected, in the presence of 4.0 mM ATP, photocatalytic hydrogen production by the Fl (4.0 mM)/Cu–OBP (10.0 μM)/Et3N (15%) system dropped to 20% of the original value under the same experimental conditions. Thus, the competitive inhibition behaviour was enzymatic-like and suggested that hydrogen production occurred within the cavity of Cu–OBP.51,52
To further investigate the potential factors that influence the photocatalytic hydrogen evolution process, a mononuclear copper complex Cu–OMP (where H2OMP = 2-((phenylimino)methyl)phenol) resembling a corner of the molecular triangular metal–organic macrocycle Cu–OBP was designed and prepared. The ligand HOMP was obtained by a simple Schiff-base reaction of salicylaldehyde with aniline in a CH3OH solution. The mononuclear copper complex Cu–OMP was synthesized in approximately 77% yield by reacting Cu(BF4)2 with HOMP in the presence of NaOH in CH3OH. ESI-MS spectrum of the solution-phase Cu–OMP complex exhibited [NaCu(OMP)2]+ and {Na[Cu(OMP)2]2}+ peaks at m/z = 478.11 and 935.22, suggesting the formation of the mononuclear species in solution. Cyclic voltammetry of Cu–OMP exhibited a broad peak at approximately −0.75 V (vs. Ag/AgCl, Fig. 2b), corresponding to the CuI/Cu0 couple. This potential is consistent with the Cu–OBP triangle and permits the exploration of redox-induced reactions near the H2/H+ couple. Single-crystal structure analysis revealed that the mononuclear Cu–OMP copper centre was affixed to two nitrogen and two oxygen atoms. These donors belonged to two bidentate six-membered ring chelators, and the copper centre possessed the same coordination environment as those in Cu–OBP (Scheme 2). Therefore, Cu–OMP was considered an ideal reference compound for Cu–OBP in the investigation of photocatalytic hydrogen evolution from water within a supramolecular system.
 | ||
| Scheme 2 Square planar coordination geometries of the copper ions in Cu–OMP and Cu–OBP. The copper, oxygen, nitrogen, and carbon atoms are drawn in cyan, red, blue, and grey, respectively. aThe numbers in parentheses represent the deviation for the front number. | ||
Photolysis of a solution containing Fl (4.0 mM) and Cu–OMP (30.0 μM) in a solvent mixture containing Et3N (15% in volume) and EtOH/H2O (1:1 in volume) resulted in hydrogen generation under the photocatalytic conditions optimized at pH = 12.5. The TON was approximately 100 moles of hydrogen per mole of redox catalyst after irradiating for 12 h, and the addition of ATP (4.0 mM) did not obviously change the produced hydrogen volume, demonstrating the normal homogeneous behaviour dynamics of the system (Fig. 3a). The generated hydrogen volume was approximately 30% of that produced by Cu–OBP under the same reaction conditions (10 μM Cu–OBP, ensuring the same concentration of copper ions), and with an equal copper ion concentration, Cu–OMP exhibited a considerably lower catalytic activity. The superiority of the Cu–OBP/Fl/Et3N system over the Cu–OMP/Fl/Et3N system is attributed to the new reaction pathways of the Cu–OBP/Fl/Et3N system and to the increased concentration of the reaction within the confined space.53
The application of supramolecular artificial photosynthetic (AP) systems is often limited by aqueous solubility, especially in photocatalytic proton and CO2 reduction. Solubility improvements are thus crucial, a problem that can be overcome by a tailor-made supramolecular flask possessing high solubility in aqueous solution. Therefore, by the introduction of six hydroxymethyl groups, a triangular analogue Cu–WBP (where H2WBP = 2,2′-(((2,2′-bis(hydroxy methyl)-[1,1′-biphenyl]-4,4′-diyl)bis(azanylylidene))bis(methan ylylidene))diphenol) with similar structural features but with a 2,2′-bis(hydroxymethyl)benzidine moiety to substitute the benzidine moiety was designed and prepared. The six hydroxymethyl groups on the periphery of Cu–WBP provided the assembly with water solubility.54,55 The hydroxymethyl substituents not only make the pocket water soluble but also form a hydrophobic environment from the outward direction of the hydroxymethyl groups, thereby allowing easier encapsulation of Fl for light-driven hydrogen production.
The triangular Cu–WBP exhibited a suitable reduction potential at −0.76 V. The solution-phase Cu–WBP structure was further characterized by ESI-MS analysis. The intense peaks at m/z = 771.65, 782.64 and 793.62 were assigned to [H2Cu3(WBP)3]2+, [HNaCu3(WBP)3]2+ and [Na2Cu3(WBP)3]2+, respectively, by a simple comparison with the simulation results based on natural isotopic abundances, suggesting the formation and stability of the M3L3 species in solution. The ESI-MS spectrum of the Cu–WBP solution containing an equimolar amount of Fl also exhibited a new peak at m/z = 937.67, which was assigned to a [H2Cu3(WBP)3(Fl)]2+ species (Fig. 4), implying the formation of a 1:1 stoichiometric complex species Fl ⊂ Cu–WBP in solution. In addition, loading an identical amount of the water-soluble Cu–WBP analogue with the Cu–OBP provided the same catalytic efficiency for light-driven hydrogen production. The results of the catalytic experiments suggest that the introduction of water-soluble functional groups does not influence the photo-catalytic properties of the AP system. Even upon increasing the EtOH/H2O volume ratio to 2:8, the hydrogen evolution reaction nearly maintained the original reaction rate (Fig. 3), providing a strategy for the construction of water-soluble systems as efficient homogeneous catalysts for photocatalytic proton reduction.
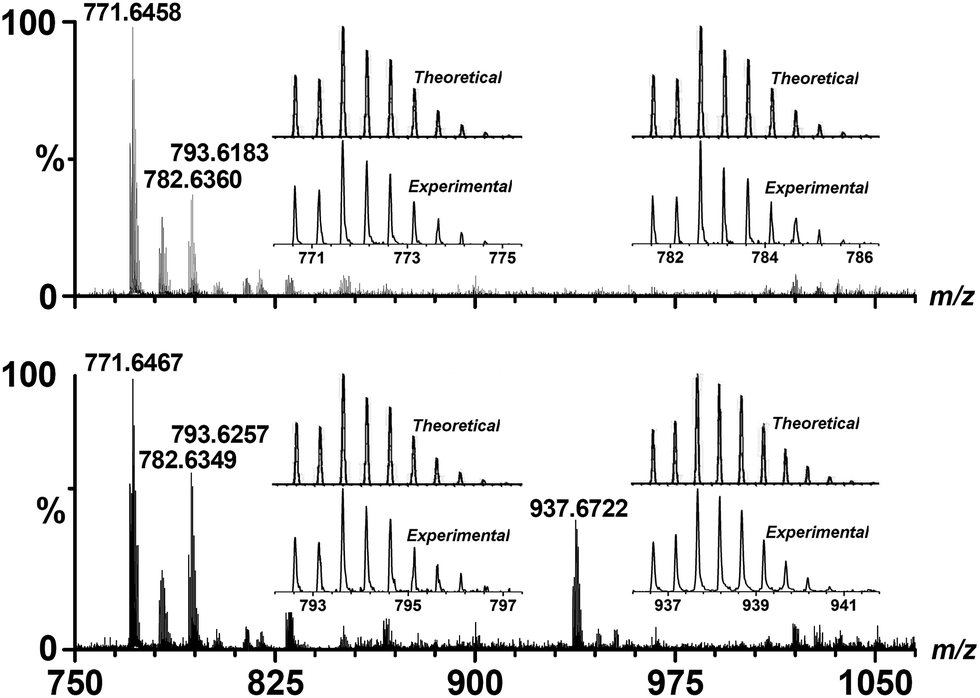 | ||
| Fig. 4 ESI-MS spectra of Cu–WBP in EtOH solution (top) and of Fl in the aforementioned solution (bottom). The insets show the measured and simulated isotopic patterns at m/z = 771.64, 782.63, 793.62 and 937.67, respectively. | ||
Conclusions
In summary, we applied our strategy for the construction of Cu-based AP systems for hydrogen generation from water by encapsulating an organic photosensitizer in the pocket of a metal–organic triangle. For the first time, copper ions were introduced into a metal–organic macrocycle as a highly effective visible-light photocatalyst for homogeneous splitting water to hydrogen. The copper triangle exhibited a suitable redox potential for hydrogen production. The results provide an opportunity for the development of highly efficient copper-based supramolecular catalysts for photocatalytic proton reduction. The introduction of hydroxymethyl groups provides aqueous solubility for the supramolecular catalyst without influencing the original photocatalytic properties. The superior activity and stability suggest that this novel approach for the construction of water-soluble supramolecular AP systems as efficient homogeneous catalysts is promising and could be extended to other aqueous chemical transformations.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the National Natural Science Foundation of China (21501019, 21531001, and 21421005).Notes and references
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed
.
- K. Yan and M. Fujita, Science, 2015, 350, 1165–1166 CrossRef CAS PubMed
- R. J. Hooley, Nat. Chem., 2016, 8, 202–204 CrossRef CAS PubMed
- D. Zhang, A. Martinez and J. P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS PubMed
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed
- D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235–1238 CrossRef CAS PubMed
- Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed
- X. Jing, C. He, Y. Yang and C. Y. Duan, J. Am. Chem. Soc., 2015, 137, 3967–3974 CrossRef CAS PubMed
- D. Luo, X. Zhou and D. Li, Angew. Chem., Int. Ed., 2015, 54, 6190–6195 CrossRef CAS PubMed
- L. Yang, C. He, X. Liu, J. Zhang, H. Sun and H. Guo, Chem.–Eur. J., 2016, 22, 5253–5260 CrossRef CAS PubMed
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez, L. Guy and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 6574–6577 CrossRef CAS PubMed
- J. Ayme, J. E. Beves, C. J. Campbell, G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, J. Am. Chem. Soc., 2015, 137, 9812–9815 CrossRef CAS PubMed
- H. B. Gray and A. W. Maverick, Science, 1981, 214, 1201–1205 CAS
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed
- D. M. Dalton, S. R. Ellis, E. M. Nichols, R. A. Mathies, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 10128–10131 CrossRef CAS PubMed
- D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 9202–9205 CrossRef CAS PubMed
- J. Zhang, H. Yu, C. Zhang, C. He and C. Y. Duan, New J. Chem., 2014, 38, 3137–3145 RSC
- C. He, J. Wang, L. Zhao, T. Liu, J. Zhang and C. Y. Duan, Chem. Commun., 2013, 49, 627–629 RSC
- X. Jing, P. Wu, X. Liu, L. Yang, C. He and C. Y. Duan, New J. Chem., 2015, 39, 1051–1059 RSC
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef CAS
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y. G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef CAS PubMed
- A. Bhagi-Damodaran, M. A. Michael, Q. Zhu, J. Reed, B. A. Sandoval, E. N. Mirts, S. Chakraborty, P. Moënne-Loccoz, Y. Zhang and Y. Lu, Nat. Chem., 2017, 9, 257–263 CrossRef CAS PubMed
- C. J. K. Wijekoon, S. R. Udagedara, R. L. Knorr, R. Dimova, A. G. Wedd and Z. Xiao, J. Am. Chem. Soc., 2017, 139, 4266–4269 CrossRef CAS PubMed
- T. Zhang, C. Wang, S. Liu, J. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed
- M. T. Zhang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed
- R. C. Walroth, K. C. Miles, J. T. Lukens, S. N. MacMillan, S. S. Stahl and K. M. Lancaster, J. Am. Chem. Soc., 2017, 139, 13507–13517 CrossRef CAS PubMed
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. C. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed
- M. K. Coggins, M. T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed
- A. Zarkadoulas, E. Koutsouri and C. A. Mitsopoulou, Coord. Chem. Rev., 2012, 256, 2424–2434 CrossRef CAS
- D. Guo, C. Q. Qian, C. Y. Duan, K. L. Pang and Q. J. Meng, Inorg. Chem., 2003, 42, 2024–2030 CrossRef PubMed
- M. Schmittel and K. Mahata, Inorg. Chem., 2009, 48, 822–824 CrossRef CAS PubMed
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed
- Z. Jiang, Y. Li, M. Wang, D. Liu, J. Yuan, M. Chen, J. Wang, G. R. Newkome, W. Sun, X. Li and P. Wang, Angew. Chem., Int. Ed., 2017, 56, 11450–11455 CrossRef CAS PubMed
- J. Zhuang, C. H. Kuo, L. Y. Chou, D. Y. Liu, E. Weerapana and C. K. Tsung, ACS Nano, 2014, 8, 2812–2819 CrossRef CAS PubMed
- H. Yu, C. He, J. Xu, C. Duan and J. N. H. Reek, Inorg. Chem. Front., 2016, 3, 1256–1263 RSC
- B. Gómez, V. Francisco, F. Fernández-Nieto, L. Garcia-Rio, M. Martín-Pastor, M. R. Paleo and F. J. Sardina, Chem.–Eur. J., 2014, 20, 12123–12132 CrossRef PubMed
- S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC
- W. Zhao, C. Wang, L. Chen, R. Lin, X. Cui, Q. Zhu, Z. Tao and J. Liu, RSC Adv., 2016, 6, 11937–11942 RSC
- X. Y. Dong, M. Zhang, R. B. Pei, Q. Wang, D. H. Wei, S. Q. Zang, Y. T. Fan and T. C. W. Mak, Angew. Chem., Int. Ed., 2016, 55, 2073–2077 CrossRef CAS PubMed
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed
- K. M. K. Swamy, S. Ko, S. Kwon, H. N. Lee, C. Mao, J. Kim, K. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 44, 5915–5917 RSC
- A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5, 1397–1406 CrossRef CAS
- L. Zhao, J. Wei, J. Lu, C. He and C. Duan, Angew. Chem., Int. Ed., 2017, 56, 8692–8696 CrossRef CAS PubMed
- C. J. Hastings, D. Fiedler, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 10977–10983 CrossRef CAS PubMed
- L. Yang, X. Jing, C. He, Z. Chang and C. Duan, Chem.–Eur. J., 2016, 22, 18107–18114 CrossRef CAS PubMed
- S. Zarra, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 4837–4840 CrossRef CAS PubMed
- T. F. Beltrán, R. Llusar, M. Sokolov, M. G. Basallote, M. Jesus Fernández-Trujillo and J. ÁngelPino-Chamorro, Inorg. Chem., 2013, 52, 8713–8722 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, as well as additional tables and figures. CCDC 1483351. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09285g |
| This journal is © The Royal Society of Chemistry 2017 |