
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards the development of multifunctional hybrid fibrillary gels: production and optimization by colloidal electrospinning†
Jaime Faria,
Coro Echeverria*,
João P. Borges *,
Maria H. Godinho and
Paula I. P. Soares*
*,
Maria H. Godinho and
Paula I. P. Soares*
i3N/CENIMAT, Departamento de Ciência dos Materiais, Faculdade de Ciências e Tecnologia, FCT/UNL, 2829-516 Caparica, Portugal. E-mail: coro@fct.unl.pt; jpb@fct.unl.pt; pi.soares@fct.unl.pt
First published on 17th October 2017
Abstract
The incorporation of thermosensitive microgels that can act as active sites into polymeric fibers through colloidal electrospinning originates multifunctional, highly porous, and biocompatible membranes suitable for biomedical applications. The use of polyvinylpyrrolidone (PVP), a biocompatible, water-soluble polymer as a fiber template, not only allows the use of a simple set-up to produce composite membranes, but also avoids the use of organic solvents to prepare such systems. Further crosslinking with ultraviolet (UV) radiation avoids membrane dissolution in physiological conditions. Highly porous, UV crosslinked composite membranes with monodisperse mean fiber diameters around 530 nm were successfully produced. These composite membranes showed a Young Modulus of 22 MPa, and an ultimate tensile strength of 3 MPa, accessed in the mechanical tests. Furthermore, the same composite membranes were able to swell about 30 times their weight after 1 hour in aqueous medium. In this work composite multifunctional membranes were designed and extensively studied. PVP, a biocompatible water-soluble polymer, was used as a fiber template to incorporate thermoresponsive poly-(N-isopropylacrylamide) (PNIPAAm)-based microgels into the composite membrane using colloidal electrospinning. The design of multifunctional membranes can be further tailored to several biomedical applications such as temperature-controlled drug delivery systems.
Introduction
The design, research and development of new and improved smart structures and systems is currently a very hot topic in Materials Science and Engineering. One versatile and easy to scale-up strategy to produce such systems is by using electrospinning. Electrospinning is a cost-effective and simple process capable of forming polymer fibres in the submicron range with high surface to volume ratio, flexibility and tuneable porosity.1,2 The resultant fibres have been used in a variety of applications, ranging from sensors to drug delivery and tissue scaffolds.3–6 Fibre's surface topography, morphology and orientation are highly dependent on a number of parameters: (i) solution parameters, (ii) process parameters and (iii) environmental conditions.2Colloidal or emulsion electrospinning is similar to the traditional electrospinning but uses a colloidal system of two or more immiscible phases as fibre precursor instead of an homogeneous solution.7 Since this technique allows the production of multicomponent nanofibers with controlled morphology using a simple nozzle, colloidal electrospinning comes as a good replacement for co-axial electrospinning, which uses coaxial nozzles for the same purpose.8 Furthermore, this approach is environmentally friendly since it enables the minimization of the amount of organic solvents used during the production process.9
The main advantages that colloidal electrospinning possesses is the different fibre morphologies that can be obtained, regarding dispersed phase placement inside the fibre. Taking this in consideration, there are two main types of morphologies that appear in colloidal electrospinning fibres: ‘bead-on-a-string’10 and ‘core–shell’.11
The incorporation of stimuli-sensitive microgels, which can act as active sites, into polymeric fibres by means of colloidal electrospinning can originate multifunctional fibres with fast thermosensitive behaviour and tuneable surfaces. Díaz et al.12 produced composite fibres of poly-(N-isopropylacrylamide) (PNIPAAm)-based microgels using polyvinylpyrrolidone (PVP) as fibre template, obtaining fibres with a mean diameter of 0.9 μm and a water uptake of 17 times their dry weight. Poly-(ethylene oxide) (PEO) fibres confining PNIPAAm–chitosan (PNIPAAm–Cs) microgels with a mean diameter of 63 nm and bead-on-a-string morphology were also reported.13
PVP is a neutral hydrophilic water-soluble polymer with low toxicity and high biocompatibility with living tissues, capable of binding reversibly to various molecules (dyes, metals and some polymers) in solution.14,15 PVP can easily be crosslinked using ultraviolet (UV) radiation, a cheap and high-energy radiation, thus allowing its application in biological systems without polymer dissolution.15–19
We have previously reported the use of colloidal electrospinning to produce multifunctional membranes with thermosensitive microgels as active sites using PEO, also a water-soluble polymer, as a fibre template.13 Therefore we could not apply such multifunctional membranes in physiological systems because of the risk of polymer/membrane dissolution. In the current work we will use PVP as a fibre template to perform colloidal electrospinning, followed by UV crosslinking to prevent polymer dissolution. During UV irradiation, the pyrrolidone substituents and cyclic amides on the PVP chains generate macroradicals – produced by UV direct photolysis of PVP – whose recombination could lead to intermolecular crosslinking PVP.20 The results of PVP's UV direct photolysis are depicted in Fig. S1.† To achieve such purpose we report the optimization of PVP as a fibre template for the production of composite non-woven mats (or membranes) by confining thermosensitive PNIPAAm microgels in PVP fibres using colloidal electrospinning. UV irradiation was used to crosslink the PVP matrix, thus avoiding its dissolution in physiological conditions, but still maintaining its swelling capability. Both mechanical and swelling properties of the membranes were studied and their behaviour with different UV crosslinking times analysed. The novelty of this work is the development of a production technique that allows tailoring a multifunctional composite system simply by changing the composition/cargo of the confined microgels. The simple and easy-to-use technique here used (colloidal electrospinning) is versatile enough to produce a system on demand for the desired application.
Experimental
Materials
N-Isopropylacrylamide (NIPAAm, Aldrich Chemistry, 97%) was used as a monomer and N,N-methylene bis-acrylamide (MBA, Sigma-Aldrich, 99%) as cross linker, ammonium persulfate was chosen as initiator (APS, Sigma-Aldrich, 99%) and sodium bisulfite (SBS, Acrös Organics) as catalyst. Acrylic acid 90% (Alfa Aesar) was used in order to study the effects in the microgels' LCST. PVP (Sigma-Aldrich,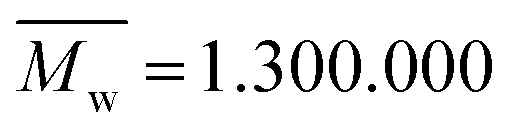 ) was used as a fibre template to produce the non-woven mats.
) was used as a fibre template to produce the non-woven mats.
All reagents were used as received without any further purification.
Synthesis of PNIPAAm microgels
PNIPAAm and PNIPAAm–AAc crosslinked microgels were synthesized by surfactant-free emulsion polymerization (SFEP) as previous described.13 Acrylic acid was used in order to study the effects in the microgels' LCST. Further information can be found in Table 1.| Reagents | wt% monomer | Mass (g) | Volume (mL) |
|---|---|---|---|
| a Since acrylic acid comes in liquid form and its density is 1.05 g cm−3, upon synthesis of PNIPAAm–AAc microgels the volume of AAc corresponding to 6 and 10 wt% (regarding PVP mass) is 57.1 and 95.2 μL, respectively. | |||
| NIPAAm | 100 | 1 | 20 |
| MBA | 5 | 0.05 | 20 |
| APS | 10 | 0.1 | 20 |
| SBS | 5 | 0.05 | 20 |
| 80 + 20 mL H2O | |||
Electrospinning
PVP solutions in different weight percentages in ethanol (10 wt%, and 14 wt%) were used as fibre template. Microgels were added to PVP solutions to achieve 10 wt%, regarding PVP total mass.UV crosslinking
PVP and composite membranes were crosslinked with 254 nm UV light in a BIO-LINK® irradiation system for 5, 10, 15, 20, 30, 40 and 60 minutes.Characterization
Scanning electron microscopy (SEM) technique was used to evaluate the morphology of the obtained fibres. Samples were prepared with a sputtered thin layer of gold/palladium using a Q300T D Quorum sputter coater and analysed using a Zeiss DSM 962 SEM or a SEM apparatus fitted with a Carl Zeiss Auriga CrossBeam system (SEM-FIB). The diameter and distribution of the overall electrospun fibres were determined using ImageJ software.Transmission electron microscopy (TEM) images were obtained using a Hitachi H-8100 II with thermo-ionic emission LaB6. TEM analysis was performed in a small piece of a membrane placed in a Kevlar 25 mesh grid.
Tensile tests were performed with a tensile testing machine from Rheometric Scientific (Minimat Firmware Version 3.1). PVP and composite membranes were cut into 10 × 5 mm rectangles and mounted onto a pair of clamps and stretched at a speed of 0.1 mm min−1.
Swelling properties of the membranes were performed using 6 rectangular samples (10 × 5 mm) of each sample immersed in water for 0.5, 1, 2, 3 and 5 h. Between each immersion time the water was changed in order to minimize any possible single polymer chains resulting from the dissolution to reattach to the PVP molecule.
Results and discussion
Morphology of the non-woven mats
In order to obtain fibres with a monodisperse distribution of diameters as well as a minimum mean fibre diameter (MFD), a study where electrospinning parameters such as polymer solution concentration, process flow rate, applied voltage and TCD were varied in specific ranges was performed. There is an increase of MFD with the increase of the solution's concentration (Fig. 1A): if the solution is too dilute (low viscosity) the dominant factor is the surface tension of the droplet, which results in the inability of forming a continuous fibre and mainly drops of solution or beaded fibres will reach the collector; on the other hand, if the solution is highly viscous, the electric field may not be high enough to disrupt the viscoelastic forces, which can lead to the formation of fibres with several different diameters.21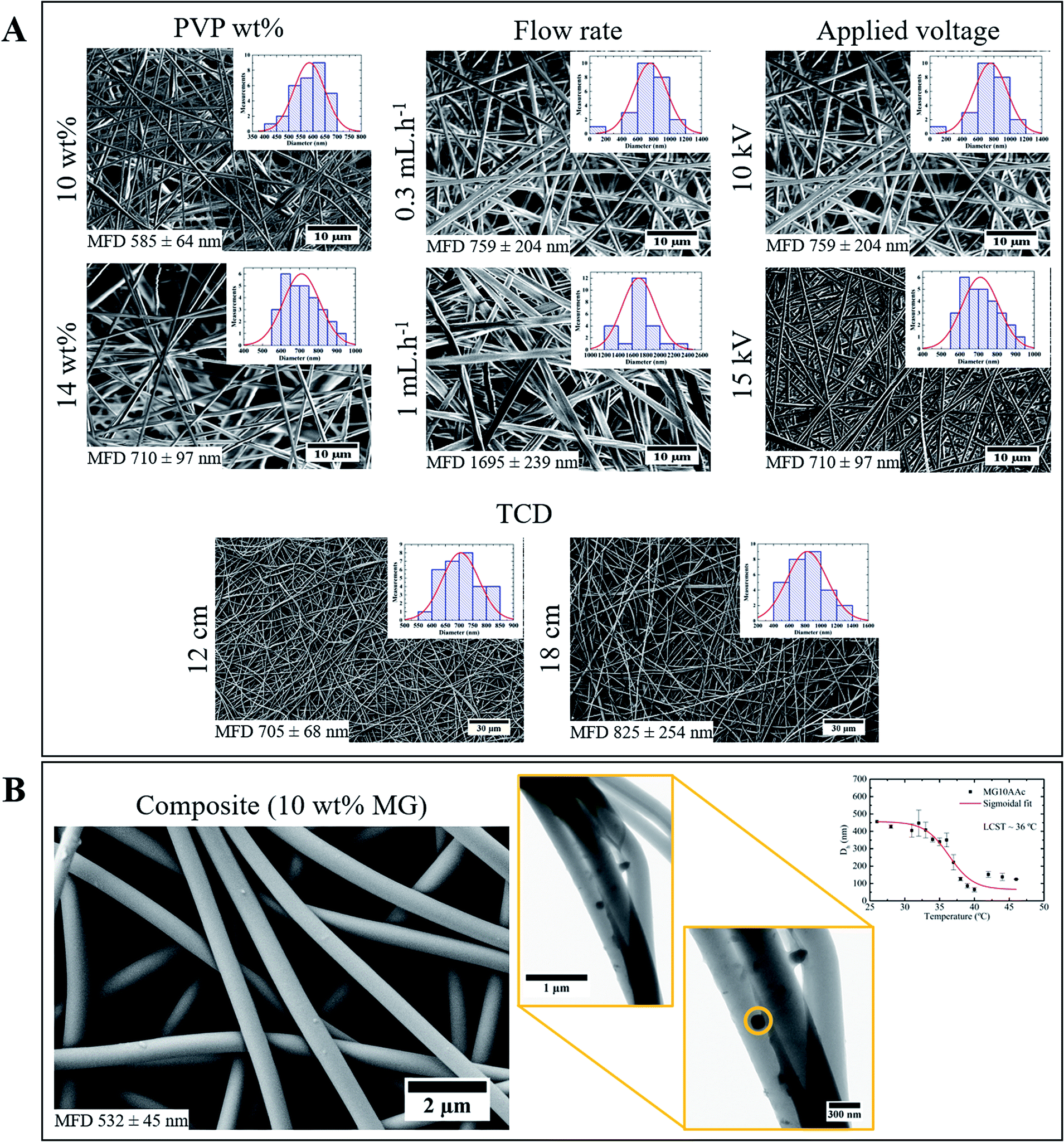 | ||
| Fig. 1 (A) Representative SEM images of electrospinning optimization study; (B) SEM (left) and TEM (right) images of composite membrane produced with the optimized parameters. | ||
Since the flow rate determines how much solution is available at the tip of the needle for fibre formation, for a given voltage there is a lower limit of flow rate for the Taylor cone to be stabilized.22 If the flow rate is increased above this limit the fibre diameter increases because there is a greater volume of solution at the tip than the one that can be ejected; therefore, MFD increases with the increasing flow rate (Fig. 1A).
When high voltages are applied, the polymer jet suffers a greater stretching due to the greater columbic forces and stronger electric field, which leads to thinner fibres. However, applying lower voltages results in the reduction of the polymer jet's acceleration, which increases its time of flight and consequently its stretching ability.23 In this study the applied voltage did not affect the fibres MFD, in the tested range (Fig. 1A).
A small increase in MFD was observed with the increase of TCD (Fig. 1A). The non-woven mat produced with 12 cm TCD appears to be denser than the 18 cm TCD one. Moreover, TCD of 18 cm originates a wider dispersion of diameters, from 400 nm to 1400 nm. Therefore, the TCD of 12 cm is more appropriate for the desired optimization.
Using the optimized electrospinning parameters, composite membranes were electrospun from a 14 wt% PVP/EtOH solution with 10 wt% of microgels, using a flow rate of 0.7 mL h−1, 15 kV and a TCD of 12 cm. Although 0.7 mL h−1 was not an obtained parameter on the electrospinning optimization study, it is well known that a decrease in flow rate leads to fibres with smaller diameters.24 The decrease in process flow rate from 1 to 0.7 mL h−1 was made in order to bring the fibres' diameter closer to the microgels one.
Using the optimized parameters a composite membrane with MFD of 497 nm was obtained, versus a PVP membrane with 817 nm (Fig. 1B). Several factors could be influencing this decrease: the used flow rate was lower (0.7 mL h−1) which gives the solvent more time to evaporate, resulting in fibres with lower diameter,2 the solution's viscosity could also influence the MDF. To better understand the influence of this parameter, rheological studies were made to the solutions used in the electrospinning and colloidal electrospinning experiments. Both solutions had a fairly similar value of viscosity (3778.7 Pa s for the 14 wt% PVP/EtOH solution and 3023.3 Pa s for the 14 wt% PVP/EtOH with 10 wt% microgels solution), which means that the viscosity is not an influencing parameter in the composite fibres' MFD. Moreover, in the TEM image (Fig. 1B) it is possible to identify a microgel incorporated inside a PVP fibre. The size of the microgel is found to be close to 150 nm. However, as suggested by the dynamic light scattering (DLS) plot (Fig. 1B), the microgel's hydrodynamic diameter (Dh) at 25 °C (temperature used when electrospinning) corresponds to 450 nm. This value of diameter decreases with temperature due to the microgels' collapse. The diameter of the microgels in the collapsed state corresponds to 150 nm which fits with the diameter measured for the microgels observed by TEM. The difference in microgel size observed by TEM compared to DLS could be related to the solvent evaporation process that the membrane suffers: during this process, the solvent evaporates from both membrane and microgel, leading to a possible collapse by the microgel.
UV crosslinking
UV irradiation had the purpose of inducing crosslinks in PVP which prevents its further dissolution in water. Therefore, a comparative study between PVP and composite membranes was made. Both membrane' types were irradiated for 5, 10, 15, 20, 30, 40 and 60 minutes.Regarding PVP membranes (Fig. 2), there are no great changes in the MFD with the increase of irradiation time, since all membranes have a MFD of approximately 830 nm (Table 2). However, with 60 minutes of UV there are some visible cracks on the fibres' surface, possibly indicating excessive irradiation time, which could be related to an increase of crosslinking degree. The same effect was observed in composite membranes, where no significant differences were found in the MFD, although some cracks were also observed after 60 minutes of UV (Fig. 2 and Table 2).
 | ||
| Fig. 2 Representative SEM images of PVP (14 wt% PVP/EtOH) and composite membranes (14 wt% PVP/EtOH solution with 10 wt% of microgels) UV irradiated for 5 and 60 minutes. The red arrows indicate the cracks on the fibres' surface, a possible indication of excessive UV irradiation time. | ||
| UV irradiation (min) | PVP MFD (nm) | Composite MFD (nm) |
|---|---|---|
| 5 | 823 ± 50 | 554 ± 64 |
| 10 | 856 ± 111 | 511 ± 65 |
| 15 | 833 ± 69 | 579 ± 109 |
| 20 | 665 ± 49 | 543 ± 69 |
| 30 | 741 ± 82 | 532 ± 45 |
| 40 | 871 ± 64 | 509 ± 60 |
| 60 | 817 ± 46 | 497 ± 78 |
Mechanical performance of the non-woven mats
The mechanical tests were performed in order to evaluate the influence of UV irradiation time (and consequently, in PVP crosslinking) in the Young's Modulus and Ultimate tensile strength (UTS).As for PVP membranes (Fig. 3), an equilibrium for the Young's Modulus and UTS is reached after 20 minutes of UV, which means that irradiating the mats for more than 20 minutes does not bring a visible advantage to the system's mechanical properties. Although the mechanical properties do not change, the morphology is in fact affected with the increase of irradiation time (appearance of cracks for 60 minutes of UV). The same mechanical behaviour was observed for composite membranes. As for the influence of the confined microgels, it seems that it is not too evident through the mechanical properties: composite membranes have a ‘maximum’ Young's Modulus of approximately 22 MPa, while PVP membranes round 25 MPa; the same happens for the UTS values (Fig. 3). Once again, this lack of influence may be due to the reduced size of the microgels or to the small load confined inside the PVP fibres.
 | ||
| Fig. 3 Mechanical properties of PVP vs. composite membranes: (A) Young's modulus (E), and (B) ultimate tensile strength (UTS). | ||
Swelling ability of the non-woven mats
Observing the swelling ratio curves in Fig. 4, a significant difference between the PVP membranes irradiated for 5 minutes and 30 minutes stands out. This could be explained using the crosslinking density data, calculated from the swelling ratio (Fig. 4 and Table 3). When a membrane is crosslinked, a polymeric network is formed. In our case, with an increase of irradiation time there is an increase in the crosslinking density of the formed network. At the same time that the network gets denser its polymeric chains suffer a decrease in mobility, which ultimately results in a lower uptake of water, thus making the membranes irradiated for 5 minutes to have a higher swelling ratio than those irradiated for 30 minutes. The difference in swelling ratio between the 30 and 60 minutes can be considered as negligible. The slight difference can also indicate that the ideal irradiation time may be 30 minutes, considering both the mechanical properties and the morphology study.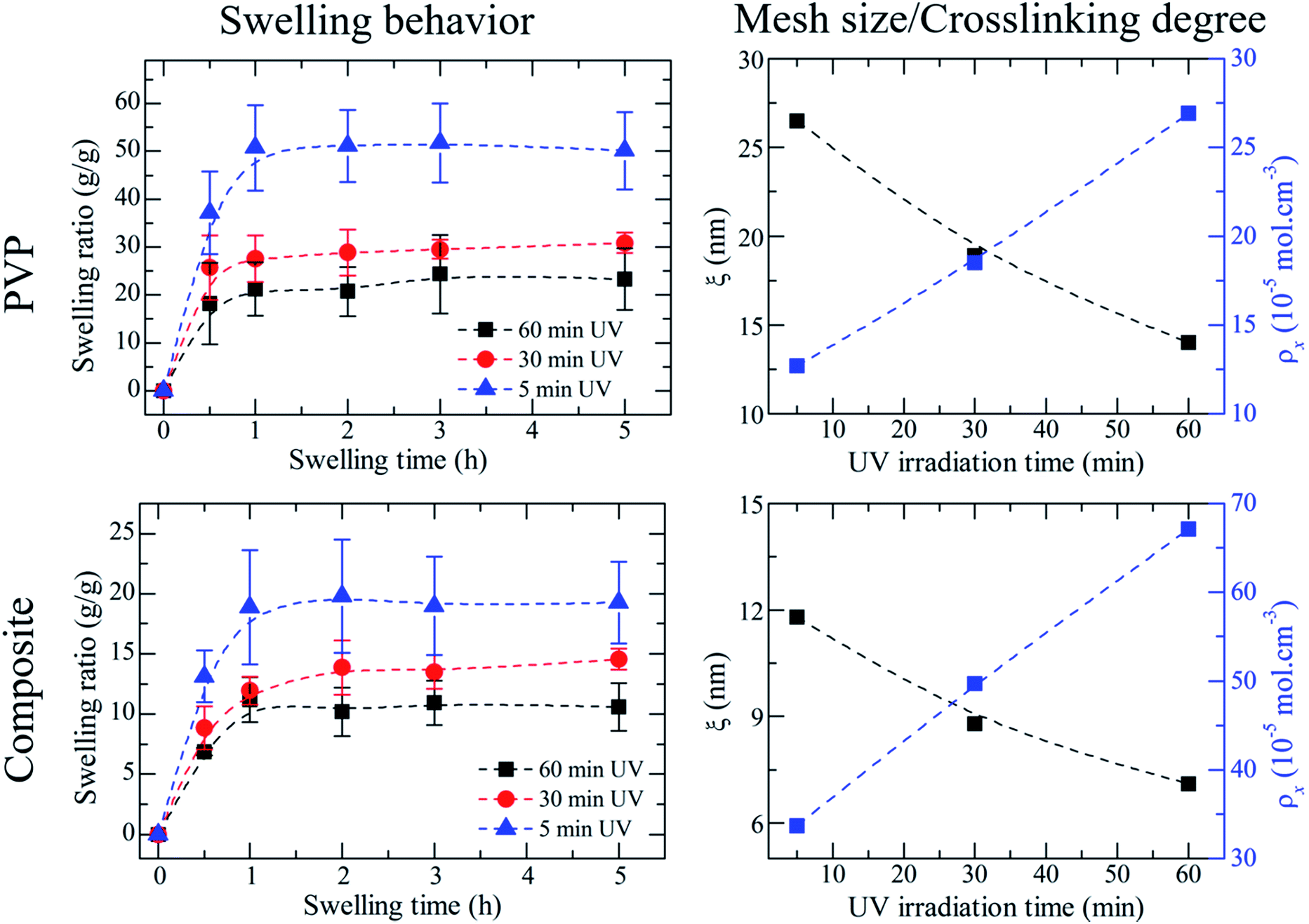 | ||
| Fig. 4 Swelling behaviour of UV crosslinked PVP vs. composite membranes, and their respective variation of mesh size and crosslinking degree with increasing UV irradiation time. | ||
| UV irradiation (min) | PVP | Composite | ||||
|---|---|---|---|---|---|---|
| 5 | 30 | 60 | 5 | 30 | 60 | |
 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 168 168 |
27.2 | 12.3 | 3704 | 11.8 | 33.7 |
| ξ (nm) | 6468 | 18.2 | 19.3 | 2516 | 8.8 | 49.7 |
| ρx (10−5 mol cm−3) | 4723 | 14.2 | 26.5 | 1863 | 7.1 | 67.1 |
From the analysis of the swelling properties it is possible to calculate the network mesh size, which is directly related to the crosslinking degree of the polymer network after UV crosslinking process by the application of the Equilibrium Swelling theory25 by assuming that UV crosslinking gives rise to the formation of a hydrogel. When studying a hydrogel's behaviour in a liquid medium there are three important parameters for the characterization of the network structure: (i) polymer volume fraction in the swollen state (υ2,s), (ii) molecular weight of the polymer chain between two neighbouring crosslinking nodes  and (iii) mesh size (ξ). Both
and (iii) mesh size (ξ). Both 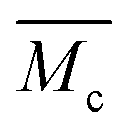 and mesh size can only be average values due to the fact that the crosslinking process is completely random.26
and mesh size can only be average values due to the fact that the crosslinking process is completely random.26
These three parameters can be determined using the Equilibrium Swelling theory that states that a crosslinked polymer gel, when immersed in a fluid and allowed to reach equilibrium with its surroundings, is subject only to two opposing forces: the thermodynamic force of mixing and the retractile force of the polymer chains. However, this theory cannot be applied to hydrogels prepared in the presence of water. Peppas and Merrill modified the original Flory–Rehner theory to consider the volume fraction of the chains in the relaxed state (υ2,r), i.e. immediately after crosslinking (eqn (1)):26
 | (1) |
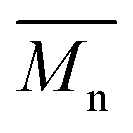 is the molecular weight of the polymer chains prepared without a cross linker,
is the molecular weight of the polymer chains prepared without a cross linker, ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) is the specific volume of the polymer (PVP – 0.785 cm3 g−1,27), V1 is the molar volume of water (18 cm3 mol−1) and χ1 is the Flory interaction parameter (0.48). The polymer volume fraction in the swollen state (υ2,s) can be determined by dividing the weight of the polymer in the dry state for the weight in the swollen state. The
is the specific volume of the polymer (PVP – 0.785 cm3 g−1,27), V1 is the molar volume of water (18 cm3 mol−1) and χ1 is the Flory interaction parameter (0.48). The polymer volume fraction in the swollen state (υ2,s) can be determined by dividing the weight of the polymer in the dry state for the weight in the swollen state. The 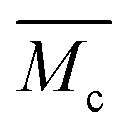 is especially important because it influences both the polymer's mesh size and the crosslinking density (ρx), which can be calculated using eqn (2) and (3) respectively.
is especially important because it influences both the polymer's mesh size and the crosslinking density (ρx), which can be calculated using eqn (2) and (3) respectively.
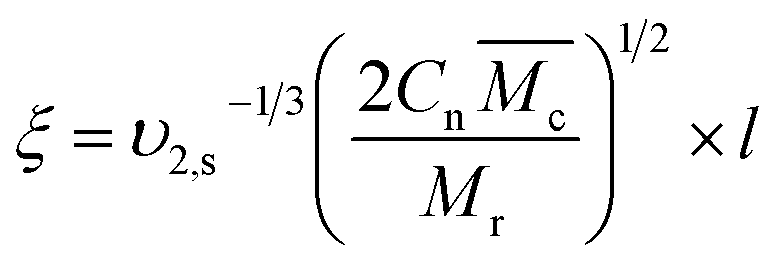 | (2) |
 | (3) |
Interestingly the composite membranes present a smaller swelling ratio at equilibrium, than the PVP ones (approximately 10 vs. 30, at 30 minutes of UV). This could be explained by the presence of microgels. Theoretically, when a fluid penetrates a network it chooses the path of least resistance cross the network, i.e. a straight line. In composite membranes the addition of microgels creates obstacles (the microgels themselves) that interfere with the normal diffusion of water, forcing it to work around the obstacles. This means that the actual (average) path traversed by the species while diffusing in the interstitial fluid (in our case, water) in a porous medium (composite membranes) is longer than the path traversed in a solid medium (PVP membranes).
The ratio of the actual distance travelled by the species (Δl) per unit length of the medium (Δx) is commonly denominated as tortuosity (τ),29 which is a parameter that strongly influences the diffusion coefficient 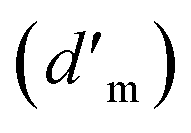 of a fluid through a membrane. The relation between tortuosity and diffusion coefficient is presented in eqn (4).
of a fluid through a membrane. The relation between tortuosity and diffusion coefficient is presented in eqn (4).
 | (4) |
Another possible explanation for the composite membranes' reduced swelling can be the insufficient crosslinking of the microgels. After the microgels' synthesis, if the percentage of crosslinking agent is insufficient, some unbonded polymer chains remain on the surface of the microgel. When submitting the final composite membrane to UV treatment, those free polymer chains may bond to the PVP during the irradiation process, turning the microgels into anchoring points that hinder the swelling of the composite system.
Conclusions
Current research in materials development is focused in producing multifunctional systems using biocompatible materials for biomedical applications. Electrospinning is a simple and versatile tool to produce multifunctional membranes that can be tailored for the desired application. In this work we were able to successfully produce multifunctional, highly porous, crosslinked polymeric membranes with active sites (thermosensitive microgels) incorporated for potential biomedical applications. We have extensively studied the production parameters to create a membrane that can incorporate a certain amount of water and still be able to maintain a somewhat elastic behaviour by reaching a commitment between structural stability, mechanical properties and swelling capability. The most suitable studied UV irradiation time to produce such membrane would be between 20 and 30 minutes, since samples with this irradiation time show mechanical properties identical to the ones that were irradiated for longer periods of time. Above 30 minutes of irradiation, the swelling properties are also similar.Although the confinement of microgels did not show a noticeable change in the mechanical properties of the membranes, changes in the swelling properties were evident. To further investigate the influence of the microgels, a higher load should be confined into the fibres, since with a 10 wt% load the microgels are not homogeneously distributed throughout the entire membrane, which could be affecting the properties of the overall membrane.
This work gives a step forward towards the design and development of new and improved smart systems that can be tailored for the desired application, whether it is biomedical or industrial. For instance, one possibility is loading different stimuli-responsive microgels with distinct types of drugs, thus creating a multiresponsive membrane able to prevent different symptoms; another approach would be loading the microgels with anti-corrosion agents to produce a self-healing membrane. The versatility of the used technique, as well as the different components of the proposed composite are what make this system a very interesting and promising topic of research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by National Funds through FCT-Portuguese Foundation for Science and Technology, Reference UID/CTM/50025/2013 and FEDER funds through the COMPETE 2020 Program under the project number POCI-01-0145-FEDER-007688. We also acknowledge support grant from the Portuguese Science and Technology Foundation through grant SFRH/BPD/88779/2012 (CE).References
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325–347 CrossRef CAS PubMed.
- G. Buschle-Diller, A. Hawkins and J. Cooper, in Modified Fibers with Medical and Specialty Applications, ed. J. V. Edwards, G. Buschle-Diller and S. C. Goheen, Springer Netherlands, Dordrecht, 2006, pp. 67–80, DOI:10.1007/1-4020-3794-5_5.
- M. Scampicchio, A. Bulbarello, A. Arecchi, M. S. Cosio, S. Benedetti and S. Mannino, Electroanalysis, 2012, 24, 719–725 CrossRef CAS.
- D. G. Yu, X. X. Shen, C. Branford-White, K. White, L. M. Zhu and S. W. Bligh, Nanotechnology, 2009, 20, 055104 CrossRef PubMed.
- A. J. Meinel, O. Germershaus, T. Luhmann, H. P. Merkle and L. Meinel, Eur. J. Pharm. Biopharm., 2012, 81, 1–13 CrossRef CAS PubMed.
- D. Crespy, K. Friedemann and A.-m. Popa, Macromol. Rapid Commun., 2012, 33, 1978–1995 CrossRef CAS PubMed.
- A. L. Yarin, Polym. Adv. Technol., 2011, 22, 310–317 CrossRef CAS.
- S. Agarwal and A. Greiner, Polym. Adv. Technol., 2011, 22, 372–378 CrossRef CAS.
- H. Qi, P. Hu, J. Xu and A. Wang, Biomacromolecules, 2006, 7, 2327–2330 CrossRef CAS PubMed.
- F. Elahi, G. Guoping, F. Khan and W. Lu, J. Bioeng. Biomed. Sci., 2013, 3, 1–14 Search PubMed.
- J. E. Díaz, A. Barrero, M. Marquez, A. Fernandez-Nieves and I. G. Loscertales, Macromol. Rapid Commun., 2010, 31, 183–189 Search PubMed.
- S. C. S. Marques, P. I. P. Soares, C. Echeverria, M. H. Godinho and J. P. Borges, RSC Adv., 2016, 6, 76370–76380 RSC.
- G. D'Errico, M. De Lellis, G. Mangiapia, A. Tedeschi, O. Ortona, S. Fusco, A. Borzacchiello and L. Ambrosio, Biomacromolecules, 2008, 9, 231–240 CrossRef PubMed.
- X. F. Zhu, P. Lu, W. Chen and J. A. Dong, Polymer, 2010, 51, 3054–3063 CrossRef CAS.
- S. Chuangchote, T. Sagawa and S. Yoshikawa, J. Appl. Polym. Sci., 2009, 114, 2777–2791 CrossRef CAS.
- Q. Yang, Z. Li, Y. Hong, Y. Zhao, S. Qiu, C. Wang and Y. Wei, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3721–3726 CrossRef CAS.
- G. P. Kumar, A. R. Phani, R. G. Prasad, J. S. Sanganal, N. Manali, R. Gupta, N. Rashmi, G. S. Prabhakara, C. P. Salins, K. Sandeep and D. B. Raju, Int. J. Pharm., 2014, 471, 146–152 CrossRef CAS PubMed.
- J. Rosiak, J. Olejniczak and W. Pȩkala, Int. J. Radiat. Appl. Instrum. C Radiat. Phys. Chem., 1990, 36, 747–755 CAS.
- Q. G. Zhang, W. W. Hu, A. M. Zhu and Q. L. Liu, RSC Adv., 2013, 3, 1855–1861 RSC.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef CAS PubMed.
- T. E. Newsome and S. V. Olesik, J. Appl. Polym.Sci., 2014, 131, 40966 CrossRef.
- M. J. Chen, H. L. Qu, J. H. Zhu, Z. P. Luo, A. Khasanov, A. S. Kucknoor, N. Haldolaarachchige, D. R. Young, S. Y. Wei and Z. H. Guo, Polymer, 2012, 53, 4501–4511 CrossRef CAS.
- S. Agarwal, A. Greiner and J. H. Wendorff, Prog. Polym. Sci., 2013, 38, 963–991 CrossRef CAS.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 512–520 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- L. C. Lopérgolo, A. B. Lugão and L. H. Catalani, Polymer, 2003, 44, 6217–6222 CrossRef.
- D. A. Carr and N. A. Peppas, Macromol. Biosci., 2009, 9, 497–505 CrossRef CAS PubMed.
- L. Shen and Z. X. Chen, Chem. Eng. Sci., 2007, 62, 3748–3755 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07166c |
| This journal is © The Royal Society of Chemistry 2017 |