
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
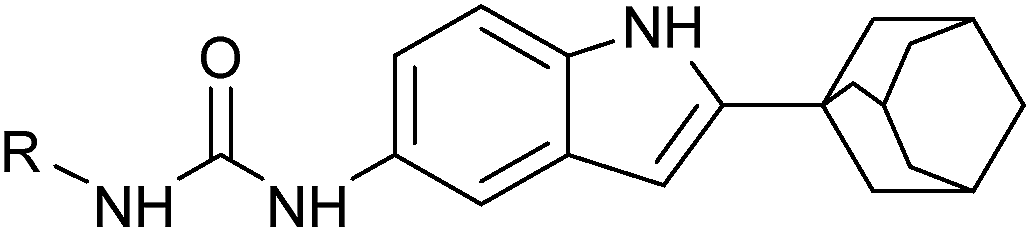
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of 1-(2-(adamantane-1-yl)-1H-indol-5-yl)-3-substituted urea/thiourea derivatives as anticancer agents†
Hongyu Hu‡
ab,
Chunrong Lin‡a,
Mingtao Ao‡a,
Yufen Jia,
Bowen Tanga,
Xiaoxiao Zhoua,
Meijuan Fang *a,
Jinzhang Zeng*a and
Zhen Wu*a
*a,
Jinzhang Zeng*a and
Zhen Wu*a
aFujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, South Xiang-An Road, Xiamen, 361102, China. E-mail: wuzhen@xmu.edu.cn; jzzeng@xmu.edu.cn; fangmj@xmu.edu.cn; Fax: +86-592-2189868; Tel: +86-592-2189868
bCollege of Xingzhi, Zhejiang Normal University, Jinhua 321004, China
First published on 8th November 2017
Abstract
The indole ring, adamantane, and urea groups are important components of bioactive molecules. The orphan nuclear receptor Nur77 as a unique transcription factor encoded by an immediate early gene is a potential therapeutic target for cancer treatment. We synthesized a series of 1-(2-(adamantane-1-yl)-1H-indol-5-yl)-3-substituted urea/thiourea derivatives and identified which of these potential anticancer candidates could modulate the expression and activity of Nur77. The synthesized compounds were initially evaluated for their anti-proliferative activity against H460 lung cancer cells, HepG2 liver cancer cells, and MCF-7 breast cancer cells. Major compounds were found to be active against these tested cancer cell lines. The compounds with IC50 values down to 20 μM exhibited selective cytotoxicity effects on the human lung cancer cell line (H460) and the normal lung cell line (MCR-5). Compounds 7n, 7s, and 7w induced Nur77-expression in a time- and dose-dependent manner in H460 cells. Compounds 7n and 7s strongly induced Parp cleavage in H460 cells, but 7w resulted in a slight induction of apoptosis. The apoptotic effect of 7s was largely inhibited when the Nur77 was knocked down by shRNA. This indicated that Nur77 served as a critical mediator for the anticancer action of 7s. The molecular docking study between Nur77 and 7s revealed that compound 7s exhibited a promising binding affinity with Nur77. These findings will provide a direction for the developing Nur77 regulator as anticancer agents.
1. Introduction
Nur77 (also known as NR4A1, NGFI-B, or TR3) is an orphan member of the nuclear receptor super-family that regulates the expression of genes involved in multiple physiological and pathological processes.1 As an immediate-early response gene, Nur77 expression could be induced by various stimuli, including mitogenic and apoptotic signaling.2–4 Nur77 plays a role in cell survival, cell death, and is implicated in various malignancies.3–11 Nur77 is upregulated in gastric tumorsphere cells,12 non-small cell lung cancer (NSCLC) tissues and cells,13 and colon tumors.14,15 Previous studies have found that Nur77 is often overexpressed in tumour tissues, when compared to the para-carcinoma normal tissues.16 Nur77 is a potentially promising drug target for cancer treatment. A number of anticancer compounds with diverse chemical structures, such as acetylshikonin analogue SK07,17 DIM-C-pPhOCH3,18 Vit K2,19 diindolylmethane analog,20 Csn-B,21 cardenolides H9 and ATE-i2-b4,22 AmB-NC,23 CD437,10,24 n-butylidenephthalide,25 and norcantharidin26 have been identified as critical modulators of the genomic or the nongenomic pathways of Nur77. These findings suggest that Nur77 is a critical and meaningful drug target. Thus, we are interested in exploring a novel Nur77 regulator.The indole ring has been proven to be a critical pharmacophore in many chemotherapeutics.27–29 The introduction of different side chains around the indole nucleus has resulted in the development of various therapeutic drugs, including obatoclax,30 and sunitinib,31 as well as many lead compounds that boast a wide variety of biological activities. Modifying the 2nd and 5th positions of the indole moiety has been shown to be crucial for receptor binding and activation.32 The adamantane derivatives were initially used as an antiviral drug (adamantane) to combat various strains of the flu,33 to treat parkinson's disease (adapalene, dopamantine, memantine, rimantadine and tromantadine), and as dipeptidyl peptidase-4 (DPP-4) inhibitors (saxagliptin, and vildagliptin) for the treatment of type 2 diabetes.34 Some other clinical drugs like sorafenib are urea derivatives of the indole pharmacophore.
Based on the biological profiles of the indole ring, the adamantane, the urea group, and the continuation of our research work on synthesis of indole derivatives,32 we initially synthesized a series of novel 1-(2-(adamantan-1-yl)-1H-indol-5-yl)-3-substituted urea/thiourea derivatives and evaluated their anticancer activity on cancer cell line (HepG2), and breast cancer cell line (MCF-7) by MTT assay. The compounds that exhibited prominent activity against tested cancer cell lines with IC50 down to 20 μM were subsequently tested for their cytotoxicity on normal cell lines (MCR-5 and LO2) as well as their effect on Nur77 activation in H460 cells. Those compounds that had Nur77 activation ability in H460 cells and also exhibited selective cytotoxicity on tumours and normal cell lines which were isolated from the same tissue, were used to further explore their induction of apoptosis in various tumour cell lines and normal NIH-3T3 cells. Compounds 7s, 7n and 7w induced apoptosis in H460 cells. This ability for apoptosis induction was closely associated with their Nur77 activation. The RNA interference technology and the molecular docking study were performed to explore the molecular mechanisms that mediate the induction of apoptosis by this specific class of compounds.
2. Results and discussion
2.1. Syntheses of the designed compounds
The general chemistry for the synthesis of 7a–7y and 9a–9o is outlined in Scheme 1. First, we synthesized 2-adamantane-1H-indol-5-amine (5). The treatment of 1-adamantanecarboxylic acid chloride (1) with o-toluidine resulted in N-o-tolylcycloadamantanecarboxamide (2),32 and 2.5 M of n-butyl lithium in hexane was then added dropwise to obtain the corresponding 2-adamantane-1H-indole (3).32 Compound 5 was formed through the nitrification and catalytic hydrogenation of compound 3.35 5-Isocyanato-1H-indole-adamantane (6) was obtained by reacting compound 5 with triphosgene.36 The intermediate 6 was subjected to reactions with various amines in order to produce compounds 7a–7y. The reaction of compound 5 with triethylenediamine (Dabco), CS2, and bis(trichloromethyl)carbonate (BTC) resulted in 5-isothiocyanato-1H-indole-adamantane (8).37 Compound 8 was converted into compounds 9a–9o by treatment with the corresponding amine. The structures of the target compounds were confirmed with 1H-NMR, 13C-NMR, and high-resolution mass spectrometry (HRMS). The purities of the target compounds were evaluated with high-performance liquid chromatography with diode-array detection (HPLC-DAD) performed at 254 nm and 280 nm. | ||
| Scheme 1 Synthesis of 1-(2-(substituted hydrazinecarbonyl group)-1H-indol-5-yl)-3-substituted urea. Reagents and conditions: (a) toluene, o-toluidine, K2CO3, r.t., 4 h; (b) n-BuLi, THF, 0 °C, N2, 3 h; (c) NH4NO3, H2SO4, 0 °C, 1 h; (d) 10% Pd/C, MeOH, 70 °C, 2 h. (e) Triphosgene, THF, trimethylamine, 0 °C, 4 h; (f) R–NH2, toluene, 60 °C, 4 h. (g) Dabco, CS2, toluene, BTC, DCM. (h) R–NH2, toluene, 60 °C, 4 h. | ||
2.2. Anti-proliferative activities of synthesized compounds in various cancer cells and their structure–activity relationship
The synthesized compounds were evaluated for their anti-proliferative activities against various human cancer cell lines including H460, HepG2 and MCF-7. The concentration required for 50% inhibition of cell viability (IC50) was calculated. The IC50 values of the compounds are summarized in Tables 1 and 2. The results indicated that a majority of the synthesized compounds had a moderate anti-proliferative activity against the three human cancer cell lines. Exceptions were found in compounds 7c, 7d, 7e, 7g, 7h, 7i, 9c, and 9d. Attempts were made to establish the structure–activity relationship (SAR) among the tested compounds based on data collected from three independent experiments.
|
||||
|---|---|---|---|---|
| Compound | R | IC50 (μM) ± S.D. | ||
| H460 | HepG2 | MCF-7 | ||
| a Each compound was tested in triplicate. All error bars represent mean ± SD from three independent experiments. | ||||
| 7a |  |
5.77 ± 0.12 | 5.93 ± 0.15 | 5.16 ± 0.20 |
| 7b |  |
25.25 ± 1.45 | 15.19 ± 1.00 | 13.78 ± 0.98 |
| 7c | 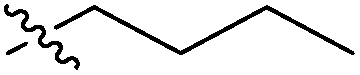 |
>100 | 34.06 ± 1.25 | >100 |
| 7d | 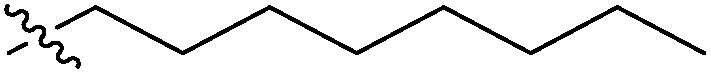 |
>100 | >100 | >100 |
| 7e | 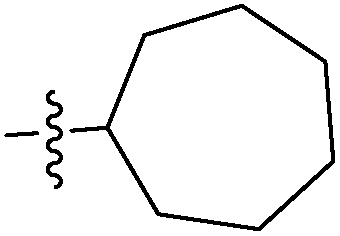 |
>100 | >100 | >100 |
| 7f | 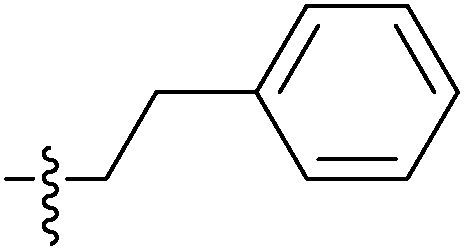 |
0.42 ± 0.09 | 0.28 ± 0.08 | 6.96 ± 0.12 |
| 7g | 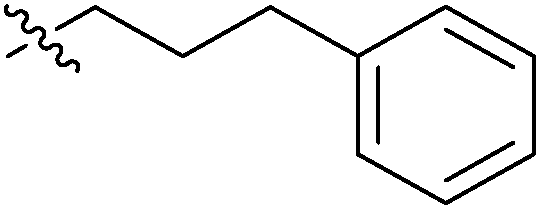 |
13.21 ± 0.15 | 1.42 ± 0.10 | >100 |
| 7h | 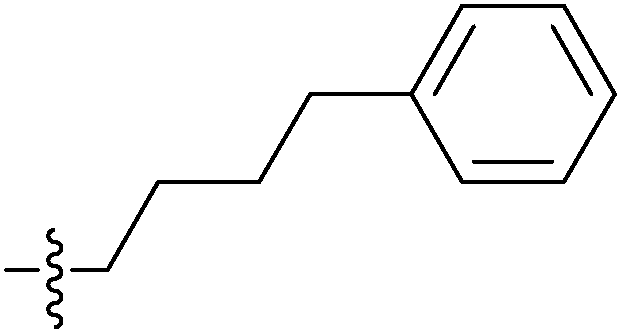 |
>100 | >100 | >100 |
| 7i | 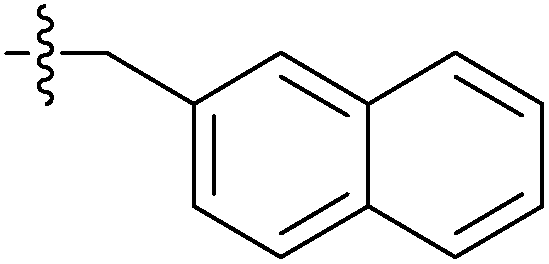 |
>100 | >100 | 23.11 ± 0.52 |
| 7j | 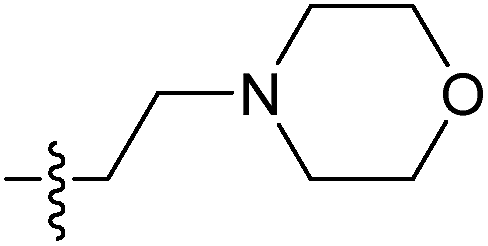 |
11.75 ± 0.85 | 22.10 ± 1.00 | 16.01 ± 1.02 |
| 7k | 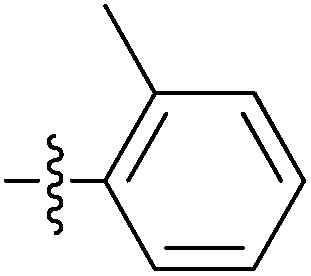 |
11.99 ± 0.95 | 13.06 ± 1.10 | 8.07 ± 0.85 |
| 7l | 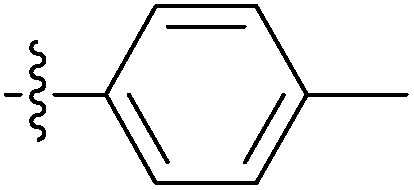 |
41.82 ± 2.55 | 75.74 ± 2.65 | 40.47 ± 2.89 |
| 7m | 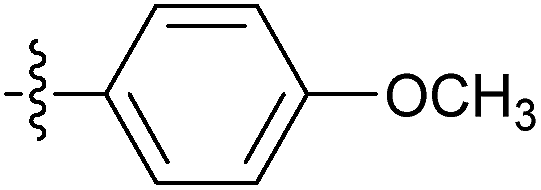 |
60.89 ± 4.55 | 18.32 ± 1.45 | 13.96 ± 0.65 |
| 7n | 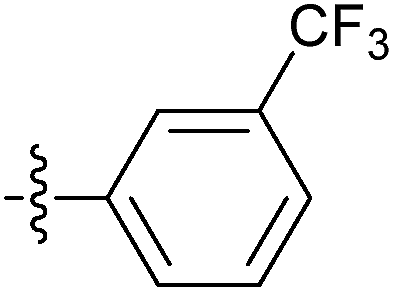 |
12.89 ± 1.41 | 22.24 ± 1.41 | 18.1 ± 2.18 |
| 7o | 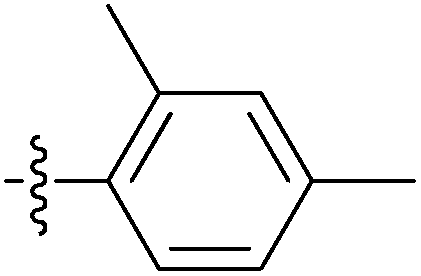 |
54.13 ± 2.40 | 17.22 ± 1.36 | 11.60 ± 0.65 |
| 7p | 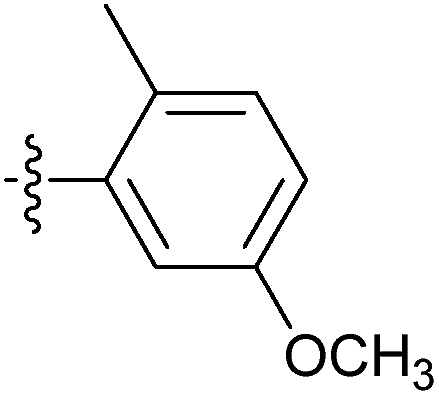 |
50.33 ± 2.70 | 72.46 ± 3.40 | 51.09 ± 1.70 |
| 7q | 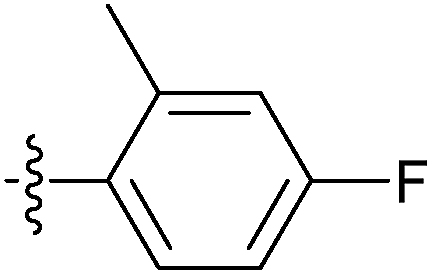 |
62.82 ± 3.50 | 77.37 ± 3.12 | 56.84 ± 2.54 |
| 7r | 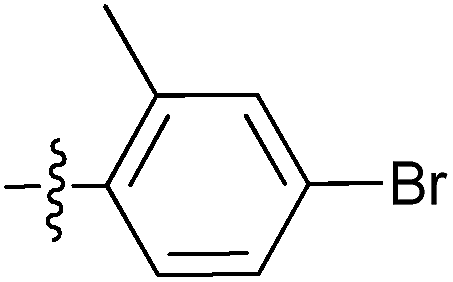 |
32.30 ± 1.53 | 16.99 ± 1.20 | 12.61 ± 1.00 |
| 7s | 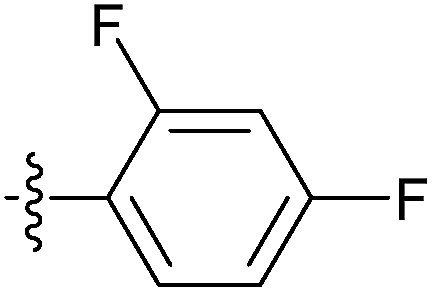 |
10.51 ± 0.30 | 15.50 ± 0.13 | 19.04 ± 0.27 |
| 7t | 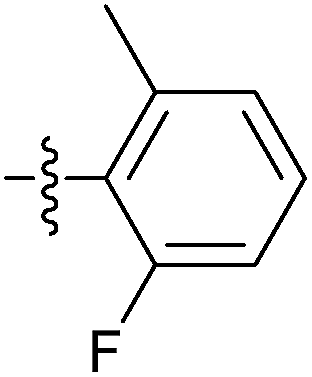 |
9.75 ± 0.50 | 8.98 ± 1.10 | 9.79 ± 0.91 |
| 7u | 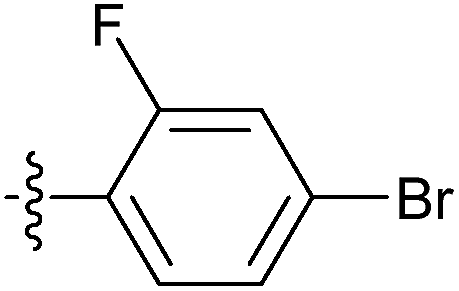 |
9.92 ± 1.20 | 6.99 ± 0.50 | 6.78 ± 0.78 |
| 7v | 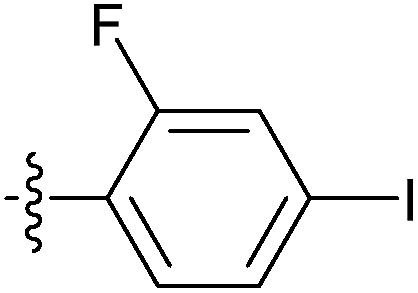 |
10.94 ± 1.50 | 4.56 ± 0.57 | 5.97 ± 0.67 |
| 7w | 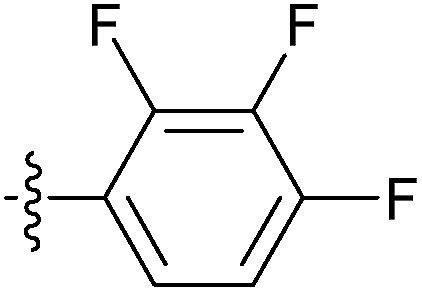 |
14.51 ± 0.17 | 13.15 ± 0.23 | 24.75 ± 0.31 |
| 7x | 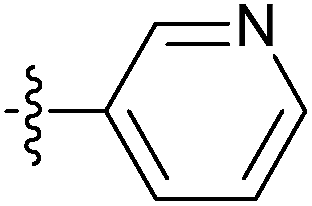 |
6.25 ± 0.47 | 6.38 ± 0.50 | 4.99 ± 0.45 |
| 7y | 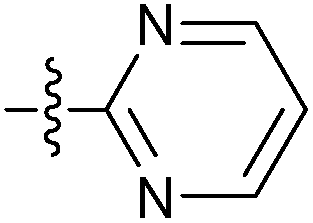 |
13.58 ± 0.30 | 10.47 ± 0.09 | 7.59 ± 0.09 |
| DDP | 3.50 ± 0.10 | 6.10 ± 0.09 | 4.90 ± 0.05 | |
|
||||
|---|---|---|---|---|
| Compound | R | IC50 (μM) ± S.D. | ||
| H460 | HepG2 | MCF-7 | ||
| a Each compound was tested in triplicate. All error bars represent mean ± SD from three independent experiments. | ||||
| 9a |  |
20.23 ± 0.12 | 15.93 ± 0.17 | 25.89 ± 0.20 |
| 9b |  |
27.25 ± 1.40 | 18.02 ± 1.00 | 21.13 ± 0.98 |
| 9c |  |
>100 | >100 | >100 |
| 9d |  |
>100 | >100 | >100 |
| 9e |  |
30.20 ± 0.88 | 35.50 ± 0.85 | 25.00 ± 0.20 |
| 9f | 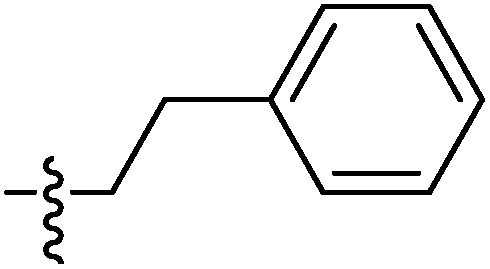 |
14.52 ± 0.10 | 22.20 ± 0.18 | 16.96 ± 0.17 |
| 9g | 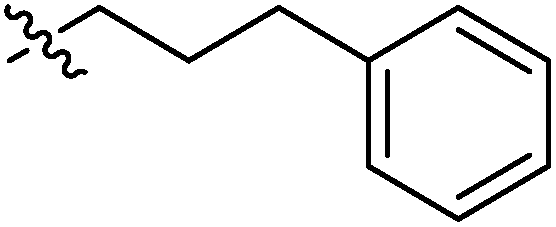 |
25.36 ± 0.15 | 26.12 ± 0.10 | 19.62 ± 0.10 |
| 9h | 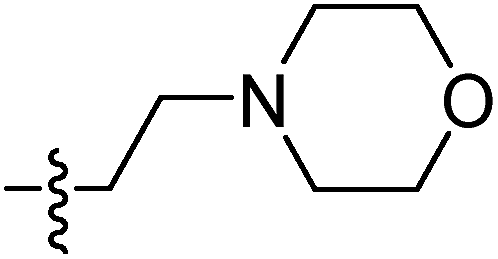 |
22.23 ± 0.18 | 22.36 ± 0.15 | 26.36 ± 0.19 |
| 9i | 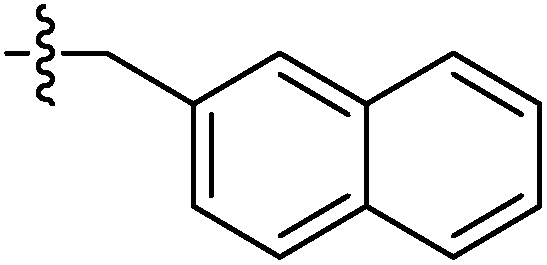 |
38.54 ± 0.18 | 32.56 ± 0.25 | 28.11 ± 0.52 |
| 9j | 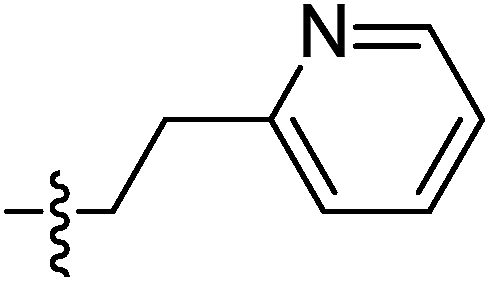 |
31.80 ± 0.85 | 22.10 ± 1.00 | 26.01 ± 1.00 |
| 9k | 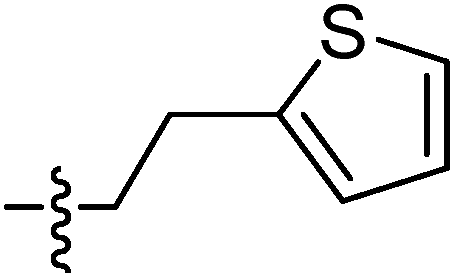 |
25.19 ± 0.95 | 29.06 ± 1.10 | 18.07 ± 0.85 |
| 9l | 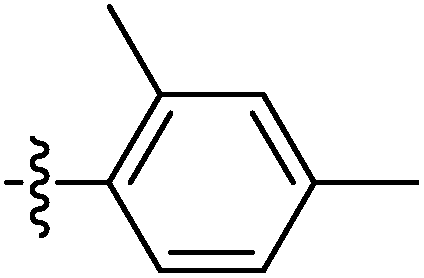 |
21.80 ± 1.55 | 15.74 ± 0.65 | 20.47 ± 1.89 |
| 9m | 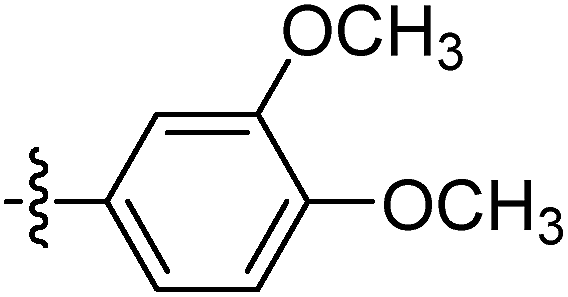 |
25.80 ± 1.55 | 28.48 ± 1.45 | 23.80 ± 0.65 |
| 9n | 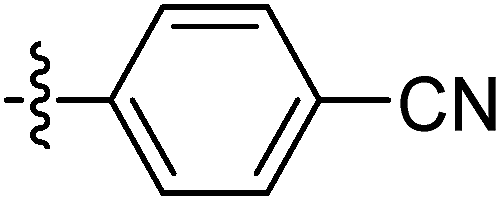 |
41.89 ± 1.20 | 22.20 ± 1.40 | 28.1 ± 2.10 |
| 9o | 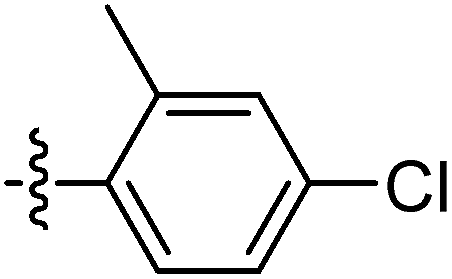 |
20.13 ± 1.40 | 17.22 ± 1.30 | 21.60 ± 0.60 |
| DDP | 3.50 ± 0.10 | 6.10 ± 0.09 | 4.90 ± 0.05 | |
2.3. In vitro cytotoxicity of urea derivatives against two normal cell lines
Twelve urea derivatives that showed prominent activity within all three of the human cancer cell lines with IC50 values down to 20 μM were assessed within the in vitro cytotoxic activity against the human normal lung MCR-5 cell line and the normal hepatocytes LO2 cell line. These urea derivatives' IC50 values for the MCR-5 cells were above 45.0 μM. They exhibited good anti-proliferative activity against the LO2 cells with IC50 values (after 48 h of treatment) ranging from 4.0 μM to 35.0 μM, as shown in Table 3. This demonstrated these compounds were less toxic for the MCR-5 cells than for the LO2 cells. The IC50 ratio of the normal cells to the cancer cells was termed the “relative toxicity index” (RTI). The RTI was estimated to investigate their sensitivity between the normal and the tumour cells in the two human tissue systems (lung and liver). In the lung system, the tested urea derivatives were sensitivity to cancer H460 cells, with RTI values up to 4.70. Most of the urea derivatives in the liver system showed lower selective between the human liver cancer HepG2 cells and the normal hepatocytes LO2 cell line (RTI < 2). This excluded compounds 7f, 7t and 7v. The results indicated that the normal lung MCR-5 cells were more resistant to the toxic activity of these compounds than the lung cancer H460 cells. A majority of the compounds did not possess a selective resistance against the HepG2 cancer cells or the LO2 normal hepatocytes cells.| Compound | MRC-5 | LO2 | ||
|---|---|---|---|---|
| IC50 (μM) ± S.D. | RTIb | IC50 (μM) ± S.D. | RTIc | |
| a Each compound was tested in triplicate. All error bars represent mean ± SD from three independent experiments.b The IC50 ratio is between lung normal MRC-5 cells to cancer H460 cells.c The IC50 ratio is between liver normal LO2 cells to cancer HepG2 cells. | ||||
| 7a | 77.37 ± 0.07 | 13.41 | 5.43 ± 0.09 | 0.92 |
| 7f | >100 | >200 | 5.52 ± 0.04 | 19.71 |
| 7j | 68.25 ± 0.11 | 5.80 | 33.32 ± 0.09 | 1.51 |
| 7k | 56.35 ± 0.08 | 4.70 | 7.71 ± 0.07 | 0.59 |
| 7n | 85.76 ± 0.13 | 6.65 | 13.95 ± 0.14 | 0.63 |
| 7s | 73.86 ± 0.11 | 7.03 | 3.44 ± 0.10 | 0.22 |
| 7t | >100 | >10.26 | 29.64 ± 0.13 | 3.30 |
| 7u | 50.34 ± 0.12 | 5.07 | 5.12 ± 0.09 | 0.73 |
| 7v | 63.71 ± 0.06 | 5.82 | 14.73 ± 0.07 | 3.23 |
| 7w | >100 | >6.89 | 5.19 ± 0.06 | 0.39 |
| 7x | 46.38 ± 0.09 | 7.42 | 4.46 ± 0.05 | 0.71 |
| 7y | 67.35 ± 0.12 | 5.96 | 7.64 ± 0.05 | 0.73 |
| DDP | >100 | >28.57 | 39.69 ± 0.16 | 6.51 |
2.4. The Nur77 activation ability of compounds with IC50 values below 20 μM in H460
A popular research model has showed that H460 cancer cells are sensitive to the Nur77 dependent apoptotic effect.10,40 Our synthesized urea derivatives had a good sensitivity between the normal MCR-5 cells and the tumour H460 cells that were isolated from human lung tissue. So, we investigated whether our synthesized urea derivatives with IC50 values below 20 μM could activate Nur77 in the H460 cancer cells. There were three compounds (7n, 7s, and 7w) that activated Nur77. Results showed that Nur77 was activated upon treatment with 10 μM of the compounds (7n, 7s, and 7w) in a time-dependent manner (Fig. 1(A)). Nur77 activation was observed in H460 cells treated with 7n/7s for 0.5 h. This activation was delayed for 1 h in compound 7w. Nur77 was activated further when treated for 3 h and 5 h with any of the compounds. The activation was maintained at a high level at 7 h. We examined whether Nur77 activation was also dose-dependent. H460 cancer cells were treated with various concentrations of each compound for 3 h (5 μM, 10 μM, and 15 μM) (Fig. 1(B)). As the concentration of each compound increased, the activation of Nur77 improved. Note, when equal amounts of protein lysates (40 μg) was added, 7s was more effective in up regulating Nur77 than 7n and 7w. 7w was the weakest compound.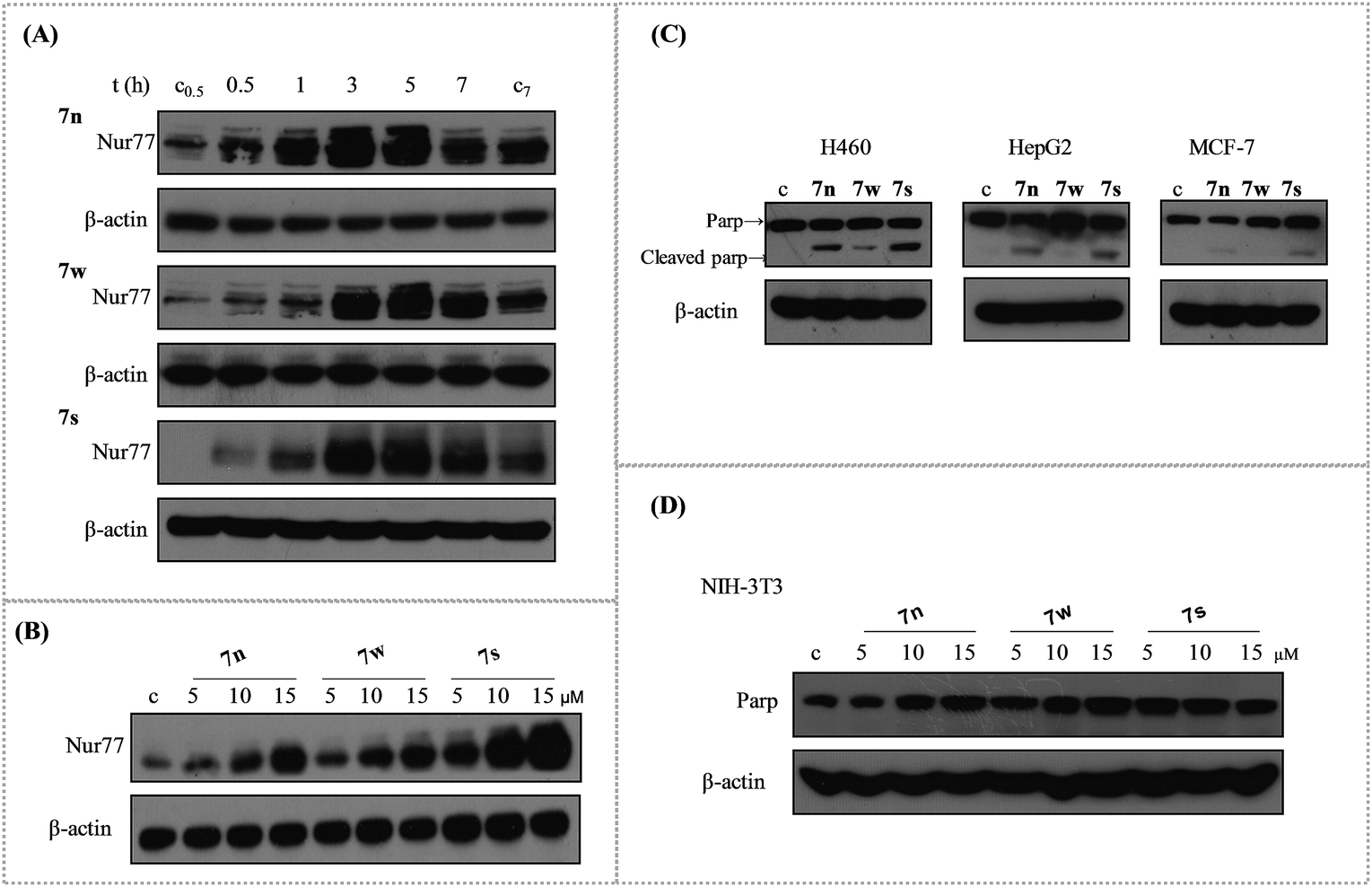 | ||
| Fig. 1 The Nur77 activation of compounds 7n, 7w, and 7s in H460 cells and their apoptosis induction in H460, HepG2, MCF-7 and NIH-3T3. (A) Time-course analysis. H460 cancer cells treated with compounds (10 μM) for 0.5 h, 1 h, 3 h, 5 h, and 7 h were analysed to detect Nur77 levels via western blotting. (B) Dose dependent effect of compounds to active Nur77. H460 cancer cells treated with compounds (5 μM, 10 μM, and 15 μM) for 3 h were analysed via western blotting. (C) The apoptotic effects of compounds 7n, 7s, and 7w in H460, HepG2 and MCF-7 cancer cells. These three cancer cell lines were initially treated with 10 μM compounds for 24 h and then analysed for the cleavage of Parp protein via western blotting. (D) The apoptotic effects of compounds in NIH-3T3 cells. | ||
2.5. The apoptosis induction of 7n, 7w and 7s in H460, HepG2, MCF-7, and NIH-3T3
Apoptosis is a popular approach used by a majority of the anticancer drugs to kill tumour cells.38 Apoptosis stimulation is considered standard and is the best strategy for anticancer therapy.39 Compounds 7n, 7s, and 7w were selected to study the apoptotic effects in various human cancer cells. These cancer cell lines were treated with the three compounds at 10 μM for 24 h to induce apoptosis. The cleavage of PARP protein was used as a sensitive apoptotic marker that occurred early in the apoptotic response. It was then examined by western blotting. The PARP cleavage was observed in various degrees in the tested cell lines (Fig. 1(C)). The induction of PARP cleavage of all three compounds was apparent in H460 cancer cells. Compounds 7n and 7s had better activity than 7w. Compounds 7n and 7s induced the cleavage of PARP protein in HepG2 cancer cells, but 7w did not have an effect. Compound 7s was the only compound that slightly induced PARP cleavage in MCF-7 cancer cells. These results suggested that 7n, 7s, and 7w were more potent inducers of apoptosis in H460 cancer cell line than the other two cell lines. Compound 7s showed higher apoptosis against these three human cancer cell lines than compounds 7w and 7n. The apoptotic effect of these compounds at various concentrations was tested in NIH-3T3 mouse embryo cells over 24 h. The results showed that all of the three compounds did not induce PARP cleavage at various concentrations for 24 h (Fig. 1(D)). This indicated that these three compounds had little effect on normal cells.2.6. Compound 7s induced apoptosis depending the Nur77 activation and the best growth inhibition of H460 cancer cells
We used RNA interference technology to confirm that the apoptosis effect of these compounds was related with the activation of Nur77. The H460 cancer cells were transfected with shNur77 or shctr for 24 h and then treated with 10 μM of the compounds for an additional 24 h. The cleavage of PARP and the expression of Nur77 were examined (Fig. 2(A)). The compounds still induced Parp cleavage in cells transfected with the shctr. In cells transfected with the shNur77, the Nur77 was effectively blocked. The degree of PARP cleavage of 7s and 7n was significantly down regulated, particularly in 7s. There was little change for 7w. This suggested that Nur77 could play a key role in the apoptosis effect of 7s in H460 cells.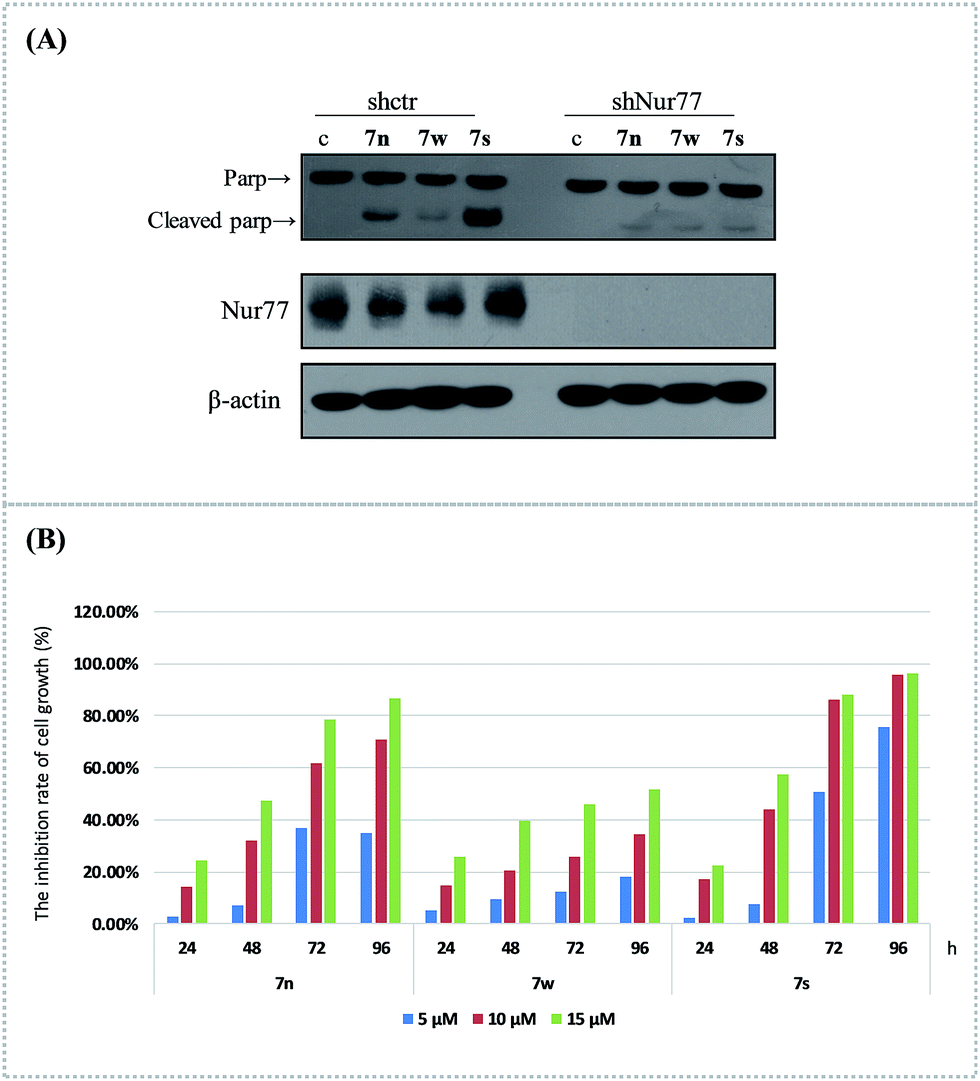 | ||
| Fig. 2 The compounds induced apoptosis and inhibited growth of H460 cells. (A) Nur77 expression was required for the apoptotic effect of 7n, 7s, and 7w in H460 cells. H460 cancer cells were transfected with control or Nur77 shRNA for 24 h, then treated with or without 10 μM of the compounds for an additional 24 h. The Parp cleavage and Nur77 expression were analysed via western blotting. (B) Growth inhibition activities of compounds on H460 cells were assessed with MTT assay. H460 cancer cells treated with these three compounds at various concentrations (5 μM, 10 μM and 15 μM) and tested at 24 h, 48 h, 72 h, and 96 h time points. DMSO was used as the negative control. | ||
The cell growth inhibition of 7n, 7s, and 7w in H460 cancer cells was confirmed with MTT assays. The effects of these compounds on the growth of H460 cells at various concentrations (5 μM, 10 μM, and 15 μM) were tested at 24 h, 48 h, 72 h, and 96 h (Fig. 2(B)). The results showed that these compounds inhibited the proliferation of the H460 cell line in a dose- and time-dependent manner. Compound 7s demonstrated superior growth inhibition on H460 cells. At exposure times up to 96 h, the inhibition rates of 7n at the various concentrations (5 μM, 10 μM and 15 μM) were 35.02%, 70.73%, and 86.50%. The inhibition rates for 7w were 18.02%, 34.47%, and 51.71%. The inhibition rates for 7s were 75.70%, 95.72%, and 96.05%. The anti-proliferation abilities of these three compounds were similar to their apoptosis induction and Nur77 activation effects in H460 cells.
2.7. Molecular docking study of 7s and Nur77
The in vitro bioassay revealed the high ability of compound 7s when activating Nur77 in H460 cancer cells. We performed a molecular docking study for 7s and Nur77. The docking study revealed that compound 7s exhibited a promising binding affinity with Nur77 with a docking binding energy of −7.30 kcal mol−1. The specific non-bonded interactions between 7s and the residues of Nur77 in 3D and 2D are shown in Fig. 3. The urea group of compound 7s formed a hydrogen bonding interaction with the backbone's carbonyl group of amino acid LEU175. The moiety of difluorobenzene inserted deeply into the hydrophobic pocket formed by PHE172, LEU224, VAL167, PHE158, LEU162 and LEU118. The adamantine group extended out of the pocket and interacted with the surface of ALA201, ASN198, and GLN197 via hydrophobic interactions. The hydrogen bond between 7s and Nur77 could still exist even though the LEU175 mutated to other amino acids (like an alanine). This indicated that the hydrogen bond between 7s and the backbone of LEU175 could be important for the affinity and selection of the complex 7s–Nur77.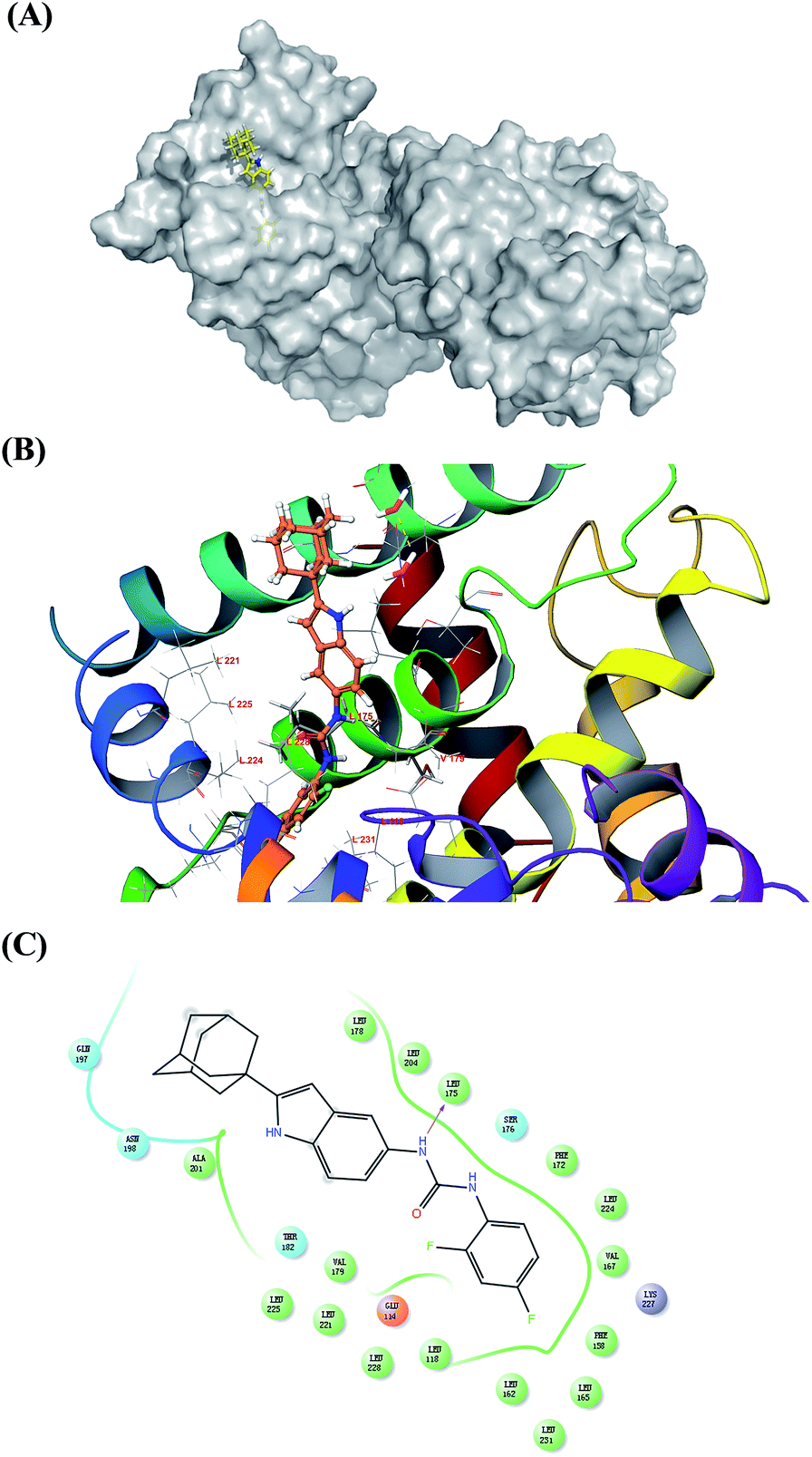 | ||
| Fig. 3 Molecular docking 3D and 2D models of 7s and Nur77. (A) The interactions between Nur77 are displayed on the grey surface presentation. The 7s is represented in the yellow stick. The protein surface conformation is the crystal structure of homodimer of the orphan nuclear receptor Nur77 (PDB ID: 4RE8). The moiety of difluorobenzene of ligand inserted deeply into the hydrophobic pocket. (B) Close-up view depiction of the superposition of Nur77 LBP bound to 7s. The Nur77 binding site is presented with a coloured ribbon. Neighbouring amino acids were displayed in lines within a distance of 5 Å approximately to 7s (the hydrogen bond between 7s and LEU 175 are shown as yellow dots). (C) 2D binding model of 7s with Nur77: hydrogen bonds are indicated with solid arrows, colour lines around 7s stand for the binding pocket and the residues in colours nearby established the pocket. The green colour denotes the hydrophobic nature of amino acids, the red colour denotes the acid amino acids, the purple denotes the alkalinity of amino acids, the cyan denotes the polar amino acids, and the grey points of ligand atoms denotes the solvent accessibility. | ||
3. Conclusion
In this study, we synthesized and conducted a biological evaluation of two new series of 1-(2-(adamantan-1-yl)-1H-indol-5-yl)-3-substituted urea/thiourea derivatives as potential anticancer agents. The synthetic method was relatively simple. The target compounds were easily purified and produced in high yields. Among of synthesized compounds, most showed moderate anti-proliferative activity against all three of the tested cancer cell lines. The compounds with prominent anticancer activity also exhibited good sensitivity between the normal (MRC-5) and the tumour (H460) cell lines that were isolated from lung tissue. Three compounds that could active Nur77 were obtained: 7n, 7s, and 7w. They were evaluated for their abilities to induce apoptosis in various cells and the inhibition of cell growth in H460 cancer cells. The results showed that 7n and 7s achieved better anticancer activity than 7w. The shRNA experiment revealed that the apoptosis effect of 7n and 7s was dependent on Nur77, particularly in 7s. Compound 7w could have an alternative mechanism. The molecular docking suggested that compound 7s exhibited a promising binding affinity with Nur77. These results are helpful in the development of a Nur77 targeted drug. Further studies are also required to determine their roles in preventing cancer.4. Experimental section
4.1. Chemistry
All reagents were commercially available and used without further purification, unless otherwise indicated. Reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) on Merck silica gel 60F-254 by fluorescence. The target compounds were purified by column chromatography. The purities of the target compounds were assessed by high performance liquid chromatography (HPLC) with a COSMOSIL C18-MS-II column (250 mm × 4.6 mm i.d., 5 μm). The HPLC analysis was performed on an Agilent Technologies 1100 Series HPLC system. The mobile phase was acetonitrile (A) and water (B), eluted as the following gradient program: A from 80% to 100% and B from 20% to 0% during 0–20 min. The flow rate was 1 mL min−1 and the detection wavelengths were 254 nm and 280 nm. 1H-NMR and 13C-NMR spectra were obtained using a Bruker AV2 600 Ultra shield spectrometer at 600 and 150 MHz respectively. Chemical shifts were given in parts per million (ppm) relative to tetramethyl-silane (TMS) as an internal standard. Multiplicities were abbreviated as follows: single (s), doublet (d), doublet–doublet (dd), doublet–triplet (dt), triplet (t), triplet–triplet (tt), triplet–doublet (td), quartet (q), quartet-doublet (qd), multiplet (m), and broad signal (br s). High-resolution mass spectral (HRMS) data were acquired on a Q Exactive. Melting points were measured on a SGWX-4 micro-melting point spectrometer and were uncorrected.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ONH and Ar–H), 7.17 (d, J = 7.5 Hz, 1H, Ar–H), 7.05 (dt, J = 1.1, 7.4 Hz, 1H, Ar–H), 2.26 (s, 3H, Ar–CH3), 1.99–2.13 (m, 3H, Ad–H), 1.97–2.01 (m, 6H, Ad–H), 1.73–1.80 (m, 6H, Ad–H); ESI-HRMS (+): m/z [M + H]+ calculated for C18H24NO+, 270.1852, found 270.1853; [M + Na]+ calculated for C18H23NONa+, 292.1672, found 292.1669.CO), 118.1 (Ar–CH), 115.5 (Ar–CH), 111.1 (Ar–CH), 96.4 (C-3′), 42.5 (Ad–C), 36.7 (s, 3C, Ad–C), 33.8 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C).
ONH and Ar–H), 7.17 (d, J = 7.5 Hz, 1H, Ar–H), 7.05 (dt, J = 1.1, 7.4 Hz, 1H, Ar–H), 2.26 (s, 3H, Ar–CH3), 1.99–2.13 (m, 3H, Ad–H), 1.97–2.01 (m, 6H, Ad–H), 1.73–1.80 (m, 6H, Ad–H); ESI-HRMS (+): m/z [M + H]+ calculated for C18H24NO+, 270.1852, found 270.1853; [M + Na]+ calculated for C18H23NONa+, 292.1672, found 292.1669.CO), 118.1 (Ar–CH), 115.5 (Ar–CH), 111.1 (Ar–CH), 96.4 (C-3′), 42.5 (Ad–C), 36.7 (s, 3C, Ad–C), 33.8 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C).4.1.6.1 Compound 7n. White solid; yield: 87.3%; HPLC purity: 95.5% (tR = 21.05 min); mp: 265–267 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.74 (s, 1H, NH-1′), 8.90 (s, 1H, C
ONH-1), 8.46 (s, 1H, CONH-3), 8.04 (s, 1H, H-2′), 7.56 (d, J = 1.5 Hz, 1H, H-4′), 7.54–7.56 (m, 1H, Ar–H), 7.46–7.51 (m, 1H, Ar–H), 7.27 (d, J = 7.7 Hz, 1H, Ar–H), 7.21 (d, J = 8.4 Hz, 1H, H-7′), 7.01 (dd, J = 2.0, 8.4 Hz, 1H, H-6′), 6.03 (d, J = 1.7 Hz, 1H, H-3′), 2.03–2.08 (m, 3H, Ad–H), 1.92–1.99 (m, 6H, Ad–H), 1.72–1.80 (m, Ad–H); 13C NMR (150 MHz, DMSO-d6): δ 153.4 (CO), 150.8 (C-2′), 141.5 (C-1′′), 133.0 (C-8′), 131.1 (C-5′), 130.3 (C-5′′), 129.96 (q, J = 30.8 Hz, CF3C), 128.4 (C-9′), 124.8 (q, J = 271.8 Hz, CF3), 122.0 (C-6′′), 118.0 (q, J = 4.4 Hz, C-2′′), 114.35 (C-6′), 114.30 (q, J = 4.4 Hz, C-4′′) 111.1 (C-7′), 110.5 (C-4′), 95.4 (C-3′), 42.3 (Ad–C), 36.8 (s, 3C, Ad–C), 33.9 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C); ESI-HRMS (+): m/z [M + H]+ calculated for C26H27F3N3O+, 454.2101, found 454.2101; [M + Na]+ calculated for C26H26F3N3ONa+, 476.192, found 476.1922.
4.1.6.2 Compound 7s. White solid; yield: 89.6%; HPLC purity: 96.4% (tR = 17.89 min); mp: 178–181 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.73 (d, J = 1.3 Hz, 1H, NH-1′), 8.72 (s, 1H, C
ONH-1), 8.36 (d, J = 2.0 Hz, 1H, CONH-3), 8.12–8.17 (m, 1H, 6′′), 7.56 (d, J = 1.8 Hz, 1H, H-4′), 7.26–7.30 (m, 1H, 5′′), 7.20 (d, J = 8.4 Hz, 1H, 7′), 7.01–7.05 (m, 1H, H-3′′), 6.99 (dd, J = 2.1, 8.5 Hz, 1H, H-6′), 6.02 (d, J = 1.7 Hz, 1H, H-3′), 2.03–2.08 (m, 3H, Ad–H), 1.93–1.98 (m, 6H, Ad–H), 1.72–1.80 (m, 6H, Ad–H); 13C NMR (150 MHz, DMSO-d6): δ 156.8 (dd, J = 12.1, 241.0 Hz, CF-2′′), 153.1 (CO), 152.3 (dd, J = 12.1, 241.5 Hz, CF-4′′), 150.8 (C-2′), 132.9 (C-8′), 131.2 (C-5′), 128.4 (C-9′), 125.1 (dd, J = 3.3, 11.0 Hz, C-1′′), 121.9 (dd, J = 3.3, 8.8 Hz, C-6′′), 113.9 (C-6′), 111.4 (dd, J = 23.1, 24.2 Hz, C-5′′), 111.1 (C-7′), 110.0 (C-4′), 104.1 (dd, J = 3.3, 21.3 Hz, C-3′′), 95.4 (C-3′), 42.3 (Ad–C), 36.8 (s, 3C, Ad–C), 33.9 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C); ESI-HRMS (+): m/z [M + H]+ calculated for C25H26F2N3O+ 422.2038, found 422.2030; [M + Na]+ calculated for C25H25F2N3ONa+, 444.1850, found 444.1852. (ESI Fig. S1†)
4.1.6.3 Compound 7w. White solid; yield: 85.8%; HPLC purity: 94.8% (tR = 20.80 min); mp: 271–273 °C; 1H NMR (600 MHz, DMSO-d6): δ 10.74 (s, 1H, NH-1′), 8.76 (s, 1H, C
ONH-1), 8.55 (s, 1H, CONH-3), 7.88–7.98 (m, 1H, H-6′′), 7.56 (d, J = 1.7 Hz, 1H, H-4′), 7.23–7.27 (m, 1H, H-5′′), 7.21 (d, J = 8.6 Hz, 1H, 7′) 6.99 (dd, J = 2.0, 8.6 Hz, 1H, H-6′), 6.03 (d, J = 1.7 Hz, 1H, H-3′), 2.03–2.08 (m, 3H, Ad–H), 1.93–1.98 (m, 6H, Ad–H), 1.72–1.80 (m, 6H, Ad–H); 13C NMR (150 MHz, DMSO-d6): δ 152.9 (CO), 150.8 (C-2′), 145.3 (ddd, J = 2.2, 9.9, 242.1 Hz, CF-2′′), 141.7 (ddd, J = 3.3, 11.0, 245.4 Hz, CF-4′′), 139.5 (dt, J = 14.3, 246.5 Hz, CF-3′′), 126.5 (dd, J = 3.3, 7.7 Hz, C-6′′), 133.0 (C-8′), 131.0 (C-5′), 128.4 (C-9′), 115.2 (m, C-1′′), 114.0 (C-6′), 112.1 (dd, J = 3.3, 17.1 Hz, C-5′′), 111.1 (C-7′), 110.1 (C-4′), 95.4 (C-3′), 42.3 (Ad–C), 36.8 (s, 3C, Ad–C), 33.9 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C); ESI-HRMS (+): m/z [M + H]+ calculated for C25H25F3N3O+, 440.1944, found 440.1938; [M + Na]+ calculated for C25H24F3N3ONa+, 462.1764, found 462.1760.At the series of urea derivatives, the other target compounds were synthesized following the general procedure as described above with the yield of 84–90%. Their structures were confirmed by the spectroscopic data including 1H NMR, 13C NMR, and ESI-HRMS (see ‘ESI†’).
CS), 119.0 (C-6′), 117.3 (C-7′), 111.2 (C-4′), 96.8 (C-3′), 42.5 (Ad–C), 36.7 (s, 3C, Ad–C), 33.9 (s, 3C, Ad–C), 28.4 (s, 3C, Ad–C).4.1.8.1 Compound 9a. White solid; yield: 84.6%; mp: 285–287 °C; HPLC purity: 97.3% (tR = 21.07 min); 1H NMR (600 MHz, DMSO-d6): δ 10.89 (s, 1H, NH-1′), 9.15 (s, 1H, C
SNH-1), 7.32 (d, J = 1.7 Hz, 1H, H-4′), 7.25 (d, J = 8.4 Hz, 1H, H-7′), 6.87 (dd, J = 1.9, 8.5 Hz, 1H, H-6′), 6.73 (br s, 1H, CSNH-3), 6.07 (d, J = 1.5 Hz, 1H, H-3′), 2.03–2.09 (m, 3H, Ad–H), 1.93–1.98 (m, 6H, Ad–H), 1.72–1.80 (m, 6H, Ad–H), 1.45 (s, 9H, (CH3)3C); 13C NMR (150 MHz, DMSO-d6): δ 180.3 (CS), 151.1 (C-2′), 134.3 (C-8′), 130.2 (C-5′), 128.4 (C-9′), 118.9 (C-6′), 116.5 (C-7′), 111.3 (C-4′), 95.7 (C-3′), 53.0 ((CH3)3C), 42.2 (Ad–C), 36.8 (s, 3C, Ad–C), 33.9 (s, 3C, Ad–C), 29.2 (s, 3C, (CH3)3C), 28.4 (s, 3C, Ad–C); ESI-HRMS (+): m/z [M + H]+ calculated for C23H32N3S+, 382.2311, found 382.2307; [M + Na]+ calculated for C23H31N3SNa+, 404.2131, found 404.2126.At the series of thiourea derivatives, the other final compounds were synthesized following the general procedure as described above with the yield of 84–92%. Their structures were characterized by NMR, ESI-HRMS and HPLC analyses. (see ‘ESI†’).
4.2. Biology materials and methods
4.3 Molecular docking
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81773600, 81302652, 31471273, 41376172 and 31461163002), the Project of South Center for Marine Research (No. 14GYY023NF23). This research was also financially supported by Fujian Science and Technology project (No. 2014N5012). We also thank the College of Pharmaceutical Sciences, Xiamen University and the Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University for providing us with the Shrodinger 2015-1 software and Gaussian 09 software.Notes and references
- A. Chawla, J. J. Repa, R. M. Evans and D. J. Mangelsdorf, Science, 2001, 294, 1866–1870 CrossRef CAS PubMed.
- Z. Xia, X. Cao, E. Rico-Bautista, J. Yu, L. Chen, J. Chen, A. Bobkov, D. A. Wolf, X.-K. Zhang and M. I. Dawson, MedChemComm, 2013, 4, 332–339 RSC.
- Q. Wu, S. Liu, X. F. Ye, Z. W. Huang and W. J. Su, Carcinogenesis, 2002, 23, 1583–1592 CrossRef CAS PubMed.
- S. O. Lee, X. Li, S. Khan and S. Safe, Expert Opin. Ther. Targets, 2011, 15, 195–206 CrossRef CAS PubMed.
- M. A. Maxwell and G. Muscat, Nucl. Recept. Signaling, 2006, 4, e002 Search PubMed.
- H. M. Mohan, C. M. Aherne, A. C. Rogers, A. W. Baird, D. C. Winter and E. P. Murphy, Clin. Cancer Res., 2012, 18, 3223–3228 CrossRef CAS PubMed.
- U. Moll, N. Marchenko and X. Zhang, Oncogene, 2006, 25, 4725–4743 CrossRef CAS PubMed.
- L. De Léséleuc and F. Denis, Cell Death Differ., 2006, 13, 293–300 CrossRef PubMed.
- F. Weih, R. P. Ryseck, L. Chen and R. Bravo, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5533–5538 CrossRef CAS.
- Y. Li, B. Lin, A. Agadir, R. Liu, M. I. Dawson, J. C. Reed, J. A. Fontana, F. Bost, P. D. Hobbs and Y. Zheng, Mol. Cell. Biol., 1998, 18, 4719–4731 CrossRef CAS PubMed.
- H. Uemura and C. Chang, Endocrinology, 1998, 139, 2329–2334 CrossRef CAS PubMed.
- Y. Y. Zhan, J. P. He, H. Z. Chen, W. J. Wang and J. C. Cai, Cancer Lett., 2013, 329, 37–44 CrossRef CAS PubMed.
- S. O. Lee, T. Andey, U. H. Jin, K. Kim, M. Singh and S. Safe, Oncogene, 2012, 31, 3265–3276 CrossRef CAS PubMed.
- H. Wu, Y. Lin, W. Li, Z. Sun, W. Gao, H. Zhang, L. Xie, F. Jiang, B. Qin and T. Yan, FASEB J., 2011, 25, 192–205 CrossRef PubMed.
- S. To, W. Zeng, J. Zeng and A. Wong, Br. J. Cancer, 2014, 110, 935–945 CrossRef CAS PubMed.
- S. O. Lee, M. Abdelrahim, K. Yoon, S. Chintharlapalli, S. Papineni, K. Kim, H. Wang and S. Safe, Cancer Res., 2010, 70, 6824–6836 CrossRef CAS PubMed.
- J. Liu, W. Zhou, S. S. Li, Z. Sun, B. Lin, Y. Y. Lang, J. Y. He, X. Cao, T. Yan and L. Wang, Cancer Res., 2008, 68, 8871–8880 CrossRef CAS PubMed.
- S. D. Cho, S. O. Lee, S. Chintharlapalli, M. Abdelrahim, S. Khan, K. Yoon, A. M. Kamat and S. Safe, Mol. Pharmacol., 2010, 77, 396–404 CrossRef CAS PubMed.
- T. Sibayama-Imazu, Y. Fujisawa, Y. Masuda, T. Aiuchi, S. Nakajo, H. Itabe and K. Nakaya, J. Cancer Res. Clin. Oncol., 2008, 134, 803–812 CrossRef CAS PubMed.
- K. Yoon, S. O. Lee, S. D. Cho, K. Kim, S. Khan and S. Safe, Carcinogenesis, 2011, 32, 836–842 CrossRef CAS PubMed.
- H. Z. Chen, Q. F. Liu, L. Li, W. J. Wang, L. M. Yao, M. Yang, B. Liu, W. Chen, Y. Y. Zhan, M. Q. Zhang, J. C. Cai, Z. H. Zheng, S. C. Lin, B. A. Li and Q. Wu, Gut, 2012, 67, 714–724 CrossRef PubMed.
- S. Zhe, X. H. Cao, M. M. Jiang, Y. K. Qiu, H. Zhou, L. Q. Chen, B. Qin, H. Wu, F. Q. Jiang, J. B. Chen, J. Liu, Y. Dai, H. F. Chen, Q. Y. Hu, Z. Wu, J. Z. Zeng, X. S. Yao and X. K. Zhang, Oncogene, 2012, 31(21), 2653–2667 CrossRef PubMed.
- L. Hao, J. Luan, D. Zhang, C. Li, H. Guo, L. Qi, X. Liu, T. Li and Q. Zhang, Colloids Surf., B, 2014, 117, 258–266 CrossRef CAS PubMed.
- B. Liang, X. Song, G. Liu, R. Li, J. Xie, L. Xiao, M. Du, Q. Zhang, X. Xu and X. Gan, Exp. Cell Res., 2007, 313, 2833–2844 CrossRef CAS PubMed.
- P. C. Lin, Y. L. Chen, S. C. Chiu, Y. L. Yu, S. P. Chen, M. H. Chien, K. Y. Chen, W. L. Chang, S. Z. Lin and T. W. Chiou, J. Neurochem., 2008, 106, 1017–1026 CrossRef CAS PubMed.
- S. Liu, H. Yu, S. M. Kumar, J. S. Martin, Z. Bing, W. Sheng, M. Bosenberg and X. Xu, Cancer Biol. Ther., 2011, 12, 1005–1014 CrossRef CAS PubMed.
- M. Z. Zhang, N. Mulholland, D. Beattie, D. Irwin, Y. C. Gu, Q. Chen, G. F. Yang and J. Clough, Eur. J. Med. Chem., 2013, 63, 22–32 CrossRef CAS PubMed.
- M. Z. Zhang, Q. Chen and G. F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed.
- R. Álvarez, P. Puebla, J. F. Díaz, A. C. Bento, R. García-Navas, J. de la Iglesia-Vicente, F. Mollinedo, J. M. Andreu, M. Medarde and R. Peláez, J. Med. Chem., 2013, 56, 2813–2827 CrossRef PubMed.
- M. Nguyen, R. C. Marcellus, A. Roulston, M. Watson, L. Serfass, S. R. M. Madiraju, D. Goulet, J. Viallet, L. Belec, X. Billot, S. Acoca, E. Purisima, A. Wiegmans, L. Cluse, R. W. Johnstone, P. Beauparlant and G. C. Shore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19512–19517 CrossRef CAS PubMed.
- O. I. Parisi, C. Morelli, L. Scrivano, M. S. Sinicripi, M. G. Cesario, S. Candamano, F. Puoci and D. Sisci, RSC Adv., 2015, 5, 65308–65315 RSC.
- H. Y. Hu, X. D. Yu, F. Wang, C. R. Lin, J. Z. Zeng, Y. K. Qiu, M. J. Fang and Z. Wu, Molecules, 2016, 21, 530 CrossRef PubMed.
- T. H. Maugh, Science, 1979, 206, 1058–1060 Search PubMed.
- T. A. Blanpied, R. J. Clarke and J. W. Johnson, J. Neurosci., 2005, 25, 3312–3322 CrossRef CAS PubMed.
- M. W. Robinson, J. H. Overmeyer, A. M. Young, P. W. Erhardt and W. A. Maltese, J. Med. Chem., 2012, 55, 1940–1956 CrossRef CAS PubMed.
- L. L. Yang, G. B. Li, S. Ma, C. Zou, S. Zhou, Q. Z. Sun, C. Cheng, X. Chen, L. J. Wang, S. Feng, L. L. Li and S. Y. Yang, J. Med. Chem., 2013, 56, 1641–1655 CrossRef CAS PubMed.
- J. Yao, J. Chen, Z. He, W. Sun and W. Xu, Design, synthesis and biological activities of thiourea containing sorafenib analogs as antitumor agents, Bioorg. Med. Chem., 2012, 20(9), 2923–2929 CrossRef CAS PubMed.
- Y. C. Duan, Y. C. Zheng, X. C. Li, M. M. Wang, X. W. Ye, Y. Y. Guan, G. Z. Liu, J. X. Zheng and H. M. Liu, Eur. J. Med. Chem., 2013, 64, 99–110 CrossRef CAS PubMed.
- M. Hassan, H. Watari, A. AbuAlmaaty, Y. Ohba and N. Sakuragi, BioMed Res. Int., 2014, 1–23 Search PubMed.
- Y. Han, X. Cao, B. Lin, F. Lin, S. Kolluri, J. Stebbins, J. Reed, M. Dawson and X. Zhang, Oncogene, 2006, 25, 2974–2986 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Grapics System, 2002 Search PubMed.
- Schrödinger Release 2016-1: Maestro, Schrödinger, LLC, New York, NY, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08149a |
| ‡ The first two authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |