
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of Au dotted Co3O4 nanosheets as an efficient bifunctional catalyst for Li–oxygen batteries†
Juan Xiang‡
,
Taeseup Song‡ and
Ungyu Paik
Department of Energy Engineering, Hanyang University, Seoul 133-791, South Korea. E-mail: upaik@hanyang.ac.kr
First published on 7th November 2017
Abstract
Two-dimensional Co3O4 nanosheets dotted with Au nanoparticles were synthesized on the carbon gas diffusion layer as a bifunctional catalyst for Li–O2 batteries by thermal evaporation and low-temperature calcination. The two-dimensional Co3O4 nanosheets improved the catalytic activity and Au nanoparticles provided additional nucleation sites for the Li2O2 growth in the process of discharge, thus allowing the uniform formation of Li2O2. Moreover, the size and distribution of Au nanoparticles were tuned by evaporating Au in different thicknesses. The catalytic performance of the Co3O4–Au hybrid was improved due to the synergetic effects of both materials and the improvements were closely associated with the size and distribution of Au nanoparticles. When the rationally designed catalyst was used as a cathode catalyst in Li–oxygen batteries, it lowered the polarization effect during cycling and realized the stable cyclability for 70 cycles at a limited capacity of 1000 mA h g−1.
Introduction
Li–oxygen batteries (Li–O2 batteries) are a promising power source with a high theoretical energy density compatible with gasoline (∼13 W h g−1).1–3 However, the application scope of Li–O2 batteries is limited by the low energy efficiency and poor cycle stability caused by the low electronic conductivity of insoluble discharging products.4–6 Various carbon materials including commercial graphene, carbon, carbon nanotubes and metal-based catalyst were proposed as the cathode.7–10 However, carbon electrode can react with Li2O2 to form Li2CO3.11,12 Indecomposable side products (e.g. Li2CO3) formed during charging/discharging process could cover the metal based catalyst, which leads to the degradation of the catalytic activity. Therefore, the development of the catalyst that can significantly reduce the polarization or carbonaceous material free electrode is necessary. Moreover, nonconductive binders such as polyvinylidene fluoride (PVDF) increased the impedance of cells.13 So it is necessary to develop a binder-free cathode with well-designed catalysts.Nanostructured metal oxide catalysts including MnO2, Co3O4, and RuO2 were reported as efficient OER and ORR catalysts in Li–O2 batteries.14–17,24 Compared with other transition metal oxides, Co3O4 shows the better capacity retention, initial capacity, and the superior activity towards OER and ORR.18–22 Kim et al. obtained carbon nanotube/Co3O4 nanocomposites selectively coated with polyaniline via an electropolymerization method. Co3O4 particles on the CNT surface facilitated the dissociation of reaction products such as Li2O2 and reduced the overpotential.23 Au is one of the most active ORR catalysts in non-aqueous media by promoting ORR through the peroxide route.4,25–30 Fan et al. anchored gold nanoparticles to vertically aligned carbon nanotubes, in which Au acted as additional nucleation sites for Li2O2 growth.27 Besides, by improving the conduction property of the carbonate species, Au nanoparticles (Au NPs) can effectively reduce overpotentials of Li–O2 batteries and extend the life cycle of the batteries.27 In this regard, rationally designed Au NPs and superior-active Co3O4 composite can improve electrochemical properties of non-aqueous Li–O2 batteries by taking the advantages of both Co3O4 and Au.
In this study, two-dimensional (2D) Co3O4 nanosheets (Co3O4 NSs) decorated with different thicknesses of Au NPs growing directly on the carbon gas diffusion layer (GDL) were prepared by the combination of thermal evaporation and low-temperature calcination. Commercially ordered GDL was employed as a conductive support with high electronic conductivity and electrocatalytic activity.28–30 The as-prepared hybrid cathode had many tailored properties. Firstly, the 2D geometry of Co3O4 NSs provided sufficient space for Li2O2 as well as the large surface area for uniform Au nanoparticle loading. Secondly, the conformal coating of binder-free Co3O4 NSs–Au catalyst on GDL could prevent the direct contact between carbon support and Li2O2, thus solving the problems caused by carbon oxidation.31 Thirdly, the direct growth of Co3O4 NSs–Au on GDL could effectively avoid by-products generated during binder decomposition and enhance the electronic conductivity. Moreover, by controlling the evaporation conditions, the size and distribution of Au NPs were tuned to obtain the optimal ratio of Au to Co3O4. With the rationally tailored cathode architecture and synergistic effect between the hybrid catalysts, the Li–O2 battery with GDL-Co3O4 NSs–Au cathode exhibited the excellent electrochemical performance of the low polarization and stable cyclability.
Experimental
Material synthesis
The carbon gas diffusion layer used in the experiment was obtained from Sigma Aldrich without further treatment. The synthesis method of Co3O4 ultrathin NSs on GDL substrate (GDL-Co3O4 NSs) involved two steps: thermal evaporation and subsequent thermal treatment (Scheme 1 in ESI†). First, a layer of cobalt seeds were deposited on a GDL substrate by thermal evaporation technique. High-purity cobalt target was mounted on a tungsten boat at a distance of around 60 cm to the GDL substrate. The system was evacuated by a cryopump up to a pressure of 5 × 10−6 Torr for evaporation. The cobalt layer grew by evaporating cobalt with the voltage of 115 to 135 kV applied on the tungsten boat and the film deposition rate was increased from 0.1 to 0.3 Å s−1 until a film thickness of the cobalt layer reached 100 nm. The deposition rate of the individual source was controlled by Inficon quartz crystal rate monitor. After evaporation, the as-made cobalt film was thermally treated in a quartz tube furnace at 310 °C for 1 h with a ramping rate of 5 °C min−1 in air atmosphere. To synthesize gold-dotted Co3O4 ultrathin NSs, gold nanoparticles with the thickness of 10 nm, 30 nm and 50 nm were deposited on the as-synthesized GDL-Co3O4 NSs substrate by thermal evaporation and denoted as GDL-Co3O4 NSs–10Au, GDL-Co3O4 NSs–30Au, and GDL-Co3O4 NSs–50Au, respectively. The deposition rate was controlled to be 0.1 Å s−1 by Inficon quartz crystal rate monitor. To synthesize gold-dotted GDL, gold nanoparticles with a thickness of 30 nm were deposited directly on GDL substrate by thermal evaporation with a deposition rate of 0.1 Å s−1 and denoted as GDL-30Au.Material characterization
GDL, GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–10/30/50Au was characterized by using X-ray diffraction (XRD, Rigaku D/MAX RINT-2000), scanning electron microscopy (SEM, JEOL JSM-7600F), and high-resolution transmission electron microscopy (HR-TEM, JEOL JEM-2100F). Energy-dispersive X-ray spectroscopy (EDS spectra) were analyzed by EDX attached to the TEM instrument. X-ray photoelectron spectrometer (XPS, VG microtech ESCA2000) was used to analyze the compositions of GDL-Co3O4 NSs and the discharge products of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au. The Brunauer–Emmett–Teller (BET) method was used to calculate the specific surface area of samples.Electrochemical measurements
Pure GDL, GDL-30Au, GDL-Co3O4 NSs and the as-prepared GDL-Co3O4 NSs–10Au, GDL-Co3O4 NSs–30Au, and GDL-Co3O4 NSs–50Au were used as cathodes without any additive or binder and 1 M bis(trifluoromethane)sulfonimide lithium (LiTFSi, Sigma-Aldrich, 99.5%) in tetraethylene glycol dimethyl ether (TEGDME) was used as the electrolyte. Then the cells were assembled in an argon-filled glove box by stacking Li metal as a counter electrode, polypropylene as a separator and air electrodes welded between stainless steel meshes. The electrochemical properties were evaluated with a galvanostat/potentiostat (TOSCAT 3000, Toyo Systems, Tokyo, Japan).Results and discussion
The pristine GDL was woven by carbon fiber with a diameter of about 10 μm (Fig. S1a†) and smooth surface (inset in Fig. S1a†). X-ray diffraction (XRD) measurement was conducted to identify the phase of GDL, as shown in Fig. S1b.† Two peaks at 26.3° and 54.6° are indexed to the (002) and (004) planes of the typical graphite (JCPDS no. 41-1487). The morphology of the as-prepared GDL-30Au and GDL-Co3O4 NSs was then investigated (Fig. 1a–c). After gold deposition, Au NPs were accumulated and the surface of GDL became rough. The size of Au NPs is tens of nanometers (Fig. 1a). Field-emission scanning electron microscope (FESEM) and high-magnification transmission electron microscopy (TEM) images revealed that Co3O4 NSs had the vertically aligned geometry and a smooth surface with a thickness of 5 to 10 nm and a width of 100 to 300 nm (Fig. 1b–c). Inset in Fig. 1c shows a high-resolution TEM image of the single Co3O4 NS. The lattice fringes were separated by the spacing of 0.233 nm, which was consistent with the (222) plane of Co3O4. Typical SEM images of as-prepared GDL-Co3O4 NSs–30Au are shown in Fig. 1d. It showed the similar vertically aligned morphology of the nanosheets, suggesting that the Co3O4 NSs remained stable during the thermal evaporation process. Moreover, Au NPs uniformly distributed on the surface of Co3O4 NSs were clearly observed (Fig. 1e). The size of Au NPs is about 20 nm. The interplanar spacing of the Au nanoparticle was approximately 2.35 Å, which corresponded to the (111) plane of the Au phase (JCPDS card no. 04-0784, inset in Fig. 1e). Fig. 1f displays X-ray diffraction (XRD) patterns of GDL-30Au, GDL-Co3O4 NSs, and GDL-Co3O4 NSs–30Au. After Au deposition, the XRD pattern of the GDL-30Au shows four additional peaks at 38.2°, 44.4°, 64.6°, and 77.5°, which are indexed to the (111), (200), (220) and (311) planes of Au crystalline (JCPDS card no. 04-0784). The diffraction patterns of the GDL-Co3O4 NSs in the 2θ range from 10° to 80° are indexed to cubic Co3O4 phase (JCPDS card no. 073-1701). For GDL-Co3O4 NSs–30Au, the diffraction patterns in the 2θ range from 10° to 80° are well consistent with the peaks from Au, cubic Co3O4 as well as graphite, indicating the successful fabrication of GDL-Co3O4 NSs–30Au hybrid catalyst. Additionally, X-ray photoelectron spectroscopy (XPS) was employed to analyse the chemical composition and the valence state of GDL-Co3O4 NSs. The XPS spectra of Co and O elements in GDL-Co3O4 NSs are shown in Fig. S2a and b.† Two distinct peaks at 780.1 and 795.2 eV are assigned to 2p3/2 and 2p1/2 of Co2+, whereas two small peaks at 789 and 803.9 eV are respectively ascribed to 2p3/2 and 2p1/2 of Co3+ (Fig. S2a†).32,33 A high-resolution spectrum for the O 1s region shows two peaks at 530 and 531.7 eV, which respectively correspond to the lattice oxide ions and the low coordinated oxygen ions (chemisorbed oxygen) on the surface (Fig. S2b†).34,35 Nitrogen adsorption–desorption measurement was conducted to further investigate the surface area of GDL-30Au, GDL-Co3O4 NSs, and GDL-Co3O4 NSs–30Au (Fig. S3†). The isotherms of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au are type-IV with hysteresis loops in the middle- and high-pressure regions, indicating the co-existence of micro- and mesoporous structures in the electrode. GDL-Co3O4 NSs–30Au delivered the highest surface area of 20.8 m2 g−1 compared with 3.8 and 13.5 m2 g−1 for GDL-30Au and GDL-Co3O4 NSs. The increase in the surface area might be caused by the deposition of Au on the 2D Co3O4 nanosheets.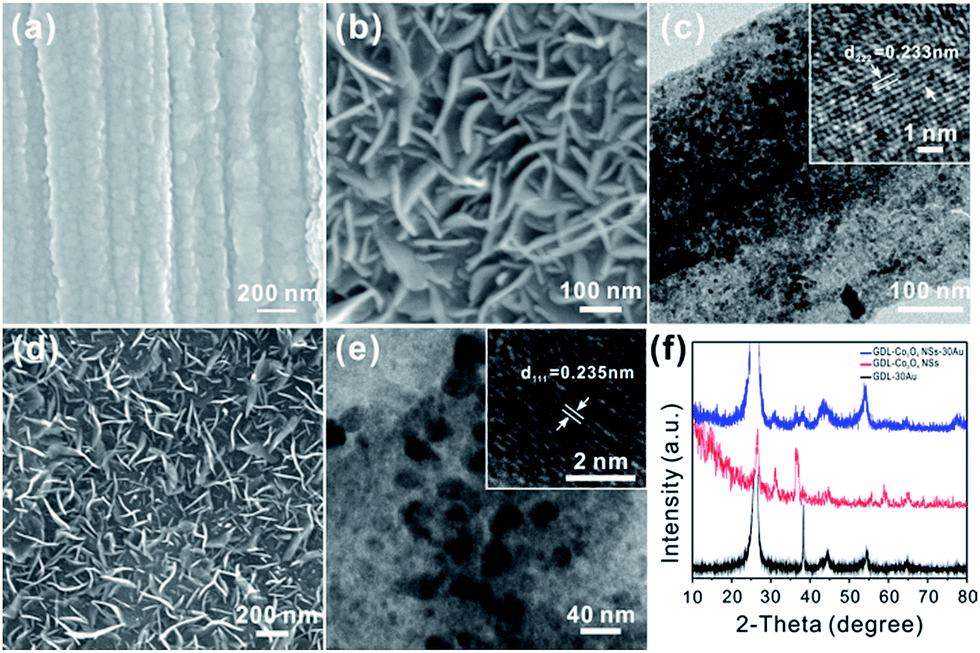 | ||
| Fig. 1 (a) SEM image of pristine GDL-30Au; (b) SEM and (c) TEM image of GDL-Co3O4 NSs; inset in (c) shows a corresponding HRTEM image of Co3O4 single nanosheet; (d) SEM images and (e) TEM images of GDL-Co3O4 NSs–30Au, inset in (e) shows a corresponding HRTEM image of Au NPs, (f) XRD pattern of GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au. | ||
To confirm whether the out-morphology Co3O4 NSs would influence the catalytic activity of GDL-Co3O4 NSs–Au, 50 nm and 200 nm thick cobalt has been deposited on GDL and transformed into Co3O4 by thermal treatment. Vertically aligned Co3O4 NSs cannot be formed on 50 nm thick cobalt (Fig. S4a†). For those with 200 nm cobalt deposition, vertically aligned Co3O4 NSs grew successfully (Fig. S4b†). The size and distribution to Co3O4 NSs were similar to those of the Co3O4 NSs formed on 100 nm thick cobalt layer.
The electrochemical properties of the GDL-Co3O4 NSs–30Au were investigated to evaluate its potential as a bifunctional composite catalyst for the oxygen electrode of Li–O2 batteries. The 1st full discharge/charge curves of GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au between 4.5 V and 2.3 V at a current density of 0.5 mA cm−2 were obtained (Fig. S5†). The GDL-Co3O4 NSs–30Au exhibited the outstanding first discharge capacity of 4585 mA h g−1, compared with GDL-Co3O4 NSs (2784 mA h g−1) and GDL-30Au (2974 mA h g−1). The GDL-Co3O4 NSs–30Au delivered the first-cycle coulombic efficiency around 67.8%, showing the improved reversibility than GDL-Co3O4 NSs (64%) and GDL-30Au (63.7%). Fig. 2a shows the first charge/discharge voltage curves with GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au at a limited capacity of 1000 mA h g−1. It is necessary to limit the capacity during cycling because Li2O2 is accumulated on the surface of the electrodes along with capacity enhancement, thus deactivating the catalytic sites and depressing the electron conductivity of the oxygen electrode.8,22,36 The tetraethylene glycol dimethyl ether (TEGDME) was used as the electrolyte solvent due to its higher stability toward O2− than carbonate-based electrolytes.37 As shown in Fig. 2a, the charge and discharge medium voltages are about 3.5 V and 2.6 V for the GDL-Co3O4 NSs–30Au catalyst, demonstrating significant improvements in both oxygen evolution reactions (OER) and oxygen reduction reactions (ORR) compared to that of GDL-30Au and GDL-Co3O4 NSs. The potential difference between the ORR and OER curves was calculated from the data in Fig. 2a (Table S1†). The GDL-Co3O4 NSs–30Au catalyst showed the lowest oxygen electrode potential differences. The potential difference of GDL-Co3O4 NSs–30Au was 0.89 V, whereas those of GDL-30Au and GDL-Co3O4 NSs were 1.27 V and 1.49 V, respectively. The lowest overpotential of GDL-Co3O4 NSs–30Au among all the synthesized hybrid catalysts for the oxygen electrode reactions confirmed that it had superior catalytic activities for both ORR and OER.38 Fig. 2b–d demonstrates the charge/discharge profiles of GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au. The GDL-Co3O4 NSs–30Au also exhibits the excellent cycling performance of 70 cycles, which are much longer than those of GDL-30Au (8 cycles) (Fig. 2b) and GDL-Co3O4 NSs (50 cycles) (Fig. 2c). In the cycles, the charging potential rises gradually, whereas the discharging potential drops gradually to near 2.3 V. This phenomenon could be ascribed to the indecomposable nature of the side products (e.g., Li2CO3) during charging/discharging.39 Fig. S6† shows the first charge/discharge voltage curves employing GDL-Co3O4 NSs from 50, 100 and 200 nm thick Co depositions at a limited capacity of 1000 mA h g−1. The charge/discharge curves of GDL-Co3O4 NSs from 100 and 200 nm thick Co deposition are almost overlapped, indicating that thicker cobalt deposition cannot further improve the catalytic activity of GDL-Co3O4 NSs. For GDL-Co3O4 NSs from 50 nm thick Co deposition, the overall potential is even larger than GDL-Co3O4 NSs from 100 nm thick Co deposition. This result reveals that the 2D geometry of Co3O4 NSs plays an important role on the improvement in the catalytic properties of the GDL-Co3O4 NSs–Au hybrid catalyst. For comparison, pure GDL without loading any other catalyst was tested under the same condition. As shown in Fig. S7,† it delivers the capacity less than 100 mA g−1 after 10 cycles, demonstrating the prominent catalytic effects of Au NPs and Co3O4 NSs. Additionally, the rate performance of GDL-Co3O4 NSs–30Au is also tested (Fig. 2e). Even at a high current density of 2 mA cm−2, the discharge capacity still reaches 1536 mA h g−1. Furthermore, the Li–O2 battery with GDL-Co3O4 NSs–30Au cathode shows the excellent performance compared to previously reported results (Table S2†).
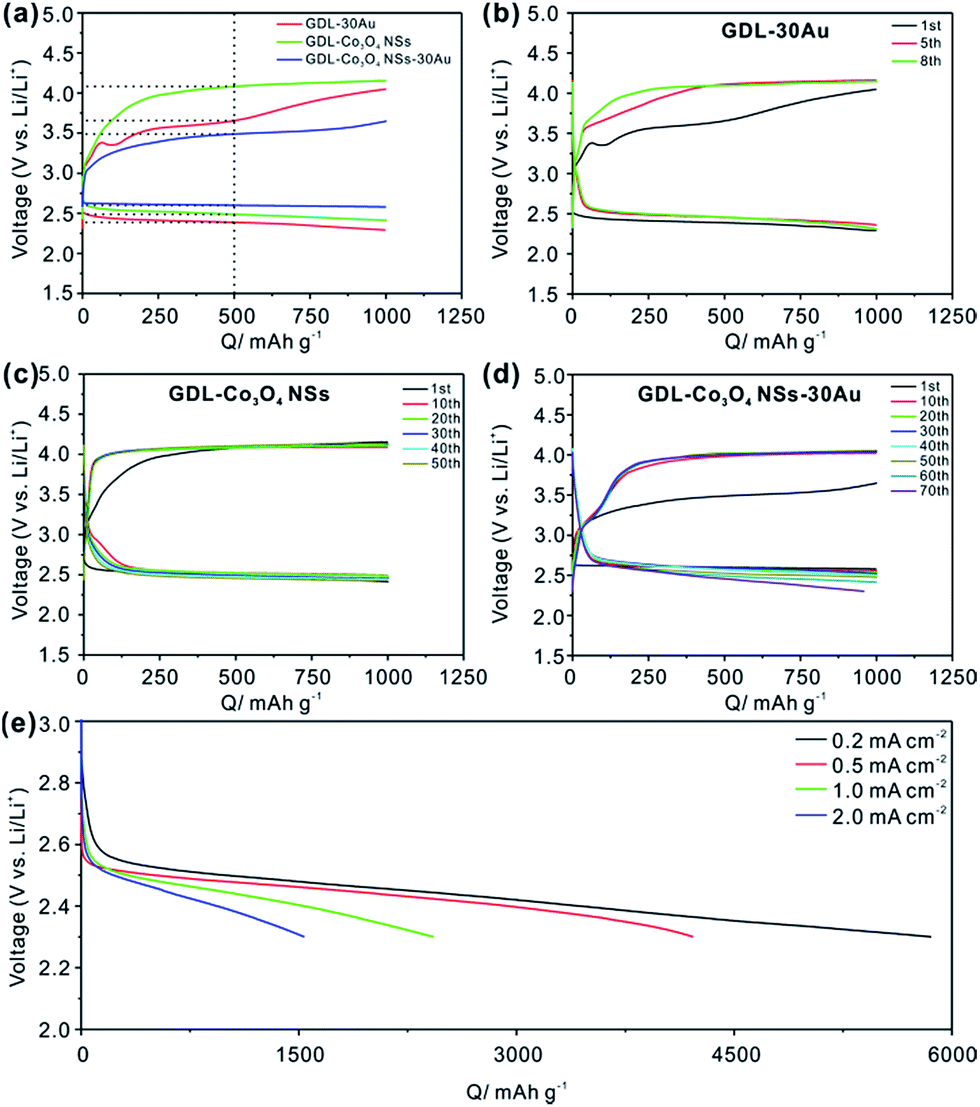 | ||
| Fig. 2 (a) Initial charge/discharge curves of GDL-30Au, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au; charge/discharge profiles of (b) GDL-30Au, (c) GDL-Co3O4 NSs and (d) GDL-Co3O4 NSs–30Au equipped Li–O2 batteries upon repeated cycles between 2.3 V to 4.5 V with the capacity limited to 1000 mA h g−1; (e) rate performance of the cell with GDL-Co3O4 NSs–30Au cathode. | ||
To study the influences of size and distribution of Au NPs on the catalytic performance of the Co3O4–Au hybrid, Au NPs with different thicknesses of 10 nm and 50 nm was deposited on the Co3O4 NSs, respectively. As shown in Fig. S8a and c,† similar vertically aligned nanosheets were maintained after Au deposition. However, the size of Au NPs increased with the increase in the Au deposition. The sizes of Au NPs were respectively tuned to be 10 nm and 40 nm for GDL-Co3O4 NSs–10Au, and GDL-Co3O4 NSs–50Au. Meanwhile, from the magnified TEM images of GDL-Co3O4 NSs–10Au and GDL-Co3O4 NSs–50Au, it revealed that the thicker Au NPs deposition layer led to the larger area covered by Au NPs for Co3O4 NSs (Fig. S8b and d†). The interplanar spacing of the Au nanoparticle for GDL-Co3O4 NSs–10Au and GDL-Co3O4 NSs–50Au was approximately 2.35 Å, which corresponded to the (111) plane of the Au phase (JCPDS card no. 04-0784) (inset in Fig. S8b and d†). Fig. S8e† displayed the XRD patterns of GDL-Co3O4 NSs–10Au and GDL-Co3O4 NSs–50Au, showing similar diffraction peaks with GDL-Co3O4 NSs–30Au, further proving the successful dotting of Au NPs on Co3O4 NSs. Fig. S8f and g† demonstrate the EDS spectra of GDL-Co3O4 NSs–10Au and GDL-Co3O4 NSs–50Au, revealing that the Au content increased when the Au NPs deposition layer became thicker.
The effects of Co3O4–Au hybrid catalyst with different distributions and sizes of Au NPs on the overpotential and cycling performance of Li–O2 batteries have been further studied with the limited capacity of 1000 mA h g−1. As shown in Fig. 3a, GDL-Co3O4 NSs–30Au catalyst still demonstrates the lowest charging plateau and the highest discharging plateau compared to GDL-Co3O4 NSs–10Au and GDL-Co3O4 NSs–50Au, indicating that the GDL-Co3O4 NSs–30Au still has the best catalytic activity toward both ORR and OER processes. Meanwhile, the variations of medium voltage of GDL-Co3O4 NSs–10/30/50Au with the cycle numbers and charge/discharge profiles of GDL-Co3O4 NSs–10/50Au are shown in Fig. 3b–d. GDL-Co3O4 NSs–30Au delivers the lowest overall overpotential and the most cycles compared to those with 10 nm and 50 nm Au NPs. The above results demonstrated that the size and distribution of Au NPs influenced the catalytic performance of Co3O4–Au hybrid greatly, indicating the importance of rational design of Au NPs and Co3O4 NSs. The improvements in electrochemical performances of GDL-Co3O4 NSs–30Au electrode can be attributed to the rationally designed electrode configuration and the synergistic catalytic activity of Au-introduced Co3O4 NSs. The 2D geometry of Co3O4 NSs provides enough space for discharging products and the large contact area between the catalyst and discharging product. Meanwhile, a proper amount of Au NPs allow the facile formation and the decomposition of discharging products, thus leading to the reduction of the polarization effect. The combination of Au NPs and Co3O4 NSs results in the enhanced catalytic effect over both OER and ORR, thus improving the cycling stability and round-trip efficiency.
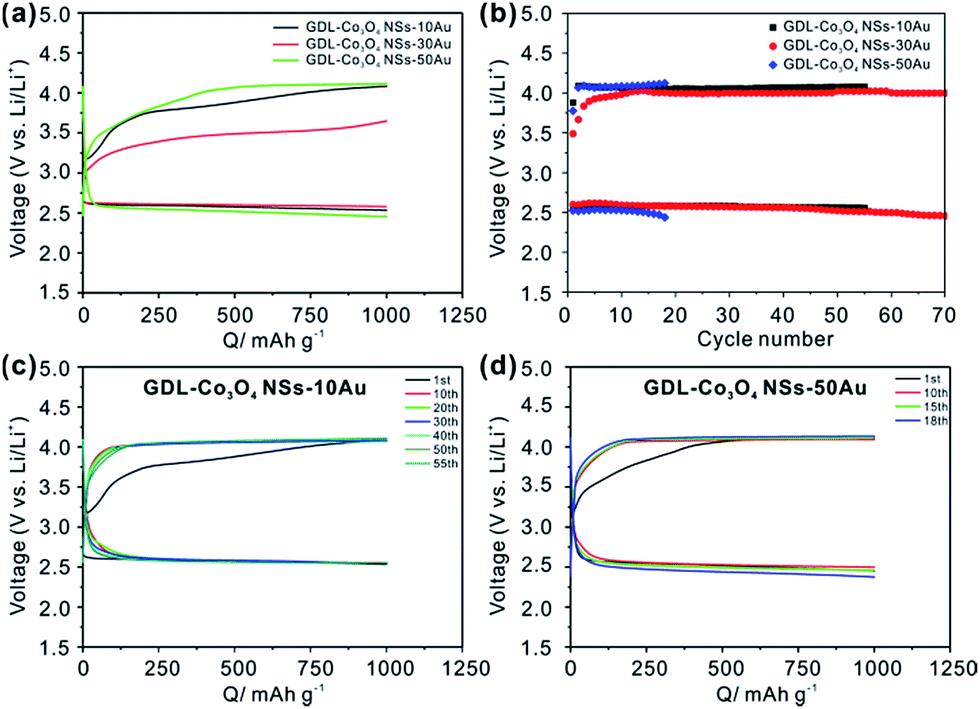 | ||
| Fig. 3 (a) Initial charge/discharge curves of GDL-Co3O4 NSs–10Au, GDL-Co3O4 NSs–30Au and GDL-Co3O4 NSs–50Au; (b) the variation of discharge and charge medium voltage with the cycle number of GDL-Co3O4 NSs–10Au, GDL-Co3O4 NSs–30Au and GDL-Co3O4 NSs–50Au; charge/discharge profile of (c) GDL-Co3O4 NSs–10Au and (d) GDL-Co3O4 NSs–50Au equipped Li–O2 batteries upon repeated cycles between 2.3 V to 4.5 V with the capacity limited to 1000 mA h g−1. | ||
To reveal the mechanism for the improved catalytic effect of GDL-Co3O4 NSs–30Au, we compared the phase composition and morphology change of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes. XRD and XPS were conducted to analyse the phase change for GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes (Fig. 4 and S9†). After the 1st discharge, newly generated peaks at 23.32°, 32.9°, 35.0°, 40.6°, and 58.68°, respectively corresponding to (002), (100), (101), (102) and (110) peaks of Li2O2 (JCPDS card no. 09-0355), was observed (Fig. 4). This result indicated that Li2O2 was the major discharging product for both GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes. Furthermore, XPS Li 1s spectra were obtained from the discharge of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au cathodes (Fig. S9†). The peaks at 54.5 eV and 55.3 eV could be assigned to Li in Li2O2 and the surface lithium carbonate species formed during the decomposition of glyme electrolyte contacting with lithium peroxide, respectively. The morphological changes in the GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes in the different states of discharge and charge were then investigated in order to understand the discharge–charge behaviors (Fig. S10a–h†). Unlike the toroid-shaped discharging product in previous reports, film-like Li2O2 was formed on the surface of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au after the 1st discharging (Fig. S10a–e†). The thin film formed after 1st discharge sticks closely to the nanosheets and becomes thicker when discharging to higher capacities.7,40–42 Subsequently, all discharging products were almost fully decomposed from the surface of both GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes after recharging (Fig. S10b–f†). The morphological changes in both electrodes were correlated with the formation and decomposition of Li2O2 during cycling. However, after 50 cycles, GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au electrodes exhibited totally different behaviours in the morphological change. For the GDL-Co3O4 NSs electrode, it was difficult to observe the 2D configuration of the electrode after discharging/recharging. The surface of the electrode was completely covered by discharging products and the void space was blocked (Fig. S10c and d†). For the case of GDL-Co3O4 NSs–30Au electrode, 2D configuration was maintained and the discharging product was removed after charging (Fig. S10h†). A possible mechanism was proposed to explain the different morphology evolution of GDL-Co3O4 NSs and GDL-Co3O4 NSs–30Au, as shown in Fig. 5. During the discharge process, Au NPs behave as additional nucleation sites for Li2O2 growth, forming Li2O2-enwrapped Co3O4 NSs–30Au (Fig. 5i). A small amount of carbonate species were formed simultaneously as proved by XPS spectra. In the charging process, the Au NPs embedded in Li2O2 promote the decomposition of carbonate species and improved the conductivity of discharging products simultaneously, thus leading to the early decomposition of the discharging layer (Fig. 5ii).27 In the following cycles, the channels formed by the decomposed discharging products enable the continuous contact between the hybrid catalysts and O2, thus resulting in the lower overpotential of the cells (Fig. 5iii). Meanwhile, the distribution and size of Au NPs influence the formation/decomposition of the discharging products greatly, indicating the importance of rational design of the hybrid structure. After continuous charging/discharging, large particles were decomposed to form a thick layer of discharging products on the surface of GDL-Co3O4 NSs–30Au surface. Consequently, the improved electrochemical performance was achieved by the combination of Au NPs with the proper size and distribution and the Co3O4 NSs. These results highlighted the power of GDL-Co3O4 NSs–30Au hybrid electrocatalyst and indicated that the performance of Li–O2 batteries could be further improved by rationally designed hybrid catalysts.
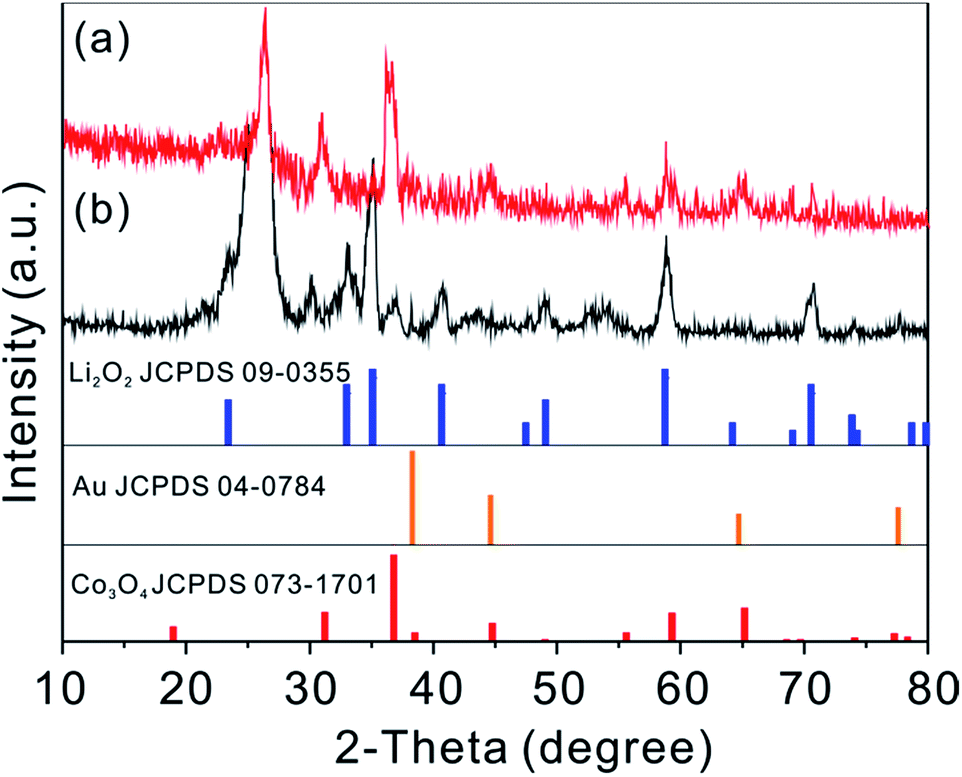 | ||
| Fig. 4 XRD patterns of (a) GDL-Co3O4 NSs and (b) GDL-Co3O4 NSs–30Au after discharge to 2.3 V. | ||
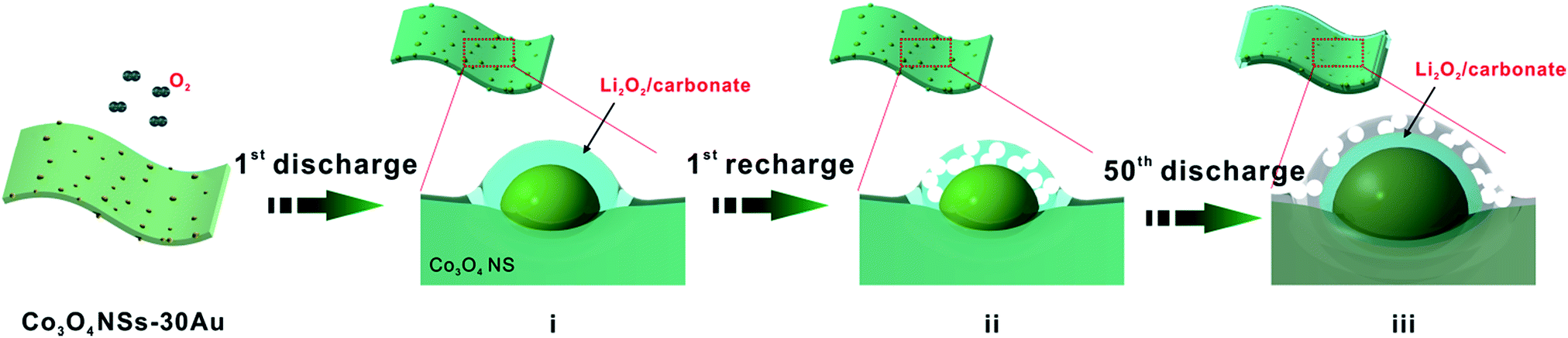 | ||
| Fig. 5 Schematic diagram of GDL-Co3O4 NSs–30Au electrode at different stages of charge and discharge. | ||
Conclusions
In summary, 3D, binder-free and free-standing Co3O4 NSs decorated with a proper amount of Au NPs growing on GDL was successfully synthesized. When it was employed as an O2 cathode, the cells exhibited the lower polarization during cycling and the improved cyclability of 70 cycles at a limited capacity of 1000 mA h g−1. The enhanced electrochemical properties of the Li–O2 cells with GDL-Co3O4 NSs–30Au electrode demonstrated that rationally designed structures and materials could further improve the catalytic properties of the hybrid catalyst. The SEM images after charging/discharging clearly present the role of gold in promoting the decomposition of carbonate species and improving the conductivity of discharging products. Furthermore, both the catalytic synergy effect from rationally designed Au NPs and Co3O4 NSs and the morphological advantages generated by the 2D geometry of Co3O4 NSs/uniformly coated small Au NPS enable the enhanced electrochemical properties. This strategy suggests a way to improve the catalytic properties of metal oxide materials for Li–O2 batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP), the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20168510050080) and the Human Resources Program in Energy Technology of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), which was granted financial resources from the Ministry of Trade, Industry & Energy, Republic of Korea (20174010201240).Notes and references
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125–1141 CAS.
- J. Lu, L. Li, J. B. Park, Y. K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- Y.-C. Lu, H. A. Gasteiger, E. Crumlin, R. McGuire and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A1016–A1025 CrossRef CAS.
- E. Yoo and H. Zhou, ACS Nano, 2011, 5, 3020–3026 CrossRef CAS PubMed.
- Y. Lei, J. Lu, X. Luo, T. Wu, P. Du, X. Zhang, Y. Ren, J. Wen, D. J. Miller, J. T. Miller, Y. K. Sun, J. W. Elam and K. Amine, Nano Lett., 2013, 13, 4182–4189 CrossRef CAS PubMed.
- R. R. Mitchell, B. M. Gallant, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2952–2958 CAS.
- H. G. Jung, J. Hassoun, J. B. Park, Y. K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
- Z.-L. Wang, D. Xu, J.-J. Xu, L.-L. Zhang and X.-B. Zhang, Adv. Funct. Mater., 2012, 22, 3699–3705 CrossRef CAS.
- M. V. Kannan and G. Gnana kumar, Biosens. Bioelectron., 2016, 77, 1208–1220 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshoj, J. K. Norskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- R. Black, S. H. Oh, J. H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef CAS PubMed.
- C. Sun, F. Li, C. Ma, Y. Wang, Y. Ren, W. Yang, Z. Ma, J. Li, Y. Chen, Y. Kim and L. Chen, J. Mater. Chem. A, 2014, 2, 7188–7196 CAS.
- E. Yilmaz, C. Yogi, K. Yamanaka, T. Ohta and H. R. Byon, Nano Lett., 2013, 13, 4679–4684 CrossRef CAS PubMed.
- R. Black, J. H. Lee, B. Adams, C. A. Mims and L. F. Nazar, Angew. Chem., Int. Ed., 2013, 52, 392–396 CrossRef CAS PubMed.
- Y. Cao, Z. Wei, J. He, J. Zang, Q. Zhang, M. Zheng and Q. Dong, Energy Environ. Sci., 2012, 5, 9765–9768 CAS.
- Q. Liu, Y. Jiang, J. Xu, D. Xu, Z. Chang, Y. Yin, W. Liu and X. Zhang, Nano Res., 2014, 8, 576–583 CrossRef.
- G. Zhao, J. Lv, Z. Xu, L. Zhang and K. Sun, J. Power Sources, 2014, 248, 1270–1274 CrossRef CAS.
- B. Li, X. Ge, F. W. Goh, T. S. Hor, D. Geng, G. Du, Z. Liu, J. Zhang, X. Liu and Y. Zong, Nanoscale, 2015, 7, 1830–1838 RSC.
- L. Leng, X. Zeng, H. Song, T. Shu, H. Wang and S. Liao, J. Mater. Chem. A, 2015, 3, 15626–15632 CAS.
- W. H. Ryu, T. H. Yoon, S. H. Song, S. Jeon, Y. J. Park and I. D. Kim, Nano Lett., 2013, 13, 4190–4197 CrossRef CAS PubMed.
- J. Y. Kim and Y. J. Park, Sci. Rep., 2017, 7, 8610 CrossRef PubMed.
- S. H. Oh and L. F. Nazar, Adv. Energy Mater., 2012, 2, 903–910 CrossRef CAS.
- Y.-C. Lu, H. A. Gasteiger, M. C. Parent, V. Chiloyan and Y. Shao-Horn, Electrochem. Solid-State Lett., 2010, 13, A69–A72 CrossRef CAS.
- Y.-C. Lu, Z. X. Xu, H. A. Gasteiger, S. Chen, K. H. Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- W. Fan, X. Guo, D. Xiao and L. Gu, J. Phys. Chem. C, 2014, 118, 7344–7350 CAS.
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 CAS.
- J. Wang, Y. Li and X. Sun, Nano Energy, 2013, 2, 443–467 CrossRef CAS.
- H. Nie, C. Xu, W. Zhou, B. Wu, X. Li, T. Liu and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 1937–1942 CAS.
- J. Lu, Y. Lei, K. C. Lau, X. Luo, P. Du, J. Wen, R. S. Assary, U. Das, D. J. Miller, J. W. Elam, H. M. Albishri, D. A. El-Hady, Y. K. Sun, L. A. Curtiss and K. Amine, Nat. Commun., 2013, 4, 2383–2391 Search PubMed.
- L. Li, S. Liu and A. Manthiram, Nano Energy, 2015, 12, 852–860 CrossRef CAS.
- H. Xia, D. Zhu, Z. Luo, Y. Yu, X. Shi, G. Yuan and J. Xie, Sci. Rep., 2013, 3, 2978–2985 CrossRef PubMed.
- D. C. Adnan Younis, X. Lin, J. Lee and S. Li, Nanoscale Res. Lett., 2013, 8, 36 CrossRef PubMed.
- Y. Feng, L. Li, S. Niu, Y. Qu, Q. Zhang, Y. Li, W. Zhao, H. Li and J. Shi, Appl. Catal., B, 2012, 111–112, 461–466 CrossRef CAS.
- G. Gnana kumar, M. Christy, H. Jang and K. S. Nahm, J. Power Sources, 2015, 288, 451–460 CrossRef CAS.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Barde and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- M. Y. Oh, J. J. Lee, A. Zahoor, G. Gnana kumar and K. S. Nahm, RSC Adv., 2016, 6, 32212–32219 RSC.
- Z. Lyu, J. Zhang, L. Wang, K. Yuan, Y. Luan, P. Xiao and W. Chen, RSC Adv., 2016, 6, 31739–31743 RSC.
- B. M. Gallant, R. R. Mitchell, D. G. Kwabi, J. Zhou, L. Zuin, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. C, 2012, 116, 20800–20805 CAS.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 CAS.
- F. Wu, X. Zhang, T. Zhao, R. Chen, Y. Ye, M. Xie and L. Li, J. Mater. Chem. A, 2015, 3, 17620–17626 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09855c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |