
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sawdust as an effective biotemplate for the synthesis of Ce0.8Zr0.2O2 and CuO–Ce0.8Zr0.2O2 catalysts for total CO oxidation†
Igor Yu. Kaplin *,
Ekaterina S. Lokteva,
Elena V. Golubina,
Konstantin I. Maslakov,
Natalia E. Strokova,
Sergei A. Chernyak and
Valery V. Lunin
*,
Ekaterina S. Lokteva,
Elena V. Golubina,
Konstantin I. Maslakov,
Natalia E. Strokova,
Sergei A. Chernyak and
Valery V. Lunin
Lomonosov Moscow State University, Chemistry Department, Leninskie Gory 1/3, Moscow, 119991, Russia. E-mail: kaplinigormsu@gmail.com
First published on 3rd November 2017
Abstract
Wood sawdust (SD) was successfully used as a biotemplate to prepare precursors of Ce0.8Zr0.2O2 (CZ) and CuO–Ce0.8Zr0.2O2 (Cu–CZ, about 25% of CuO) catalysts, which were tested in total CO oxidation at 100–400 °C after calcination at 500 or 600 °C, and compared with analogous systems prepared using cetyltrimethylammonium bromide (CTAB). The improved specific surface area and oxygen mobility of the catalysts calcined at 500 °C resulted in an increased efficiency in CO oxidation. CZ (SD-500) demonstrated much higher catalytic efficiency than the CZ (CTAB-500) sample despite having a two times less SBET due to the promoting action of Ca and K inherited from sawdust and improved concentration of superoxide surface centers. Both facilitation of CO adsorption on peroxide and superoxide centers and increased oxygen mobility provide improvement in the high-temperature efficiency of biomorphic CZ in CO oxidation. The XRD, XPS and TPR data testify that Cu in modified systems partially exists as a separate CuO phase, in small amounts as Cu+, and partially is incorporated in the surface CuyCexZr1−x−yO2−z and/or CuyCe1−yO2−x phases. Modification with Cu enhances the low-temperature efficiency of CZ systems, prepared using both templates and calcined at 500 °C. Cu–CZ (SD-500) was a bit less active than Cu–CZ (CTAB-500) only at 100–150 °C, primarily due to the decreased CuO content, and the difference in CuO interaction with CZ support, found by TPR. Presumably CuO provides low-temperature CO oxidation through the Langmuir–Hinshelwood mechanism; the Cu+/Cu2+ pair can also participate as an additional redox pair in high-temperature CO oxidation by the Mars–van-Krevelen mechanism.
1. Introduction
It is widely known that the efficiency of catalytic systems can be tuned by modification of the preparation technique. Biotemplate methods have attracted considerable interest in the past decades.1 Natural biological materials such as rattan, rice husk, cellulose, paper2–5 and widespread wood substances6,7 are used as a template for the synthesis of promising biomorphic ceramic materials of various chemical composition and texture for diverse applications. Biotemplating is an especially suitable method of mixed oxide preparation as it helps to avoid the segregation of individual components of multi-component oxides.5 The use of natural templates, especially waste materials such as wood sawdust, complies with the principles of green chemistry. Moreover, biotemplates impart to the biomorphic oxide materials the unique textural properties of natural ones, which is very promising for catalytic applications. Furthermore, minor components of biotemplates, e.g. ash admixtures, could serve as catalyst promoters. Little attention has been paid in the literature to this aspect of biotemplating.Due to their unique redox properties Ce-containing oxides are among the most important component of three-way catalysts (TWCs) which transform CO, NOx and hydrocarbons to harmless CO2, N2 and water.8 Easy electron transfer between Ce4+ and Ce3+ ions provides increased mobility of the lattice oxygen, which can be donated or accepted by oxide depending on the reaction conditions.9
The incorporation of Zr4+ into the CeO2 lattice to form mixed Ce1−xZrxO2 oxides increases thermal stability and allows the formation of a highly defective structure,10,11 which additionally improves bulk mobility of the lattice oxygen.12 This mobility is known to be a necessary condition of fast redox processes on the oxide surface.
There is some evidence in literature that the addition of CaO13–17 and alkali metals13,18 influences the catalytic properties of ceria and CZ systems. At the same time, they are among the main ash impurities in wood materials.19 Therefore, we can expect additional improvement of the catalytic action of biomorphic CZ produced using wood as a template.
Modification of CeO2 or Ce1−xZrxO2 with CuO results in a synergy in CO oxidation.20,21 Simultaneous participation of two redox pairs (Cu2+/Cu1+ and Ce4+/Ce3+) in a catalytic cycle was proposed to be a reason for positive Cu influence.22 The activity of CuO–CeO2 catalysts in CO oxidation shows a strong dependence on the preparation method,23 but to the best of our knowledge the catalytic properties of Cu–CZ prepared using wood as a biotemplate have not been studied earlier.
The present paper focuses on filling this gap in the case of ceria–zirconia double oxides, both pristine and modified with CuO. For this purpose, double Ce0.8Zr0.2O2 and triple CuO–Ce0.8Zr0.2O2 oxide systems were prepared using pine sawdust as a biotemplate. The influence of the calcination temperature of these systems and their modification with CuO additive on the physicochemical properties is discussed. Catalytic properties were tested in CO oxidation in a stoichiometric CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 mixture. Catalysts of the same composition but prepared using the organic surfactant cetyltrimethylammonium bromide (CTAB) as a template were used as comparative samples.
:O2 mixture. Catalysts of the same composition but prepared using the organic surfactant cetyltrimethylammonium bromide (CTAB) as a template were used as comparative samples.
2. Experimental
2.1. Catalyst preparation
The cerium to zirconium ratio in the catalysts (4:1) was chosen based on our previous work.24 Biomorphic Ce–Zr oxide with a Ce:Zr molar ratio of 4:1 showed better efficiency in CO oxidation in comparison with higher Zr loaded oxide systems.
Ce0.8Zr0.2O2 and CuO–Ce0.8Zr0.2O2 biomorphic systems were prepared by the impregnation of ∼15 g of pine (Pinus sylvestris) sawdust (SD, sieved fraction 0.25–0.5 mm) with the solutions of calculated amounts of metal salts ZrO(NO3)2·H2O (99.5%, Acros Organics), (NH4)2[Ce(NO3)6] (chemically pure, Reachim), and Cu(CH3COO)2·H2O (analytically pure, Vecton) for the CuO modified samples.
Impregnated sawdust was dried for 3–4 h at 120 °C to produce catalyst precursors, denoted as CZ (SD) and Cu–CZ (SD). Then the precursors were calcined at 500 °C for 3.5 h or at 600 °C for 4 h. These samples were named CZ (SD-500), Cu–CZ (SD-500), CZ (SD-600), and Cu–CZ (SD-600).
The intended molar ratio was Ce:Zr = 4:1 for CZ and Cu:Ce:Zr = 1.67:4:1 (25 mol% Cu) for Cu–CZ. The total amount of metal salts was 0.01 mol in each case.
The comparative oxide systems were produced by a surfactant-assisted method using cetyltrimethylammonium bromide (CTAB) as a template.25 Water solutions of calculated amounts of ZrO(NO3)2·H2O, (NH4)2[Ce(NO3)6] and Cu(CH3COO)2·H2O for the CuO modified samples and of 0.005 mol citric acid monohydrate (C6H8O7·H2O) (pure, Reachem) were simultaneously and slowly added to an ethanol solution of 0.005 mol CTAB (99%, BioChemica) with further continuous stirring for 1 h and evaporation of the solvents to form a gel-like substance. The gel was dried for 3 h at 120 °C to form a precursor of the catalysts. The precursor was calcined at 500 °C for 3.5 h or at 600 °C for 4 h to form CZ (CTAB-500), Cu–CZ (CTAB-500), CZ (CTAB-600), and Cu–CZ (CTAB-600). Sample designations are summarized in Table 1.
| Sample | CuO contenta (mol%) | SBETb (m2 g−1) | Lattice parameter ac (Å) | CuO crystallite sizec (nm) | XPS results | Surface content found by SEM-EDS, at% | |||
|---|---|---|---|---|---|---|---|---|---|
| Ce3+/Ce4+ | Cu+/Cu2+ | Oact (%) | Ca | K | |||||
| a Found by AAS.b SBET – the specific surface area determined by the multi-point BET method.c On the basis of XRD data. | |||||||||
| CZ (CTAB-500) | — | 165 ± 17 | 5.425 ± 0.002 | — | 0.19 | — | 15.5 | — | — |
| CZ (CTAB-600) | — | 75 ± 8 | 5.398 ± 0.005 | — | 0.11 | — | 13.9 | — | — |
| Cu–CZ (CTAB-500) | 29.0 | 89 ± 9 | 5.400 ± 0.001 | 13.0 ± 0.5 | 0.00 | 0.23 | 17.7 | — | — |
| Cu–CZ (CTAB-600) | 21.2 | 62 ± 6 | 5.399 ± 0.009 | 13.8 ± 0.6 | 0.05 | 1.70 | 15.3 | — | — |
| CZ (SD-500) | — | 45 ± 5 | 5.419 ± 0.002 | — | 0.14 | — | 17.1 | 1.2 | 0.4 |
| CZ (SD-600) | — | 35 ± 4 | 5.409 ± 0.004 | — | 0.09 | — | 15.1 | 1.4 | 0.3 |
| Cu–CZ (SD-500) | 22.6 | 93 ± 9 | 5.394 ± 0.002 | 12.0 ± 0.3 | 0.02 | 0.25 | 22.1 | 1.1 | — |
| Cu–CZ (SD-600) | 21.7 | 37 ± 4 | 5.413 ± 0.003 | 12.3 ± 0.5 | 0.01 | 1.33 | 15.8 | 2.3 | 0.4 |
2.2. Characterization
Scanning electron microscopy combined with energy dispersive X-ray spectroscopy (SEM/EDS) was performed on a JSM-6390LA (JEOL Ltd., Japan) instrument equipped with an EDS unit.The diffraction patterns were obtained using Bruker D8 Advance (Germany) and Rigaku MiniFlex 300/600 (Japan) powder diffractometers (CuKα radiation, 1.5418 Å). The XRD patterns were collected in a 2θ range of 10–70° with a step size of 0.02°. The JCPDS-PDF2 ICDD (International Center for Diffraction Data) library was used for the identification of phases.
The lattice parameters were calculated using the equation:  , where dhkl is the interplanar distance, and h, k, and l are Miller indices of crystallographic planes.
, where dhkl is the interplanar distance, and h, k, and l are Miller indices of crystallographic planes.
The average size of the crystallites was estimated using the Scherrer equation26 for the most intense reflection peaks:
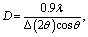
The XPS spectra were acquired on an Axis Ultra DLD spectrometer (Kratos Analytical, UK) using a monochromatic AlK* source (hν = 1486.7 eV, 150 W). The pass energy of the analyser was 40 eV. The binding energy (EB) scale of the spectrometer was preliminarily calibrated using the position of the peaks for the Au 4f7/2 (83.96 eV) and Cu 2p3/2 (932.62 eV) core levels of pure metallic gold and copper. The powder samples were mounted on a holder using double-sided adhesive tape. The Kratos charge neutraliser system was used, and the spectra were charge-corrected to give a Zr 3d5/2 peak at a binding energy of 182.1 eV typical for Zr4+ ions.
The BET specific surface area (SBET) was determined by low temperature N2 adsorption using an Autosorb-1 (Quantachrome, USA) physisorption analyzer. Samples were preliminarily degassed at 200 °C for 3 h.
Temperature-programmed reduction (TPR) was performed on a USGA-101 (UNISIT, Russia) chemisorption analyzer. Pure NiO was used for the preliminary calibration of the instrument. In a typical experiment, approx. 0.050 g of the sample was heated at 300 °C under Ar flow for 0.5 h, cooled down to 30 °C and heated again under diluted hydrogen flow (30 mL min−1, 5% H2 in Ar) from 30 to 850 °C at a 10 °C min−1 heating rate.
Thermal analysis was performed using an STA 449C (Jupiter, NETZSCH, Germany) synchronous thermal analyzer under air flow with a heating rate of 5 °C min−1. The samples produced using CTAB and SD were heated from room temperature to 800 °C or 1000 °C, respectively, at a 35 mL min−1 flow rate. The heat effects (DSC) and thermogravimetric curve (TG) were recorded simultaneously.
Cu content in the samples was measured by atomic absorption spectroscopy (AAS) on a Thermo Fisher Scientific series iCE 3000 spectrophotometer (USA) using atomization in an air–acetylene flame. Device control and processing of the results were performed using SOLAAR Data Station software. Before analysis, solid samples were dissolved by heating in a mixture of (NH4)2SO4 and concentrated H2SO4 (1:2 molar ratio).
The solution was then diluted with distilled water up to a desired volume. Standard solutions for calibration curves were prepared taking into account the background concentrations of Ce and Zr in the tested solutions.
2.3. Catalytic tests
The samples were tested in total CO oxidation using a fixed-bed pulse microcatalytic set. A weighed amount of catalyst (0.1 g) was placed in a quartz flow reactor equipped with a thermocouple and a tubular furnace. The desired temperature of the reaction was adjusted at a constant flow of He (60 mL min−1) through the reactor. Pulses of the reaction mixture (2 vol% CO and 1 vol% O2 in He) were fed into the reactor using a 6-way valve with a calibrated gas loop (1 mL). The products were analyzed by online gas chromatography (GC) using a thermal conductivity detector and 1 m packed column (Porapak Q). The stationary conversion in the isothermal mode used to plot the kinetics curves was achieved after 10–12 pulses and remained constant for at least 10 pulses.The fraction of CO2 formed (XCO2) was determined using the following equation:

3. Results and discussion
3.1. Physical and textural properties
The DSC-TG curves of Cu–CZ (SD) and Cu–CZ (CTAB) are presented in Fig. 1. The small mass loss at 50–200 °C accompanied by endothermic peaks at 90 and 190 °C in the DSC curve of Cu–CZ (SD) (Fig. 1a) results from the evaporation of physically bound absorbed water.27 The significant mass loss in the temperature range of 225–425 °C accompanied by a broad and intense exothermic peak with a maximum at about 345–400 °C corresponds to the decomposition of main wood components: cellulose, hemicellulose and lignin. Most likely the decomposition of the metal salts used as metal precursors also takes place in this temperature range. Separate exothermic peaks of the decomposition of each metal salt overlap with the broad peak of heat evolution in the range of 225–425 °C and therefore are not visible in DSC curves. Since no mass changes or thermal effects are observed at temperatures above 425 °C, the decomposition processes are accomplished below this point, and no phase transformations occur at higher temperatures.
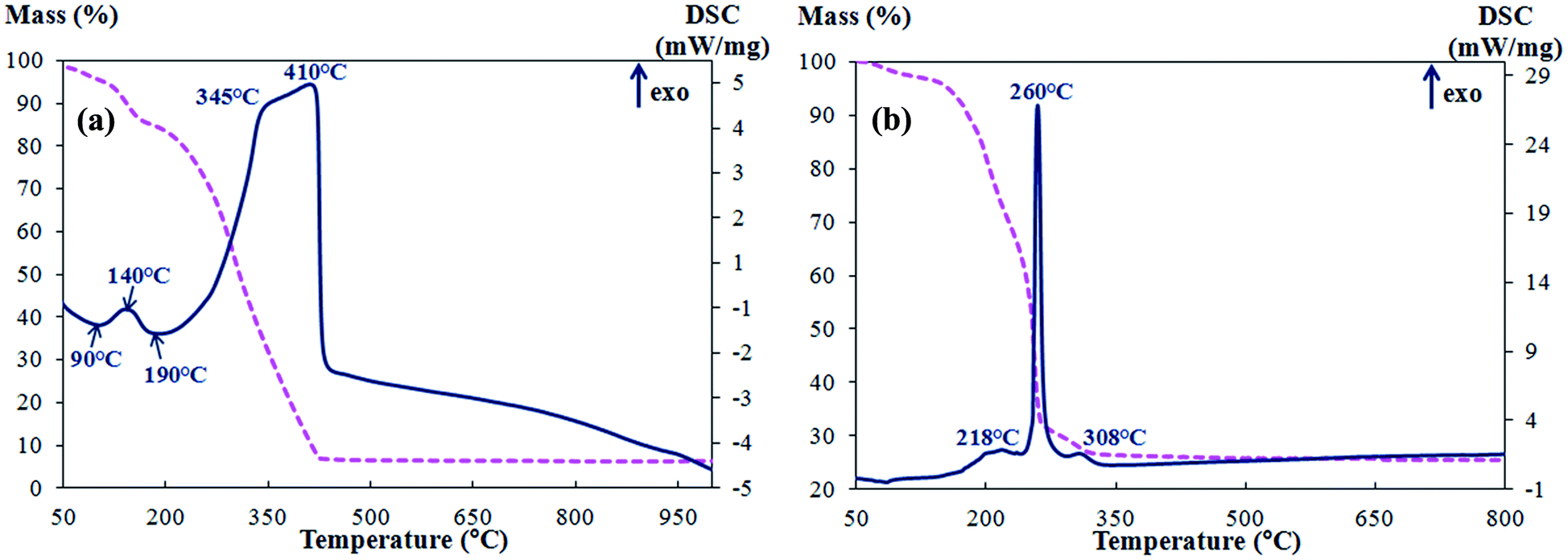 | ||
| Fig. 1 DSC-TG profiles of Cu–CZ (SD) (a) and Cu–CZ (CTAB) (b) precursors of Cu-containing catalysts. | ||
The DSC and TG curves of the Cu–CZ (CTAB) precursor are shown in Fig. 1b. The endothermic peak in the DSC curve in the temperature range of 80–100 °C and the corresponding mass loss (3.3%) result from water desorption. CTAB burning out produces a narrow intense peak of heat evolution with a maximum at 260 °C.21,25 The less intense exothermic peak at 218 °C probably arises due to the start of Cu(CH3COO)2 and citric acid decomposition. The mass loss and heat release peaks at 308 °C could also be caused by salt decomposition. In ref. 28 thermal decomposition of freeze-dried copper(II) acetate monohydrate was investigated by DSC-TG to propose a set of reactions defining the composition of the final product. It was found that calcination at 325–425 °C resulted in the formation of several coexisting phases that were identified by XRD and HT XRD as Cu0, Cu2O and CuO, but the final product formed above 525 °C was CuO. ZrO(NO3)2·H2O used in our work as a Zr source undergoes dehydration at 85–180 °C and decomposition at 180–350 °C accompanied with gaseous NOx evolution29 that proceeds in a similar temperature range of 180–280 °C.30 No mass changes or thermal effects and therefore no phase transformations were observed during further heating above 325 °C. Thus, calcination at a temperature of 325 °C is effective for the formation and crystallization of CZ and CuO from the corresponding salts.
The comparison of the DSC-TG data for the Cu–CZ (SD) and Cu–CZ (CTAB) samples reveals the difference in template removal: the organic CTAB template burns out in a narrow temperature range, whereas sawdust removal takes place in a significantly wider temperature range. This fact is consistent with the chemical nature of the templates: CTAB is an individual chemical compound whereas wood substance comprises a set of organic fragments of various compositions.
Based on the DSC-TG data for the catalyst precursors, two different temperatures of 500 and 600 °C were chosen for calcination. Incomplete sawdust decomposition is possible at temperatures below 500 °C whereas temperatures higher than 600 °C can cause excessive crystal growth and a decrease in SBET, and eventually deterioration of the catalytic efficiency of the calcined material. Such a trend was earlier observed for a CuO/Ce0.8Zr0.2O2 system prepared by a surfactant-assisted method.21
 | ||
| Fig. 2 SEM images of Cu–CZ (CTAB-500), Cu–CZ (CTAB-600), Cu–CZ (SD-500), Cu–CZ (SD-600) and pine sawdust. | ||
The surface texture of the biomorphic systems is drastically different: it reproduces the fibrous texture of wood sawdust used as a template.24 Like the CZ (CTAB) samples, the texture of the biomorphic samples does not depend on the calcination temperature.
Elemental composition of the samples was determined by EDS during SEM investigation. It was found that the surface of the CTAB-templated oxides, in addition to Ce, Zr and O, contains C and Br residues of the decomposition of CTAB and citric acid (the carbon content on the surface of all samples was between 15 and 20 at% and the Br content was between 0.5 to 1.5 at%). A similar amount of residual carbon after sawdust burning was found on the surface of the biomorphic oxides as well. These samples also contain small amounts of Ca and K (Table 1) inherited from raw sawdust. The presence of such impurities can enhance the catalytic efficiency. These elements are known to serve as promoters for Ce-containing catalysts of soot and CO oxidation.13,18
EDS element maps demonstrate the uniform distribution of Ce and Zr on the surface of all samples (see Fig. S1a and b in the ESI†), possibly due to the presence of a single phase containing both elements.
The content of copper in the modified samples was measured by AAS (Table 1). The CuO loading was 29 mol% in Cu–CZ (CTAB-500) and about 20–23 mol% in the other three samples. The difference can be explained by the separate preparation of the precursor for each sample.
The BET specific surface area of the samples is presented in Table 1. Both the increase of calcination temperature and modification with CuO lead to a decrease of SBET of the CTAB-templated samples possibly due to sintering and blocking of the pore openings with CuO agglomerates. The same trends were observed in other studies.21,31
The biomorphic systems demonstrate lower SBET than the CTAB-templated ones. It seems that the biotemplate promotes the development of macroporosity which was confirmed by SEM, but it is less effective in the micro- and mesopore formation than CTAB. In contrast with the CTAB-templated oxides, Cu modification has nearly no influence on the SBET of the biomorphic samples calcined at 600 °C, and significantly improves the SBET of the modified Cu–CZ (SD-500) sample in comparison with non-modified CZ (SD-500). Thus, the SBET values of Cu–CZ (SD-500) and Cu–CZ (CTAB-500) are comparable.
Both SEM and N2 adsorption methods reveal that the texture of the samples depends strongly on the template used, and this difference could influence the catalytic properties in CO oxidation. The presence of mesopores32 or meso–macropores31,33 in oxide catalysts usually provides high catalytic efficiency in CO oxidation. The presence of ash impurities as minor components distinguishes the biomorphic samples from the CTAB-templated ones.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and tetragonal ZrO2 (JSPDS no. 01-073-1441, space group P42/nmc) are shown in Fig. 3 for comparison.
m) and tetragonal ZrO2 (JSPDS no. 01-073-1441, space group P42/nmc) are shown in Fig. 3 for comparison.
 | ||
| Fig. 3 XRD patterns of SD- and CTAB-templated samples calcined at 500 °C (a) and 600 °C (b). | ||
There is no significant difference in the XRD patterns of the samples prepared using the SD and CTAB templates. All of them show symmetric reflexes at 2θ = 28.79, 33.36, 47.90, and 56.85° corresponding to CZ with Ce:Zr ≈ 4 crystallized in a cubic phase of fluorite structural type.21 The presence of CZ mixed oxide in all samples agrees with the SEM-EDS results which testifies the uniform distribution of Ce and Zr.
Diffraction patterns of the Cu-containing samples comprise weak reflexes at 2θ of 35.5 and 38.7°, which can be attributed to a separate CuO phase (tenorite, space group B2/b).
The significant width of the CZ mixed oxide reflexes in the diffraction patterns of all samples can be caused by the presence of a small amount of tetragonal CZ phase,34 or, more probably, by the small size of coherent scattering regions. Their size was estimated on the basis of the Scherrer equation (see Subsection 2.2.). A known trend of crystallinity improvement when raising the calcination temperature is observed in our case as a moderate increase of CZ crystal size with the increase of calcination temperature from 500 to 600 °C. No influence of the calcination temperature on the size of the CuO crystals was observed. The comparison of the average size of the CuO crystals (13–13.8 nm, see Table 1) and the average pore size for the Cu–CZ (CTAB) samples (pore width of about 3.6 nm) confirms the possibility of pore blocking by CuO.
The cell parameter a was calculated from the XRD data as well (Table 1). Modification by CuO leads to a visible decrease of the parameter both for the CTAB and SD samples calcined at 500 °C in comparison with those calcined at 600 °C, possibly due to the presence of minor amounts of triple oxide CuyCexZr1−x−yO2−z and/or double oxide CuyCe1−yO2−x.35,36
| Ce3+–VO + O2 → Ce4+–(O2−), |
Thus, ceria/soot contact, or ceria/surface carbon contact in our case, may be the starting point of the active superoxide ion formation, which then reacts with carbon monoxide to form CO2. However, additional amounts of CO2 are formed due to soot oxidation at higher temperatures (starting from 350 °C), leading to an overestimation of CO conversion in this range.
Br (0.5–1.1 at%) and K (0.2–0.4 at%) XPS lines are observed in the survey spectra of the CTAB- and SD-templated samples, correspondingly, consistent with the SEM-EDS results.
At first glance, the influence of residual bromine on the catalytic activity of the CTAB-templated samples is possible. However, similar Ni–Ce1−xZrxO2 samples produced by co-precipitation of precursor salts with ammonium bromide or KOH demonstrated nearly the same catalytic properties in dry reforming of methane.39 On the basis of this result the effect of residual bromine species can be excluded.
High resolution Ce 3d XPS spectra are presented in Fig. 4a. In these complex spectra, the intense pronounced line at EB = 916 eV testifies that the predominant oxidation state of Ce in all samples is Ce4+. However, the presence of a Ce3+ fraction cannot be excluded. To discover the Ce3+/Ce4+ ratio the intensity of the line at EB = 916 eV was calculated relative to the integral intensity of the Ce 3d region, and compared with that for the reference CeO2 spectrum. The results are shown in Table 1. The highest Ce3+/Ce4+ ratio was found in the non-modified CZ samples, while it is nearly the same for the CTAB- and SD-templated oxides and diminishes with the increase of calcination temperature.
 | ||
| Fig. 4 Ce 3d (a), O 1s (b) and Cu 2p (c) high resolution XPS spectra of the studied samples. | ||
Various reasons for the Ce3+ ion presence in CZ systems are discussed in the literature. The most probable reason is that oxygen vacancies on the surface should be balanced by the partial reduction of some Ce4+ ions to maintain general electroneutrality.40 In addition, the presence of anionic Frenkel defects, in which some oxygen ions in a tetrahedral arrangement migrate to octahedral voids forming vacancies, was demonstrated using neutron diffraction for fluorite-type crystal structures,41 e.g. ceria. Incorporation of Zr4+ into the CeO2 lattice also leads to an increase of the Ce3+ fraction in the material.40,42
According to the above mentioned reasons, it can be expected that CZ double oxide systems calcined at 500 °C and containing more Ce3+ will be more active in CO oxidation than their counterparts calcined at 600 °C, regardless of the template type.
An important trend observed for both types of system is that modification with CuO leads to a significant decrease of the Ce3+ fraction in all samples, down to zero for the Cu–CZ (CTAB-500) sample. This trend is explained by the electron transfer between Cu2+ and Ce3+ ions according to the following redox equation:
| Cu2+ + Ce3+ ↔ Cu+ + Ce4+ |
Such a process may be intrinsic to the mixed Cu–Ce oxide phase, so this fact confirms the hypothesis about partial incorporation of Cu into the CZ lattice, proposed to explain the decrease of the crystal lattice parameter a.
However, the ability of Ce4+ to fast partial reduction under XPS conditions was demonstrated in the literature.43 This fact forces us to treat the XPS data on the Ce3+/Ce4+ ratios very cautiously.
The high resolution O 1s XPS spectra of all samples presented in Fig. 4b contain a main peak with a pronounced shoulder at the high energy side. These spectra were fitted by four components. Less intensive components at 530.6 and 533.6 eV are possibly due to the oxygen in the carbon-containing surface groups, namely C–O and CO32−, or in OH−.44 A relatively high surface carbon content found by EDS and XPS confirms this assumption.
The most intensive O 1s component at 529.6 eV corresponds to the lattice oxygen surrounded by Ce4+ ions.45 The component at 531.6 eV can be associated with the presence of oxygen vacancies46 or the active surface oxygen in several forms: (1) low coordinated highly polarized oxide ions on the surface of nanocrystals;47 (2) adsorbed or weakly bound surface oxygen as a peroxide or superoxide centers which are formed on the surface during stepwise incorporation of gas phase oxygen into an oxide lattice.48 The calculated fraction of this component (Oact, %) is presented in Table 1. This value is obviously higher for the SD-templated sample than for the corresponding CTAB-templated one. Therefore, despite a nearly equal Ce3+/Ce4+ ratio in the biomorphic and CTAB-templated samples, the former contains more active oxygen, and can demonstrate higher activity in CO oxidation. This is especially true for the samples calcined at 500 °C, where the fraction of Oact is the most significant. The highest Oact content is observed for Cu–CZ (SD-500).
High resolution Cu 2p XPS spectra (Fig. 4c) prove the presence of Cu+ ions (∼932.2 eV) in addition to the predominating Cu2+ ions (∼934 eV). It should be noted that the results of the quantitative estimation of the Cu+ fraction need to be treated carefully, because partial reduction of Cu2+ to Cu+ in XPS experimental conditions was observed earlier.49 Nevertheless, comparing the acquisition time of XPS analysis and the rate of Cu2+ reduction, the presence of Cu+ is believed to be well-established.
The calculated Cu+/Cu2+ ratios are presented in Table 1. It is clearly seen that the increase of calcination temperature causes the growth of the Cu+ fraction. In ref. 50 the same effect was found by XPS for Cu–Ce–O composite catalysts prepared by co-precipitation, while the fraction of isolated Cu2+ ions decreased. STEM/EDS analyses demonstrate the migration of copper from the bulk to the surface of these composites, causing the formation of copper clusters strongly interacting with cerium oxide. Such an interaction contributes to the reduction of Cu2+. Other authors believe that Cu+ appears due to metal ion transfer between CuO and CeO2 on the phase boundary, caused by the similarity of the ionic radii of Ce4+ and Cu+.51 Cu+ is known to be an active adsorption center for CO, and therefore can strongly influence the catalytic properties of ceria or double CZ oxides in CO oxidation.50
XPS data also demonstrate that the oxygen vacancy concentration in biomorphic samples is higher that it could be expected on the basis of Ce3+ fraction estimation. The possible reason is the intercalation of K and Ca ions into the CZ crystal lattice.
 | ||
| Fig. 5 H2-TPR profiles of CZ and Cu–CZ, calcined at 500 °C (a) and 600 °C (b). | ||
| Sample | H2 consumption, μmol [g(Ce)]−1 | ||||
|---|---|---|---|---|---|
| Low-temperature range | Cu2+ reductiona | Middle-temperature range | High-temperature range | Total | |
| a Calculated value for the reduction of the amount of copper determined in the sample by AAS, providing that the sample contains only Cu2+.b N/A means that the group of peaks is absent or difficult to separate. | |||||
| CZ (CTAB-500) | N/Ab | N/A | 1307 | 954 | 2261 |
| CZ (CTAB-600) | N/A | N/A | 1290 | 703 | 1993 |
| Cu–CZ (CTAB-500) | 4003 | 2908 | N/A | 762 | 4765 |
| Cu–CZ (CTAB-600) | 3874 | 1920 | N/A | 764 | 4638 |
| CZ (SD-500) | N/A | N/A | N/A | N/A | 2467 |
| CZ (SD-600) | N/A | N/A | N/A | N/A | 2290 |
| Cu–CZ (SD-500) | 2755 | 2081 | 1068 | 1121 | 4944 |
| Cu–CZ (SD-600) | 2900 | 1977 | 962 | 1071 | 4933 |
Firstly, we will consider the TPR profiles of the CZ samples. It is known that Zr4+ is not reducible up to 1000 °C.52 The profiles of the CZ (CTAB-500) and CZ (SD-500) samples look similar to those of CZ (CTAB-600) and CZ (SD-600). They comprise three broad peaks with maxima around 450, 550 and 750 °C, which can be attributed to the reduction of CeO2 on the surface, in the subsurface layer and in the bulk.11,53,54 The maxima of the two peaks in the middle-temperature region in the TPR profile of CZ (CTAB-500) are shifted to lower temperature. Therefore, the reduction of Ce4+ proceeds easier in the surface and subsurface layers of this sample.
The ratio of hydrogen uptake at 450–550 °C (middle-temperature range) and at around 750 °C (high-temperature range) is higher for CZ (CTAB-600) than for CZ (CTAB-500) (1.8 and 1.4, respectively). It seems that due to structure ordering which is observed as a significant decrease of SBET the reduction of surface and especially bulk ceria in CZ (CTAB-600) is hindered. This guess is confirmed by the decrease in the total H2 uptake for both types of CZ sample calcined at higher temperature (Table 2).
Two peaks in the middle-temperature range of the TPR profile of CZ (SD-500) are significantly shifted to higher temperatures in comparison with CZ (SD-600). This can be explained by the decrease of SBET which hinders both the contact of H2 with the surface and as a result the removal of H2O formed during reduction. H2 uptake for both SD-templated samples is higher than that for the corresponding CTAB-templated ones, and increases remarkably in the case of CZ calcined at 500 °C (Table 2). Therefore, CZ (SD-500) is integrally more reducible than CZ (SD-600), but the reduction of surface and subsurface species proceeds more easily in CZ (SD-600). CZ (SD-600) is expected to catalyze CO oxidation at lower temperatures than CZ (SD-500).
To summarize, the transition from the organic template to the biotemplate and the decrease of calcination temperature allow an increase in the total H2 consumption by the CZ samples (Table 2) that can be explained by the increase of active oxygen content on the surface. This fact is confirmed by XPS data.
TPR profiles of both CTAB-templated Cu–CZ samples comprise the peak of bulk Ce4+ reduction with a maximum at approx. 750 °C, and a group of low-temperature peaks, which is usually attributed to copper reduction, whereas no peaks are observed around 500–550 °C. Earlier it was found that the addition of Cu completely changes the reduction behavior of CeO2.55 The reduction of surface ceria promoted by copper is observed at lower temperatures. If the same is true for CTAB-templated samples, the hydrogen consumption in the low-temperature range will be higher than that expected as a result of only copper reduction.
In addition to the total hydrogen uptake for Cu-containing samples, Table 2 shows the amount of H2 that corresponds to the complete reduction of copper, the content of which was found by AAS, providing that it is in the fully oxidized state of Cu2+. The excessive H2 uptake is observed at 30–450 °C (low-temperature range) for both CTAB-templated Cu-containing catalysts (1095 and 1954 μmol [g(Ce)]−1 for Cu–CZ (CTAB-500) and Cu–CZ (CTAB-600), respectively). These values are comparable with the H2 uptake at middle temperatures (peaks around 500–550 °C) by the corresponding double oxides CZ (CTAB-500) and CZ (CTAB-600) (1307 and 1290 μmol [g(Ce)]−1, respectively), attributed above to the reduction of Ce4+ in the surface and subsurface layers (the corresponding peaks are absent in the profiles of Cu-containing CTAB-templated samples). Such changes can be caused by the formation of double or triple Cu-containing surface oxides CuyCexZr1−x−yO2−z or CuyCexO2−z. Besides, as it was noted in ref. 53, independent of the preparation method, some copper dissolution in the CZ phase takes place providing the formation of highly dispersed copper ions that promote surface ceria reduction at lower temperatures. Note that for Cu–CZ (CTAB-600) the excessive H2 uptake in the low-temperature range is higher than the total H2 consumption during TPR of the corresponding CZ (CTAB-600) sample. This difference can be explained both by the variation in the valence state of Ce in these samples and by hydrogen spillover from copper species to the CZ support. Fundamentally, these results confirm the possibility of cerium reduction at unusually low temperatures in the presence of copper oxides.
The low-temperature group of peaks is broader for Cu–CZ (CTAB-500) (130–420 °C) than for Cu–CZ (CTAB-600) (130–320 °C). So, the reduction of bulk components, typically proceeding at higher temperatures (320–420 °C), is hindered for the sample calcined at 600 °C due to structure ordering, as it was found for corresponding CZ samples.
The TPR profiles of Cu–CZ (SD-600) and Cu–CZ (SD-500) are similar. They comprise broad low intense peak of bulk cerium reduction at about 780 °C, two peaks of surface and subsurface cerium reduction at about 460–515 °C, and a low-temperature group of three peaks at 250–400 °C. The peaks of surface and subsurface cerium reduction at about 464 and 514 °C for Cu–CZ (SD-600) nearly coincide with that for CZ (SD-600), however hydrogen uptake in this temperature range is lower for the Cu-modified sample. It can be proposed that the fraction of cerium is reduced at lower temperatures, as in the case of the CTAB-templated Cu-containing samples.
For Cu–CZ (SD-500) the discussed peaks of surface and subsurface cerium reduction are observed at the same temperatures (464 and 514 °C) as for Cu–CZ (SD-600); they are shifted to lower temperatures in comparison with the CZ (SD-500) profile. Note that the SBET of Cu-CZ (SD-500) is unusually high (93 ± 9 m2 g−1) for the SD-templated sample, and is similar to that of Cu–CZ (CTAB-500), not of CZ (SD-500). The availability of the surface to H2 due to structure disordering provides easier reduction of surface and subsurface cerium in Cu-CZ (SD-500).
The group of low-temperature peaks is shifted to higher temperatures for the Cu-modified SD-templated samples in comparison with the CTAB-templated ones. According to the discussion above, this group of peaks can comprise up to six components, corresponding to the reduction of Cu2+ to Cu+, Cu+ to Cu0, and Ce4+ to Ce3+ in the bulk and on the surface. The position of these peaks may shift depending on the material surrounding copper and cerium. For instance, it could be expected that the reduction of CuO in close vicinity to CZ or CuyCexZr1−x−yO2−z will proceed in a different temperature range. The exact attribution of all components is a very difficult task, but many considerations can be found in the literature. Thus, the intensive signal at 198 °C can be ascribed to the reduction of CuO species strongly interacting with the CZ support,56,57 to the partial reduction of surface Ce4+ to Ce3+, and to the total reduction of dispersed Cu2O particles to Cu.58 The shoulder at higher temperatures shows the reduction of large CuO particles which are not associated with the support,56,57,59 or the reduction of Ce4+ to Ce3+ and Cu+ to Cu0 in the bulk of the catalyst.58 Similar attribution of this higher-temperature component was proposed to describe the TPR profile of CuO/CeO2.60 The peaks at 250 and 330 °C can arise due to hydrogen consumption by weakly bonded O2− ions in surface oxygen vacancies of CuO, and by interphase O2− ions nearby oxygen vacancies, respectively.59
Therefore, the TPR results demonstrate the significant difference in the Cu state for the CTAB-templated and biomorphic catalysts. The Cu–CZ (CTAB-500) sample shows much more in common with samples reported early in the literature, whereas reduction of Cu species in Cu–CZ (SD-500) proceeds at a higher temperature. The probability of double Cu–Ce or triple Cu–Ce–Zr oxide surface phase formation is higher in CTAB-templated systems, demonstrating a more close interaction of Ce with Cu in the surface and subsurface layer. The total H2 consumption increases as a result of CZ modification with Cu; it is higher for SD-templated samples than for the corresponding CTAB-templated ones. The decrease of calcination temperature increases H2 uptake for Cu-containing CTAB-templated samples, and has no influence on SD-templated ones.
The presence of various Cu species in SD-samples results in the broad temperature range of copper reduction (200–400 °C) which may enhance the catalytic properties in CO oxidation.
3.2. Catalytic activity in CO oxidation
Carbon monoxide is formed in various industrial processes and during fuel burning in engines.24 It is one of the main air pollutants, and can cause mortal poisoning. CO is a colorless and odorless gas, therefore its presence in air is difficult to detect. Catalytic total oxidation is the most effective way to avoid toxic CO emissions.The catalytic efficiency of the samples was estimated on the basis of CO conversion to CO2 in the temperature range from 100 to 400 °C. The results are presented as steady-state CO conversion vs. temperature curves (Fig. 6). All tested samples were active in CO oxidation, but the temperature ranges of their catalytic action were significantly different.
 | ||
| Fig. 6 CO conversion vs. reaction temperature for the samples calcined at 500 °C (a) and 600 °C (b). | ||
According to the literature,61 the increase of the Ce3+/Ce4+ ratio leads to the increase of the amount of oxygen vacancies and unsaturated chemical bonds on the surface of a catalyst, and to the growth of the surface content of active oxygen. Lattice oxygen mobility is governed by the energy of oxygen vacancy formation and by the activation energy of oxygen migration in the lattice, and therefore significantly increases with temperature. Possibly due to this reason, CO conversion at a lower temperature (150–300 °C) doesn’t depend on the calcination temperature.
Comparing the pairs of CZ (CTAB) and Cu–CZ (CTAB) samples, calcined at the same temperature (Fig. 6a and b and S3a†), it is evident that CuO addition significantly improves the catalytic efficiency in the whole temperature range and widens the range of high CO conversion to low-temperature region in both cases. In the Cu-modified system the Ce3+ content is lower and Oact content is comparable with that in CZ (CTAB). Therefore, the improvements are possibly connected not only with the presence of bulk CuO, but also with the partial incorporation of Cu into the CZ lattice, the evidence of which for Cu–CZ (CTAB-500) was found by XRD, and with the formation of double and triple Cu–Ce and Cu–Ce–Zr oxide surface phases, which was proposed on the basis of TPR data. Recent investigations have shown36 that CO oxidation is directly catalyzed by dispersed copper ions in the mixed oxide phase. Therefore, the formation of the mixed CuyCexO2−z or CuyCexZr1−x−yO2−z phases in the bulk or on the surface of CZ will favor the catalytic activity of the Cu–CZ systems.
Despite lower Oact content, Cu–CZ (CTAB-500) provides significant improvement of CO conversion in comparison with Cu-CZ (CTAB-600). This can be explained by the higher CuO loading found by AAS (Table 1) (29.0 in Cu–CZ (CTAB-500) against 21.2 mol% in Cu–CZ (CTAB-600)) and higher SBET.
A strong influence of SBET on the catalytic performance is observed for CZ (CTAB-500). This sample shows the most developed surface (SBET = 165 m2 g−1), and provides better CO conversion at 350–400 °C, compared to modified Cu–CZ (CTAB-600) with much lower SBET (62 m2 g−1). The content of Oact in these samples is high (see Table 1), but the Ce3+ content in CZ (CTAB-500) is the highest among all the samples, providing many oxygen vacancies. The agglomerates of CuO in Cu–CZ (CTAB-600) possibly partially block the access of the CO molecules to the CZ surface making this sample less effective than CZ (CTAB-500) at high temperatures, at which high oxygen mobility in CZ plays a crucial role.
An interesting effect was found for the double CZ systems: at low temperature CZ (SD-600) provides higher CO conversion than CZ (SD-500), but starting from 250 °C the inverse relation is observed. An improved efficiency of CZ (SD-500) at higher temperatures can be caused by the higher content of oxygen vacancies, which is confirmed by the significant content of Oact and Ce3+ (17.1% and 0.14% CZ (SD-500) vs. 15.1% and 0.09% for CZ (SD-600)) estimated by XPS (Table 1). The difference in CO conversion at low temperature can be attributed to the changes in the extent of the ash admixture (Ca and K) interaction with the CZ lattice. At a high calcination temperature (600 °C) Ca and K ions more effectively penetrate the crystal lattice of CZ than at 500 °C, which increases the electron deficiency of this lattice and improves lattice oxygen mobility, even at lower Ce3+ content.
Modification of CZ by CuO leads to the improvement of the catalytic action of the biomorphic systems, calcined at 500 °C, in the whole studied temperature range. It is consistent with the increase of Oact content, decrease of lattice parameter, and significant improvement of SBET of Cu–CZ (SD-500) (Table 1).
In contrast, Cu–CZ (SD-600) is somewhat less effective than CZ (SD-600). In this case SBET, the lattice parameter and the Oact content are nearly the same for both samples. In addition, CO conversion on these samples in the range of 200–350 °C is limited by 53–56% (Fig. 6 and S3b†). Such a saturation of the CO conversion vs. temperature curves is also observed for CZ (CTAB-600) (about 55% conversion). This may be connected with the use of a stoichiometric CO:O2 mixture if the reaction proceeds predominantly via the Mars–van Krevelen mechanism on the catalyst with high lattice oxygen mobility. At high temperature, the amount of O2 in the reaction mixture is not enough to compensate for the lattice oxygen spent for CO oxidation.
As it was shown above by EDS and XPS, all biomorphic samples contain Ca and K. The intercalation of the K and Ca ions into the CZ oxide lattice is the possible reason for the significant increase of the active oxygen Oact fraction in the biomorphic samples as compared to the CTAB-templated ones, which is clearly seen from the XPS data (Table 1).
It was noted in the literature that the introduction of a small amount of CaO to CeO2 (in atomic ratios Ca/Ce = 0.1)14 or to double CZ systems15 promotes the formation of oxygen vacancies, leading to the improvement of the catalytic activity in CO oxidation.
The positive action of moderate amounts (below 20 mol%) of bulky cations, e.g. Ca2+, on the number of anionic vacancies and oxygen mobility due to a distortion of the fluorite lattice of the CZ oxide was shown in ref. 16 and 17. According to these studies the presence of Ca admixture can lead to an increase of CO conversion. Improvement of the catalytic action of CZ in CO oxidation by the deliberate addition of Ca and K to CZ during co-precipitation with a subsequent calcination at 500 °C was also observed in our previous study.13 The facilitation of the formation of strongly nucleophilic peroxide and superoxide complexes upon the chemisorption of oxygen on CZ oxides confirmed by XPS and TPR data was proposed in this study as the most probable reason for the promotion effect of potassium. The stabilization of superoxide and peroxide species in the presence of alkali ions was demonstrated early.62
Addition of CuO to both biomorphic and CTAB-templated CZ systems calcined at 500 °C (Fig. 6a) improves CO conversion in all temperature ranges studied in this work primarily due to the inclusion of a new redox pair of Cu+/Cu2+ into the catalytic cycle and the formation of mixed CuyCexO2−z or CuyCexZr1−x−yO2−z phases. The other reasons for the improvement of CO conversion are the decrease of crystallinity and the increase of surface inhomogeneity in comparison with the systems calcined at 600 °C.
On the basis of the literature data,63 the support of CuO on CeO2-containing systems can provide (depending on a temperature range of CO oxidation) both the additional adsorption sites and/or an additional redox pair of Cu+/Cu2+, according to the following scheme:
| (Cun+)surf + CO → (Cun+)surf–COads | (1) |
| 2(Cun+)surf + O2 → 2(Cun+)surf–Oads | (2) |
| (Cun+)surf–COads + (Cun+)surf–Oads → 2(Cun+)surf + CO2 | (3) |
| (Ce4+/Cu2+–O2−)surf + CO → (Ce3+/Cu+–VO)surf + CO2 | (4) |
| (Ce3+/Cu+–VO)surf + CO → (Ce3+/Cu+–VO)surf–COads | (5) |
| (Ce3+/Cu+–VO)surf–COads + O2 → (Ce4+/Cu+–O2−)surf–COads | (6) |
| (Ce3+/Cu+–VO)surf–COads + O2 → (Ce4+/Cu2+–O22−)surf + CO2 | (7) |
| (Ce4+/Cu2+–O22−)surf + (Ce3+/Cu+–VO)surf–COads → (Ce4+/Cu2+–O2−)surf + CO | (8) |
Eqn (1)–(3) describe the Langmuir–Hinshelwood mechanism of CO oxidation at relatively low reaction temperatures, when the oxygen mobility is hindered. In this mechanism, Cu2+ or Cu+ on the surface of CuO, Cu2O, CuyCexO2−z or CuyCexZr1−x−yO2−z serve as active sites.
At higher temperatures, the lattice oxygen mobility grows and the kinetic scheme of Mars–van Krevelen (reactions (4)–(8)) becomes predominant. The joint action of the redox pairs of Cu+/Cu2+ and Ce3+/Ce4+ has been described by many authors.22,62,63 The reaction mechanism comprises CO oxidation by surface oxygen species (O2−) followed by the formation of CO2 and oxygen vacancies (VO) accompanied by the reduction of a Ce4+/Cu2+ pair (reaction (4)).63,64 Then CO adsorbs an oxygen vacancy and reacts with O2 to form superoxide (O2−) and peroxide (O22−) species (reactions (6) and (7)), which are stabilized due to the promoting action of the alkali metal ions. At the final stage, the initial active site structure is recovered by reaction (8).
However, Cu modification deteriorates the catalytic efficiency of the biomorphic CZ sample calcined at 600 °C. It is possible that in this case the CuO phase blocks the active sites situated on the surface of CZ, which provides good oxygen mobility due to the lattice inclusion of Ca2+ and K+.
Cu–CZ (CTAB-500) is somewhat more active than Cu–CZ (SD-500) at 100–150 °C, mainly due to the significantly higher CuO content, and in some extent due to the stronger CuO interaction with the CZ support in the CTAB-templated system, found by TPR. However, the difference between these samples is not so prominent, if the whole temperature range is considered, considering that the SBET values for these samples are comparable. Note that on the basis of TPR data we can expect worse catalytic activity of the Cu-modified biomorphic sample, due to the weaker interaction of CuO with CZ. But it seems that the promoting action of Ca and K ions on CZ is the most important factor contributing to the improvement of the catalytic properties of Cu–CZ (SD-500). At equal CuO loading the biomorphic catalyst will probably be a good competitor to the CTAB-templated counterpart.
The results of this study confirm that sawdust, which is an industrial waste, can be effectively used instead of organic templates for the preparation of CO oxidation catalysts on the basis of ceria and zirconia. However, for the development of industrial methods the problem of the reproducibility of the catalytic properties should be addressed. Sawdust of various wood species, grown in different geographical locations, has a similar main chemical composition, but a variable content of ash impurities and other components. Considering the strong dependence of the catalytic properties on the presence of ash impurities, to provide reproducibility of the catalytic results the content of the admixture of alkali or alkaline-earth metal salts can be adjusted by mixing different sawdust batches or by the intentional addition of the desired components. Reaching an optimal method of catalyst preparation from sawdust requires the knowledge of the influence of the content of admixture on the catalytic properties, which will be the focus of the future work.
4. Conclusions
CZ and Cu–CZ oxide systems, prepared by biotemplate (wood sawdust, SD) impregnation with metal salts and subsequent calcination at 500 or 600 °C, demonstrate high efficiency in CO oxidation to CO2 at 100–400 °C. Calcination at lower temperatures provides more effective catalysts, independent of the presence of Cu. Calcination at 600 °C leads to a decrease in the Ce3+:Ce4+ ratio and excessive ordering of the crystal structure, which is confirmed by XRD data and by the decrease of the specific surface area. As a result, improved oxygen mobility and better contact of CO with the surface of the catalyst are achieved in the catalysts calcined at 500 °C.
The double oxide biomorphic sample CZ (SD-500) is much more effective in CO oxidation in comparison with the similar CTAB-templated sample CZ (CTAB-500) despite a two times less SBET value. Possible reasons for such an improvement are (1) the presence of Ca and K additives inherited from wood sawdust, and (2) the modification of the phase composition, found by XRD. Such a modification may result from the intercalation of potassium and/or calcium ions into the crystal lattice and lead to the change in lattice oxygen mobility in the mixed oxide phases. This change in oxygen mobility affects the Ce3+:Ce4+ ratio and provides an improvement of the high-temperature efficiency in CO oxidation through the Mars–van Krevelen mechanism. In addition, alkali metal ions stabilize strongly nucleophilic peroxide and superoxide complexes on the catalytic surface. These complexes can participate in CO oxidation.
Modification with Cu is very efficient for the biomorphic as well as the comparative CTAB-templated CZ systems calcined at 500 °C. Biomorphic sample Cu–CZ (SD-500) is a bit less active than Cu–CZ (CTAB-500) only at 100 and 150 °C, primarily due the decreased CuO content, and the difference in CuO interaction with the CZ support, found by TPR. At higher temperatures both samples show the same CO conversion close to 100%. The XRD, XPS and TPR data testify that Cu in modified systems exists as a separate CuO phase, in small amount as Cu+, and partially is incorporated in surface CuyCexZr1−x−yO2−z and/or CuyCe1−yO2−x phases. The formation of Cu-containing double or triple oxide surface phases is less typical for biomorphic than for CTAB-templated samples. According to the TPR data the peaks of the surface and subsurface Ce reduction move to a lower temperature group characteristic for Cu reduction. It seems that CuO provides low-temperature CO oxidation through the Langmuir–Hinshelwood mechanism; a Cu+/Cu2+ pair can also participate as an additional redox pair in high-temperature CO oxidation by the Mars–van Krevelen mechanism.
Modification with Cu of more ordered CZ (SD-600) leads to a decrease of CO conversion in the low temperature range of 100–250 °C, possibly due to the blocking of the active surface of CZ by CuO and the decrease of the accessible amount of the mobile lattice oxygen.
Therefore, the sawdust-templated samples calcined at 500 °C (CZ (SD-500) and Cu–CZ (SD-500)) showed very high efficiency in CO oxidation to CO2, which was comparable (for Cu–CZ (SD-500)) or even better (for CZ (SD-500)) than that for their CTAB-templated counterparts. It seems that the decreased specific surface area of biomorphic CZ is compensated for by the promoting action of ash impurities and structure changes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the RF State Program for maintenance of leading scientific schools NSh-8845.2016.3 and in part by the Lomonosov Moscow State University Program of Development.References
- V. Singh and S. K. Chakarvarti, American Journal of Bioengineering and Biotechnology, 2016, 2, 1–14 Search PubMed.
- C. R. Rambo and H. Sieber, Adv. Mater., 2005, 17, 1088–1091 CrossRef CAS.
- D. Yang, B. Du, Y. Yan, H. Li, D. Zhang and T. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 2377–2385 CAS.
- C. Wang, L. Jing, M. Chen, Z. Meng, Z. Chen, F. Chen and W.-C. Oh, J. Korean Ceram. Soc., 2017, 54, 23–27 CrossRef CAS.
- A. N. Shigapov, G. W. Graham, R. W. McCabe and H. K. Plummer Jr, Appl. Catal., A, 2001, 210, 287–300 CrossRef CAS.
- A. O. Turakulova, N. V. Zaletova, G. P. Murav’eva, M. V. Burova and V. V. Lunin, Russ. J. Phys. Chem. A, 2008, 82, 1403–1406 CrossRef CAS.
- X. Li, T. Fan, Z. Liu, J. Ding, Q. Guo and D. Zhang, J. Eur. Ceram. Soc., 2006, 26, 3657–3664 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- D. Duprez, C. Descorme, T. Birchem and E. Rohart, Top. Catal., 2001, 16, 49–56 CrossRef.
- C. E. Hori, H. Permana, K. Y. S. Ng, A. Brenner, K. More, K. M. Rahmoeller and D. N. Belton, Appl. Catal., B, 1998, 16, 105–117 CrossRef CAS.
- E. Aneggi, M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli, J. Alloys Compd., 2006, 408–412, 1096–1102 CrossRef CAS.
- P. Fornasiero, R. Di Monti, G. Ranga Rao, J. Kaspar, S. Meriani, A. Trovarelli and M. Graziani, J. Catal., 1995, 151, 168–177 CrossRef CAS.
- I. Y. Kaplin, E. S. Lokteva, E. V. Golubina, K. I. Maslakov, S. A. Chernyak and V. V. Lunin, Kinet. Catal., 2017, 58, 585–592 CrossRef CAS.
- A. Iglesias-Juez, A. B. Hungría, O. Gálvez, A. Martínez-Arias, M. Fernández-García, A. Guerrero-Ruiz, J. C. Conesa and J. Soria, Stud. Surf. Sci. Catal., 2001, 138, 347–354 CrossRef CAS.
- M. Fernández-García, A. Martínez-Arias, A. Guerrero-Ruiz, J. C. Conesa and J. Soria, J. Catal., 2002, 211, 326–334 CrossRef.
- T. Kuznetsova, V. Sadykov, L. Batuev, E. Moroz, E. Burgina, V. Rogov, V. Kriventsov and D. Kochubey, J. Nat. Gas Chem., 2006, 15, 149–163 CrossRef CAS.
- T. G. Kuznetsova, V. A. Sadykov, E. M. Moroz, S. N. Trukhan, E. A. Paukshtis, V. N. Kolomiichuk, E. B. Burgina, V. I. Zaikovskii, M. A. Fedotov, V. V. Lunin and E. Kemnitz, Stud. Surf. Sci. Catal., 2002, 143, 659–667 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2008, 136, 3–10 CrossRef CAS.
- C. Telmo, J. Lousada and N. Moreira, Bioresour. Technol., 2010, 101, 3808–3815 CrossRef CAS PubMed.
- X. Zheng, X. Zhang, X. Wang, S. Wang and S. Wu, Appl. Catal., A, 2005, 295, 142–149 CrossRef CAS.
- J.-L. Cao, Y. Wang, T.-Y. Zhang, S.-H. Wu and Z.-Y. Yuan, Appl. Catal., B, 2008, 78, 120–128 CrossRef CAS.
- M. Moreno, L. Bergamini, G. T. Baronetti, M. a. Laborde and F. J. Mariño, Int. J. Hydrogen Energy, 2010, 35, 5918–5924 CrossRef CAS.
- G. Avgouropoulos, T. Ioannides and H. Matralis, Appl. Catal., B, 2005, 56, 87–93 CrossRef CAS.
- I. Y. Kaplin, E. S. Lokteva, E. V. Golubina, K. I. Maslakov, S. A. Chernyak, A. V. Levanov, N. E. Strokova and V. V. Lunin, Russ. J. Phys. Chem. A, 2016, 90, 2157–2164 CrossRef CAS.
- X. B. Zhao, F. Chen, J. You, X. Z. Li, X. W. Lu and Z. G. Chen, J. Mater. Sci., 2010, 45, 3563–3568 CrossRef CAS.
- P. Scherrer, Göttinger Nachrichten Math. Phys., 1918, 2, 98–100 Search PubMed.
- A. Bryś, J. Bryś, E. Ostrowska-Ligęza, A. Kaleta, K. Górnicki, S. Głowacki and P. Koczoń, J. Therm. Anal. Calorim., 2016, 126, 27–35 CrossRef.
- J. V. Bellini, R. Machado, M. R. Morelli and R. H. G. A. Kiminami, Mater. Res., 2002, 5, 453–457 CrossRef CAS.
- M. Aghayan, M. Gasik, I. Hussainova, F. Rubio-Marcos, L. Kollo and J. Kübarsepp, Thermochim. Acta, 2015, 602, 43–48 CrossRef CAS.
- R. A. Lidin, L. L. Andreeva and V. A. Molochko, Constants of inorganic substances, Begell House, New York, 1995 Search PubMed.
- J. L. Cao, G. S. Shao, T. Y. Ma, Y. Wang, T. Z. Ren, S. H. Wu and Z. Y. Yuan, J. Mater. Sci., 2009, 44, 6717–6726 CrossRef CAS.
- H. Yen, Y. Seo, S. Kaliaguine and F. Kleitz, Angew. Chem., Int. Ed., 2012, 51, 12032–12035 CrossRef CAS PubMed.
- C. Ho, J. C. Yu, X. Wang, S. Lai and Y. Qiu, J. Mater. Chem., 2005, 15, 2193–2201 RSC.
- P. Fornasiero, G. Balducci, R. Di Monte, J. Kašpar, V. Sergo, G. Gubitosa, A. Ferrero and M. Graziani, J. Catal., 1996, 164, 173–183 CrossRef CAS.
- Y. Liu, T. Hayakawa, K. Suzuki, S. Hamakawa, T. Tsunoda, T. Ishii and M. Kumagai, Appl. Catal., A, 2002, 223, 137–145 CrossRef CAS.
- J. S. Elias, N. Artrith, M. Bugnet, L. Giordano, G. A. Botton, A. M. Kolpak and Y. Shao-Horn, ACS Catal., 2016, 6, 1675–1679 CrossRef CAS.
- E. Aneggi, D. Wiater, C. de Leitenburg, J. Llorca and A. Trovarelli, ACS Catal., 2014, 4, 172–181 CrossRef CAS.
- M. Machida, Y. Murata, K. Kishikawa, D. Zhang and K. Ikeue, Chem. Mater., 2008, 20, 4489–4494 CrossRef CAS.
- A. Wolfbeisser, O. Sophiphun, J. Bernardi, J. Wittayakun, K. Föttinger and G. Rupprechter, Catal. Today, 2016, 277, 234–245 CrossRef CAS.
- D. Prusty, A. Pathak, M. Mukherjee, B. Mukherjee and A. Chowdhury, Mater. Charact., 2015, 100, 31–35 CrossRef CAS.
- E. Mamontov and T. Egami, J. Phys. Chem. Solids, 2000, 61, 1345–1356 CrossRef CAS.
- G. Balducci, J. Kašpar, P. Fornasiero, M. Graziani, M. S. Islam and J. D. Gale, J. Phys. Chem. B, 1997, 101, 1750–1753 CrossRef CAS.
- A. Galtayries, R. Sporken, J. Riga, G. Blanchard and R. Caudano, J. Electron Spectrosc. Relat. Phenom., 1998, 88–91, 951–956 CrossRef CAS.
- X. Y. Wang, Q. Kang and D. Li, Catal. Commun., 2008, 9, 2158–2162 CrossRef CAS.
- E. Nelson and K. H. Suhulz, Appl. Surf. Sci., 2003, 210, 206–221 CrossRef.
- X. Tang, B. Zhang, Y. Li, Y. Xu, Q. Xin and W. Shen, Catal. Today, 2004, 93–95, 191–198 CrossRef CAS.
- J. P. Holgado, G. Munuera, J. P. Espinós and A. R. González-Elipe, Appl. Surf. Sci., 2000, 158, 164–171 CrossRef CAS.
- D. Mukherjee, B. G. Rao and B. M. Reddy, Appl. Catal., B, 2016, 197, 105–115 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- W. Liu and M. Flytzani-Stephanopoulos, J. Catal., 1995, 153, 317–332 CrossRef CAS.
- S. Hocevar, U. O. Krasovec, B. Orel, A. S. Arico and H. Kim, Appl. Catal., B, 2000, 28, 113–125 CrossRef CAS.
- I. Atribak, A. Bueno-López and A. García-García, J. Catal., 2008, 259, 123–132 CrossRef CAS.
- D. Terribile, A. Trovarelli, C. de Leitenburg, A. Primavera and G. Dolcetti, Catal. Today, 1999, 47, 133–140 CrossRef CAS.
- W. Mista, M. A. Malecka and L. Kepinski, Appl. Catal., A, 2009, 368, 71–78 CrossRef CAS.
- T. Caputo, L. Lisi, R. Pirone and G. Russo, Appl. Catal., A, 2008, 348, 42–53 CrossRef CAS.
- W. Liu and M. Flytzani-Stephanopoulos, Chem. Eng. J., 1996, 64, 283–294 CAS.
- L. Ma, M. F. Luo and S. Y. Chen, Appl. Catal., A, 2003, 242, 151–159 CrossRef CAS.
- V. D. Araujo, J. D. A. Bellido, M. I. B. Bernardi, J. M. Assaf and E. M. Assaf, Int. J. Hydrogen Energy, 2012, 37, 5498–5507 CrossRef CAS.
- J. L. Ayastuy, A. Gurbani, M. P. González-Marcos and M. A. Gutiérrez-Ortiz, Int. J. Hydrogen Energy, 2012, 37, 1993–2006 CrossRef CAS.
- S. Sun, D. Mao, J. Yu, Z. Yang, G. Lu and Z. Ma, Catal. Sci. Technol., 2015, 5, 3166–3181 CAS.
- Q.-F. Deng, T.-Z. Ren, B. Agula, Y. Liu and Z.-Y. Yuan, J. Ind. Eng. Chem., 2014, 20, 3303–3312 CrossRef CAS.
- P. Dolle, S. Drissi, M. Besançon and J. Jupille, Surf. Sci., 1992, 269–270, 687–690 CrossRef CAS.
- A. Martínez-Arias, M. Fernández-García, O. Gálvez, J. Coronado, J. Anderson, J. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207–216 CrossRef.
- Y.-Z. Chen, B.-J. Liaw and H.-C. Chen, Int. J. Hydrogen Energy, 2006, 31, 427–435 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10791a |
| This journal is © The Royal Society of Chemistry 2017 |