
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TiCl4/DMAP mediated Z-selective knovenagel condensation of isatins with nitroacetates and related compounds†
Luyi Zhu,
Peipei Yan,
Lvye Zhang,
Zhangtao Chen,
Xiaofei Zeng * and
Guofu Zhong*
* and
Guofu Zhong*
College of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 310036, P. R. China. E-mail: chemzxf@hznu.edu.cn; zgf@hznu.edu.cn
First published on 3rd November 2017
Abstract
A highly efficient Z-selective Knovenagel condensation reaction of isatins with nitroacetates mediated by TiCl4 and DMAP was described. The desired 2-nitro-3-ylideneoxindole acetates were obtained in good to excellent stereoselectivities and yields. Other activated methylene derivatives as well as 4-methylbenzenesulfonamide could provide good results too. This method makes it possible to obtain various unreported 3-ylideneoxindole derivatives under mild reaction conditions.
Introduction
The 3-ylideneoxindole moieties are pharmacologically important structure motifs in many natural indole alkaloids and bioactive molecules, which exhibit potent antifungal,1 anticancer,2 and antiviral activities.3 For example, indirubin (A) from a traditional Chinese herbal medicine is an inhibitor of protein kinases such as glycogen synthase kinase-3b (GSK-3b)4 and cyclin-dependent kinases (CDKs). Nintedanib (Ofev) (B) has been reported as a kinases inhibitor, launched for the treatment of idiopathic pulmonary fibrosis (IPF) and cancer.5 Oxindole compound C and related compounds also exhibited inhibitory effects on CDKs. Recently, 3-ylideneoxindole acetamides (D), as antitumor agents, displayed a similar profile to that of roscovitine.6 (Fig. 1) Furthermore, 3-ylideneoxindoles are the core structure in biologically natural products alkaloids (including neolaugerine,7 costinone A, and costinine B8). Following reports on the medicinal potential and synthetic applicability of 3-ylideneoxindole derivatives, reactions toward the synthesis of such kind of compounds have been intensely explored and several synthetic strategies have been reported. Wittig and Knoevenagel reaction, which are undoubtedly considered as some of the most effective strategies for the preparation of alkenes, have been traditionally applied for the 3-ylideneoxindoles synthesis.9 The palladium-catalyzed Heck–Suzuki–Miyaura domino reactions for the construction of substituted 3-alkylideneoxindoles from ynamides have also been reported.10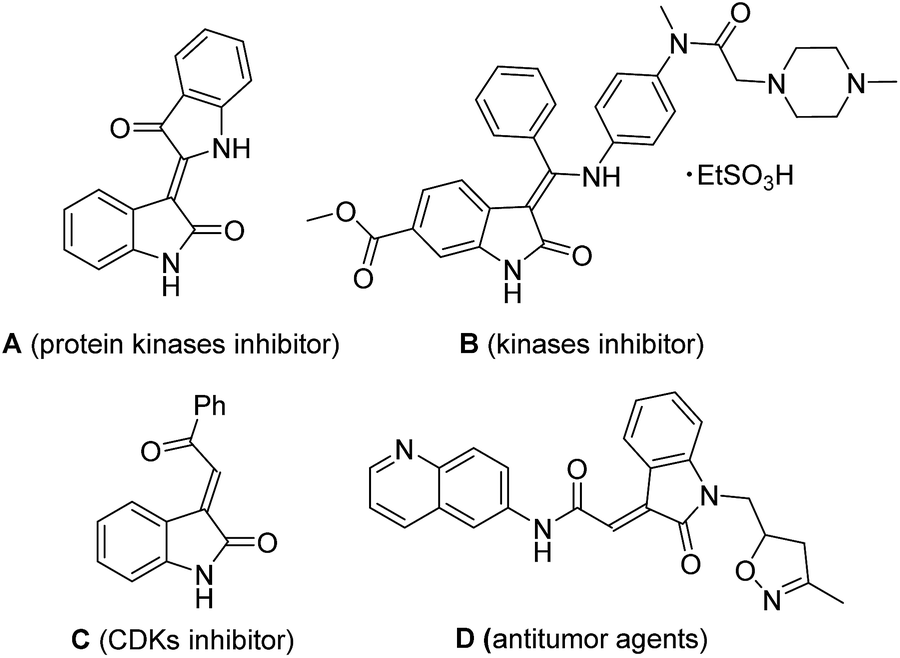 | ||
| Fig. 1 Illustrative examples of bioactive 3,2′-pyrrolidinyl spirooxindoles. | ||
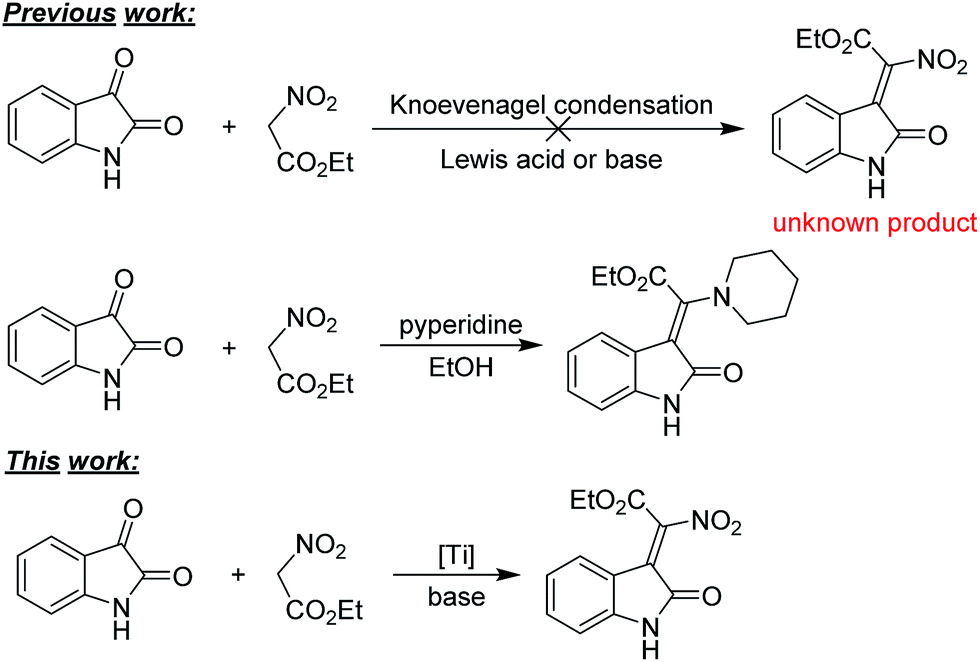
The nitro group is one of the most versatile functional groups, not only because it is essentially a masked amine, but also because its chemistry can be exploited in a number of useful ways.11 The introduction of a nitro group to 3-ylideneoxindole moiety might be of great importance. However, the synthesis of 2-nitro-2-(2-oxoindolin-3-ylidene)acetate derivatives have remained difficult, and to our knowledge, have not been reported to date. Efforts towards the synthesis of such kind of compounds failed when isatins were reacted with ethyl nitroacetate by Alencastro,12 only ethyl (Z)-2-(2-oxoindolin-3-ylidene)-2-(piperidin-1-yl)acetate was obtained (Scheme 1). The outcomes indicated that the nitro group here was unstable and could be easily substituted by the nucleophilic piperidine. Therefore, the development of a highly efficient stereoselective approach for the synthesis of biological and synthetic applicable important 2-nitro-3-ylideneoxindole acetates with regard to stereo-, regio-, and chemoselectivities and substrate generality is in high demand.
 | ||
| Scheme 1 Condensation reactions between isatin and nitroacetate. | ||
Due to the high reaction velocities, good yields, low toxic and toleration of basic labile functionalities, the TiCl4/base mediated Knoevenagel,13 Claisen14 and Dieckmann15 condensations have gained much attention and great importance over traditional methods. Very recently, Massanet and co-workers reported a condensation of acetates and formate esters employing TiCl4/Et3N system, allowing access to a variety of (E)-β-alkoxy- and (E)-β-aryloxyacrylates in good yields.16 We envisioned that TiCl4/base system might be good promoter for the condensation reaction and the non-nucleophilic condition could prevent the further transformation of the product. Herein, we report the first Knovenagel condensation reaction of isatins with nitroacetates in the presence of TiCl4/base in good yields and broad substrate scope.
Results and discussion
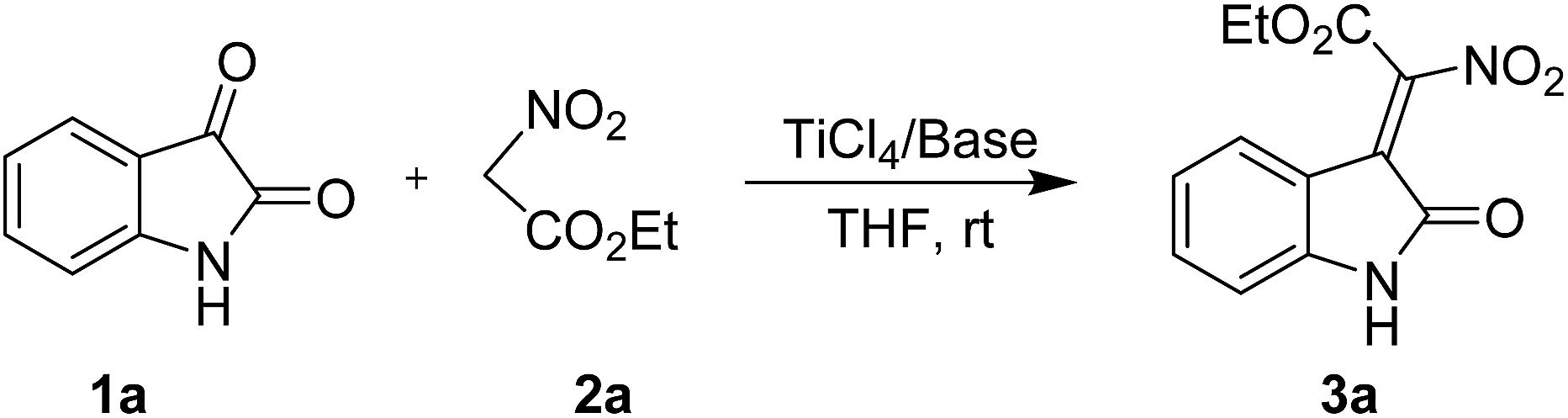
We initiated this study by choosing isatin (1a) and ethyl nitroacetate (2a) as model substrates and subjecting to a preliminary condensation condition using TiCl4 and base (Table 1). After some initial screening, we were pleased to find that the desired 2-nitro-3-ylideneoxindole acetates (3a) can be obtained in 65% yield and 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z/E selectivity in the presence of 1.5 equiv. of TiCl4 and 3.0 equiv. of Et3N at room temperature. The results of the TiCl4/Et3N system were promising, although the stereoselectivity was bad (entry 1). We then employed the organic bases, for instance n-Bu3N, DBU, DIPEA, NMM, pyridine and DMAP in the same method (entries 2–7). All organic bases tested could promote the reaction smoothly and the introduction of DMAP improved both the yield and stereoselectivity, and provided the desired 3a in 88% yield and 8:1 Z/E ratio. Unfortunately, no product could be obtained when inorganic bases were used (entries 8 and 9). After establishment of TiCl4 and DMAP as the optimal reagents in the reaction, different conditions, such as solvents, reaction temperatures, equivalents of metal source and base were subsequently investigated (entries 10–16: for more detailed optimization conditions, see the ESI†). Thus, the best result could be achieved in the presence of 1.5 equiv. of TiCl4 and 2.0 equiv. of DMAP at room temperature, affording 3a in 90% yield and 9:1 Z/E ratio.
:1 Z/E selectivity in the presence of 1.5 equiv. of TiCl4 and 3.0 equiv. of Et3N at room temperature. The results of the TiCl4/Et3N system were promising, although the stereoselectivity was bad (entry 1). We then employed the organic bases, for instance n-Bu3N, DBU, DIPEA, NMM, pyridine and DMAP in the same method (entries 2–7). All organic bases tested could promote the reaction smoothly and the introduction of DMAP improved both the yield and stereoselectivity, and provided the desired 3a in 88% yield and 8:1 Z/E ratio. Unfortunately, no product could be obtained when inorganic bases were used (entries 8 and 9). After establishment of TiCl4 and DMAP as the optimal reagents in the reaction, different conditions, such as solvents, reaction temperatures, equivalents of metal source and base were subsequently investigated (entries 10–16: for more detailed optimization conditions, see the ESI†). Thus, the best result could be achieved in the presence of 1.5 equiv. of TiCl4 and 2.0 equiv. of DMAP at room temperature, affording 3a in 90% yield and 9:1 Z/E ratio.
|
|||||
|---|---|---|---|---|---|
| Entry | TiCl4 (equiv.) | Base (equiv.) | t (h) | Yieldb (%) | Z/Ec |
| a All reactions were carried out with 1.0 equiv. (0.5 mmol) of 1a, 1.0 equiv. (0.5 mmol) of 2a, TiCl4 and base at room temperature.b Yields of isolated product after column chromatography.c Determined by 1HNMR analysis.d NMM = N-methylmorpholine.e No reaction.f CH2Cl2 was used as reaction solvent.g Diethyl ether was used as reaction solvent.h Toluene was used as reaction solvent. | |||||
| 1 | 1.5 | Et3N (3.0) | 8 | 65 | 2:1 |
| 2 | 1.5 | n-Bu3N (3.0) | 8 | 55 | 2:1 |
| 3 | 1.5 | DBU (3.0) | 8 | 67 | 1.5:1 |
| 4 | 1.5 | DIEPA (3.0) | 8 | 88 | 1.5:1 |
| 5 | 1.5 | NMMd (3.0) | 8 | 85 | 2:1 |
| 6 | 1.5 | Pyridine (3.0) | 8 | 68 | 6:1 |
| 7 | 1.5 | DMAP (3.0) | 8 | 88 | 8:1 |
| 8 | 1.5 | CsCO3 (3.0) | 24 | —e | — |
| 9 | 1.5 | Na2CO3 (3.0) | 24 | — | — |
| 10f | 1.5 | DMAP (3.0) | 24 | Trace | — |
| 11g | 1.5 | DMAP (3.0) | 24 | 32 | 2:1 |
| 12h | 1.5 | DMAP (3.0) | 24 | Trace | — |
| 13 | 1.0 | DMAP (3.0) | 8 | 43 | 9:1 |
| 14 | 1.5 | DMAP (2.0) | 8 | 90 | 9:1 |
| 15 | 1.5 | DMAP (1.5) | 12 | 72 | 9:1 |
| 16 | 2.0 | DMAP (3.0) | 12 | 90 | 9:1 |
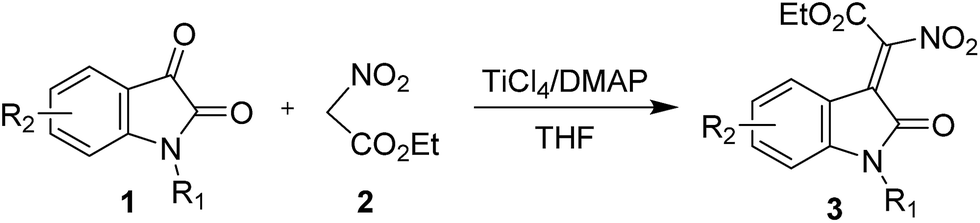
With the optimized reaction conditions in hand, we continued with evaluation of the reaction scope. We first used the ethyl nitroacetate in a variety of combinations with isatins, the corresponding products 3 were isolated in 54–95% yields (Table 2). When we use N-benzyl, N-methyl protected isatins in the reaction, we obtained 3b and 3c, respectively, in 85% and 91% yields and good Z/E ratio (entries 2 and 3); while the N-Boc protected 2d only provided the deprotected 3a in 83% yield with excellent stereoselectivity (entry 4), probably due to the strong acidity of TiCl4. The reaction of halogenated isatins proceeded smoothly and gave the desired product in good results (73–95% yields and 16:1 → 20:1 stereoselectivities, entries 5–9 and 11). However, the strong electron withdrawing group, for instance, 5-NO2 substituted isatin could not provide the desired product at all (entry 13); and isatin with electrondonating groups resulted in lower yields (entries 10, 12 and 14). Importantly, the reaction is amenable to upscaling, and we were able to prepare 1.41 g of 3a in 82% yield with 10:1 Z/E ratio (entry 15).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Yieldb (%) | Z/Ec |
| a All reactions were carried out with 1.0 equiv. (0.5 mmol) of 1, 1.0 equiv. (0.5 mmol) of 2, 2.0 equiv. (1.0 mmol) of TiCl4 and 3.0 equiv. (1.5 mmol) of DMAP at room temperature.b Yields of isolated product after column chromatography.c Determined by 1HNMR analysis.d The reaction was carried out in 10 mmol scale. | |||||
| 1 | H | H | 8 | 90 (3a) | 9:1 |
| 2 | Bn | H | 8 | 85 (3b) | 6:1 |
| 3 | CH3 | H | 8 | 91 (3c) | >20:1 |
| 4 | Boc | H | 8 | 83 (3a) | >20:1 |
| 5 | H | 5-Cl | 8 | 95 (3d) | >20:1 |
| 6 | H | 4-Br | 8 | 80 (3e) | >20:1 |
| 7 | H | 5-Br | 8 | 76 (3f) | >20:1 |
| 8 | H | 6-Br | 24 | 75 (3g) | >20:1 |
| 9 | H | 7-Br | 24 | 78 (3h) | 5:1 |
| 10 | H | 7-Me | 24 | 61 (3i) | 4:1 |
| 11 | H | 5-F | 24 | 73 (3j) | 16:1 |
| 12 | H | 5,7-CH3 | 24 | 74 (3k) | 5:1 |
| 13 | H | 5-NO2 | 8 | — | — |
| 14 | H | 5-OCH3 | 8 | 54 (3l) | >20:1 |
| 15d | H | H | 12 | 82 (3a) | 10:1 |
To determine the stereochemistry of the 2-nitro-3-ylideneoxindole acetates, the structure of 3c was confirmed by X-ray crystallographic analysis (Fig. 2, CCDC 1571225).17 The TiCl4/DMAP mediated condensation reaction provided the desired products in Z conformations (Fig. 2).
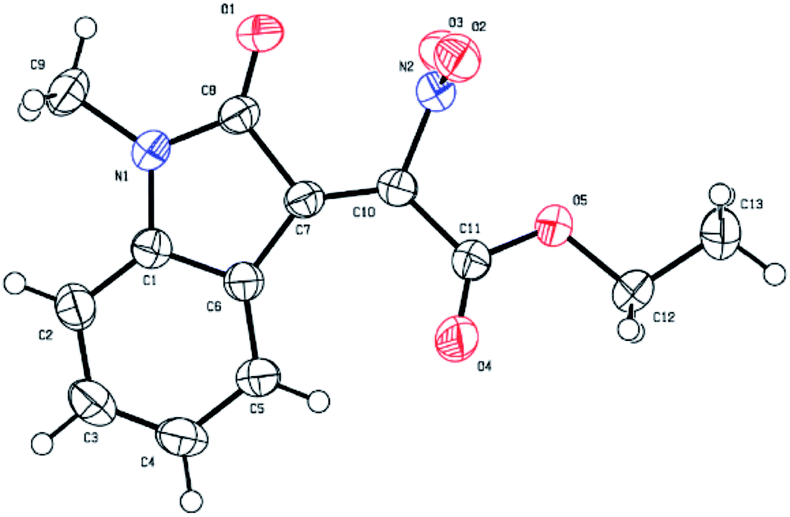 | ||
| Fig. 2 X-Ray crystal structure of product 3c. | ||
It is noteworthy that this method is equally successful with activated methylene derivatives that carry different electro-withdrawing groups, such as 2,2-dimethyl-1,3-dioxane-4,6-dione (4a), malononitrile (4b), ethyl 2-cyanoacetate (4c), and diethyl malonate (4d) (Scheme 2). The corresponding products (5a–d) were obtained in 71–97% yield and with a 7:1 Z/E ratio of 4c as determined by 1H NMR analysis. Furthermore, the reaction of isatin with 4-methylbenzenesulfonamide (4e) also proceeded smoothly and gave the desired imine 6 in 57% yield, which was not reported before (Scheme 3).
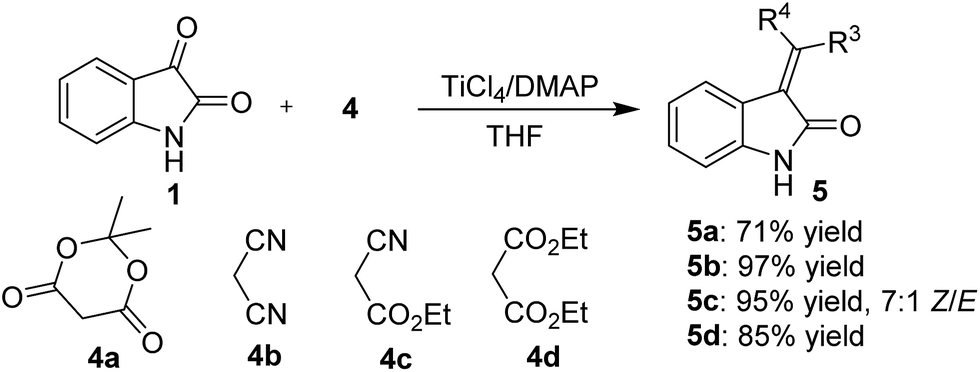 | ||
| Scheme 2 Condensation reaction of isatins with activated methylenes. | ||
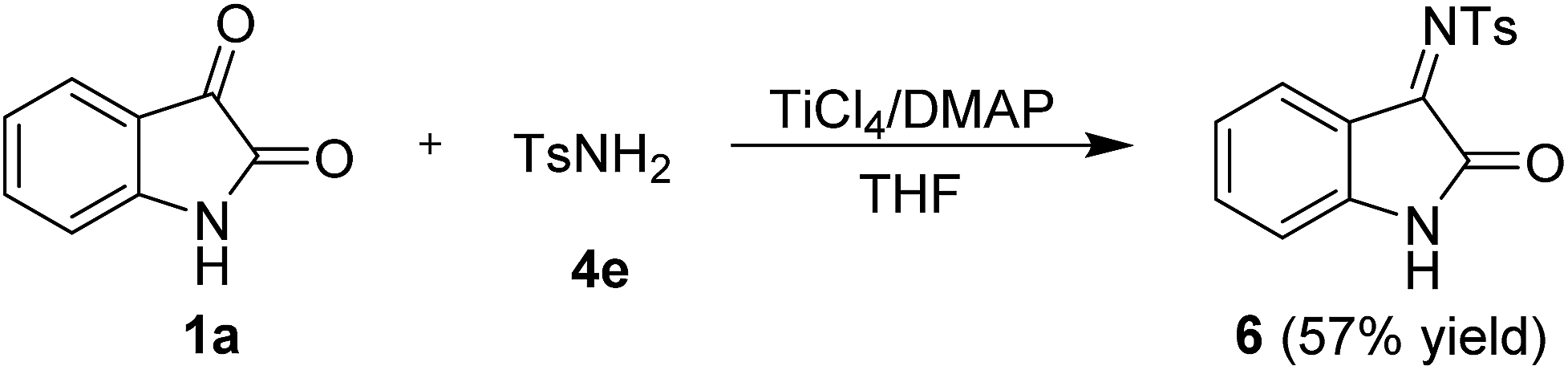 | ||
| Scheme 3 Condensation reaction of isatins with 4-methylbenzene sulfonamide. | ||
To gain insight into the reaction mechanism, we carried out several control experiments. The reaction could not proceed when TiCl4 and DMAP were used separately, which means that TiCl4 and DMAP work cooperatively in the condensation reaction (Scheme 4a and b). When the reaction was carried out in the presence of 1.5 equiv. of piperidine, a piperidine substituted product 7 was afforded in high yield, which was indicated to E configuration through NOE spectrum (see the ESI†) (Scheme 4c). We then used 3a to react with piperidine, it was found that 7 could be obtained in quantitative yield, indicating that the piperidine mediated reaction went through a Knovenagel condensation first, followed by a nucleophilic substitution reaction to yield 7 (Scheme 4d).
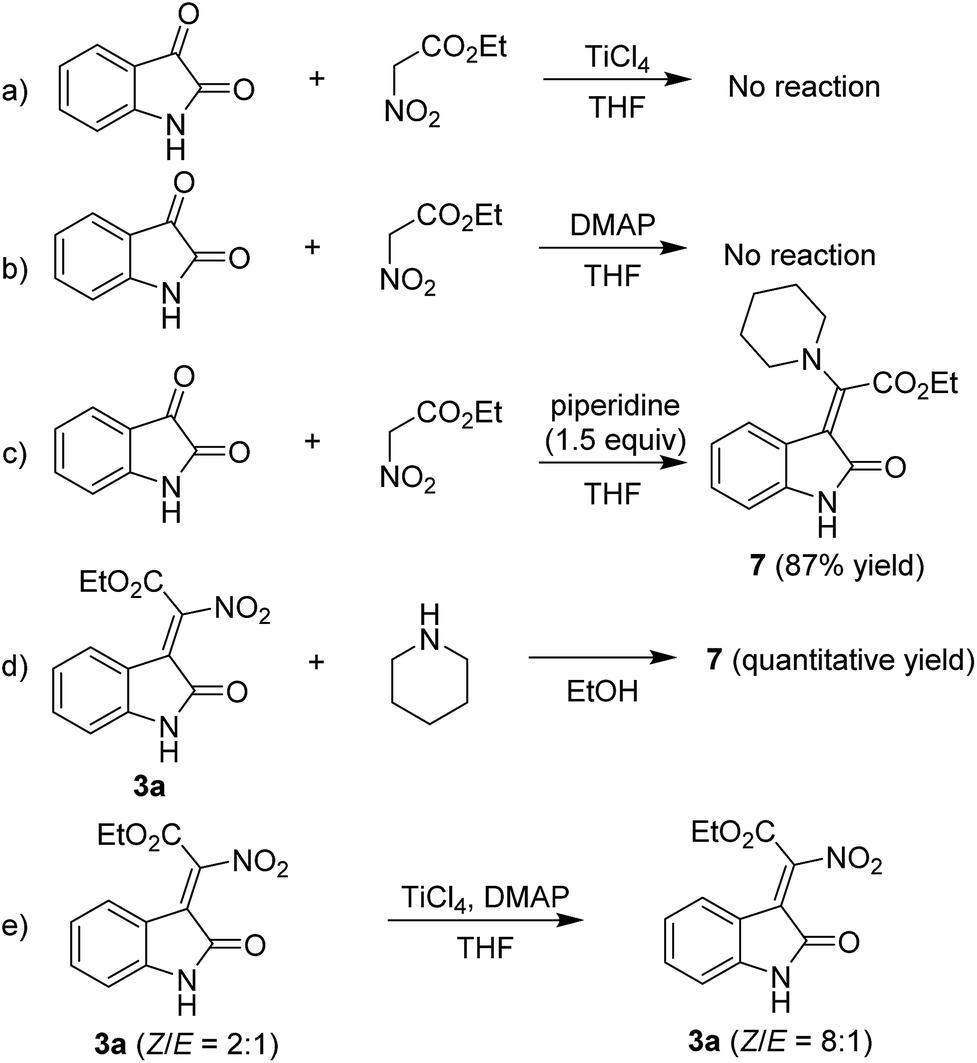 | ||
| Scheme 4 Control experiments. | ||
Based on the above results and previous literature reports, a plausible reaction mechanism is proposed and shown in Fig. 3. Firstly, the ethyl nitroacetate is activated by TiCl4 to form an anionic titanium enolate; then an aldol reaction will happen between the enolate and isatin, leading to the formation of a six membered metallacycles; next, an elimination will occur to form the new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond; further elimination of a TiOCl2 yields the desired product. The organic bases play a major role in the stereoselectivity of the condensation reaction. We found that the Z/E ratio of 3a could be changed from 2:1 to 8:1 under the TiCl4/DMAP condition, which indicated that highly nucleophilic DMAP would undergo repetitive addition–elimination onto the double bond formed to isomerize the initially formed E isomers into the Z isomers (Scheme 4e).
C double bond; further elimination of a TiOCl2 yields the desired product. The organic bases play a major role in the stereoselectivity of the condensation reaction. We found that the Z/E ratio of 3a could be changed from 2:1 to 8:1 under the TiCl4/DMAP condition, which indicated that highly nucleophilic DMAP would undergo repetitive addition–elimination onto the double bond formed to isomerize the initially formed E isomers into the Z isomers (Scheme 4e).
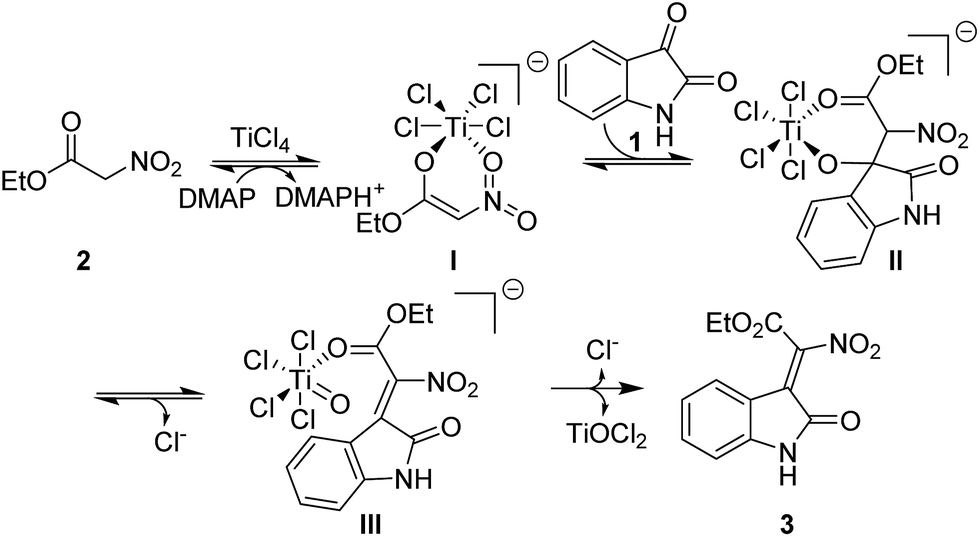 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a Z-selective Knovenagel condensation reaction of isatins with nitroacetates in the presence of TiCl4 and an organic base. Wild substrate scope was explored, and provided the corresponding 2-nitro-3-ylideneoxindole acetates in good stereoselectivities and yields. In addition, other activated methylene derivatives such as 2,2-dimethyl-1,3-dioxane-4,6-dione, malononitrile, ethyl 2-cyanoacetate, and diethyl malonate reacted under identical conditions in the same way, yielded the condensation product in good results. As an extension, condensation between isatin with 4-methylbenzenesulfonamide was also promoted by TiCl4/DMAP, gave the unreported imine in moderate yield.Experimental section
General
All starting materials were obtained from commercial sources and were used without further purification unless otherwise stated. THF was dried and distilled from sodium benzophenone prior to use. CHCl3 and CH2Cl2 were distilled from CaH2 prior to use. 1H and 13C NMR spectra were recorded on a Bruker AMX500 (500 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm), and the residual solvent peak was used as an internal reference: proton (d-chloroform δ 7.26), carbon (d-chloroform δ 77.0). Multiplicity was indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublet), br s (broad singlet). Coupling constants were reported in Hertz (Hz). All high resolution mass spectra were obtained on an Agilent 5975 spectrometer. Analytical thin layer chromatography (TLC) was performed using Merck 60 F254 precoated silica gel plate (0.2 mm thickness). Subsequent to elution, plates were visualized using UV radiation (254 nm) on Spectroline Model ENF-24061/F 254 nm. Further visualization was possible by staining with basic solution of potassium permanganate or acidic solution of ceric molybdate. Flash column chromatography was performed using Merck aluminium oxide 90 active neutral with freshly distilled solvents. Columns were typically packed as slurry and equilibrated with the appropriate solvent system prior to use.A representative procedure for TiCl4/DMAP mediated condensation reaction of isatins with ethyl nitroacetate
A stirred solution of isatin (1a, 0.5 mmol, 1.0 equiv.) in anhydrous THF was cooled to 0 °C, TiCl4 (1.0 mmol, 2.0 equiv.) and ethyl nitroacetate (2a, 0.5 mmol, 1.0 equiv.) were added slowly, then DMAP (1.5 mmol, 3.0 equiv.) was added, the resulting mixture was stirred at rt. After the reaction was complete (monitored by TLC), DCM (10 mL) was added and the mixture was filtered through a pad of celite; the celite pad was then washed by DCM (5.0 mL each) for two times; the combined organic layers were dried and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (EtOAc/hexane) to provide 3a.A representative procedure for TiCl4/DMAP mediated condensation reaction of isatins with activated methylenes (4a–4d) and 4-methylbenzenesulfonamide (4e)
A stirred solution of isatin (1a, 0.5 mmol, 1.0 equiv.) in anhydrous THF was cooled to 0 °C, TiCl4 (1.0 mmol, 2.0 equiv.) and 2,2-dimethyl-1,3-dioxane-4,6-dione (4a, 0.5 mmol, 1.0 equiv.) were added slowly, then DMAP (1.5 mmol, 3.0 equiv.) was added, the resulting mixture was stirred at rt. After the reaction was complete (monitored by TLC), DCM (10 mL) was added and the mixture was filtered through a pad of celite; the celite pad was then washed by DCM (5.0 mL each) for two times; the combined organic layers were dried and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (EtOAc/hexane) to provide 5a.Procedure for the substitution reaction of piperidine with 3a
To a stirred solution of 3a (0.2 mmol, 1.0 equiv.) in EtOH at room temperature, piperidine (0.6 mmol, 3.0 equiv.) was added. The result mixture was stirred at room temperature until the disappearance of the starting material (monitored by TLC). EtOH was removed by rotavapor under reduced pressure; the resulting residue was purified by flash chromatography (EtOAc/hexane) to provide 6.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of China (No. 21373073 and 21672048), the PCSIRT (IRT 1231), the Public Welfare Project of Zhejiang Province (2016C33088), and Hangzhou Normal University for financial support. X. Z. acknowledges a Xihu Scholar award from Hangzhou City, and G. Z. acknowledges a Qianjiang Scholar from Zhejiang Province in China.References
-
(a) M. S. C. Pedras, J. L. Sorensen, F. J. Okanga and I. L. Zaharia, Bioorg. Med. Chem. Lett., 1999, 9, 3015 CrossRef CAS
; (b) M. S. C. Pedras, P. B. Chumala and M. Suchy, Phytochemistry, 2003, 64, 949 CrossRef CAS PubMed
-
(a) C.-T. Chiou, W.-C. Lee, J.-H. Liao, J.-J. Cheng, L.-C. Lin, C.-Y. Chen, J.-S. Song, M.-H. Wu, K.-S. Shia and W.-T. Li, Eur. J. Med. Chem., 2015, 98, 1 CrossRef CAS PubMed
- T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Tuntlad, K. Zhang, D. Karanewsky and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2109 CrossRef CAS PubMed
-
(a) R. Hoessel, S. Leclerc, J. A. Endicott, M. E. M. Nobel, A. Lawrie, P. Tunnah, M. Leost, E. Damiens, D. Marie, D. Marko, E. Niederberger, W. Tang, G. Eisenbrand and L. Meijer, Nat. Cell Biol., 1999, 1, 60 CrossRef CAS PubMed
-
(a) G. J. Roth, A. Heckel, F. Colbatzky, S. Handschuh, J. Kley, T. Lehmann-Lintz, R. Lotz, U. Tontsch-Grunt, R. Walter and F. Hilberg, J. Med. Chem., 2009, 52, 4466 CrossRef CAS PubMed
-
(a) C. L. Woodard, Z. Li, A. K. Kathcart, J. Terrell, L. Gerena, M. Lopez-Sanchez, D. E. Kyle, A. K. Bhattacharjee, D. A. Nichols, W. Ellis, S. T. Prigge, J. A. Geyer and N. C. Waters, J. Med. Chem., 2003, 46, 3877 CrossRef CAS PubMed
-
(a) B. Weniger, Y. Jiang, R. Anton, J. Bastida, T. Varea and J.-C. Quirion, Phytochemistry, 1993, 32, 1587 CrossRef CAS
- I. Fatima, I. Ahmad, S. A. Nawaz, A. Malik, N. Afza, G. Luttfullah and M. I. Choudhary, Heterocycles, 2006, 68, 1421 CrossRef CAS
-
(a) R. Yanada, S. Obika, M. Oyama and Y. Takemoto, Org. Lett., 2004, 6, 2825 CrossRef CAS PubMed
-
(a) M. Chakrabarty, R. Mukherjee, S. Arima and Y. Harigaya, Hetereocycles, 2009, 78, 139 CrossRef CAS
-
(a) B. Gabriele, G. Salerno, L. Veltri, M. Costa and C. Massera, Eur. J. Org. Chem., 2001, 4607 CrossRef CAS
- S. J. Garden, C. R. W. Guimarães, M. B. Corréa, C. A. F. Oliveira, A. C. Pinto and R. B. Alencastro, J. Org. Chem., 2003, 68, 8815 CrossRef CAS PubMed
-
(a) W. Lehnert, Tetrahedron, 1973, 29, 635 CrossRef CAS
-
(a) R. K. Hill and H. N. Kbatri, Tetrahedron Lett., 1997, 19, 4337 CrossRef
-
(a) M. Patek, Collect. Czech. Chem. Commun., 1990, 1223 CrossRef CAS
- J. M. Álvarez-Calero, Z. D. Jorge and G. M. Massanet, Org. Lett., 2016, 18, 6344 CrossRef PubMed
- A CIF file for 3c was deposited with the Cambridge Crystallographic Data Centre. CCDC 1571225 contains the supplementary crystallographic data for this paper.
- D. V. Demchuk, M. N. Elinson and G. I. Nikishin, Mendeleev Commun., 2011, 21, 224 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details on the experimental procedure, characterization data of the products 3–6. CCDC 1571225. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09951g |
| This journal is © The Royal Society of Chemistry 2017 |