
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimeric alumatranes as catalysts for trimethylsilylcyanation reaction†
Yoseph Kim‡
a,
Kang Mun Lee‡ b,
So Han Kim‡a,
Jung Hee Moona and
Youngjo Kim*a
b,
So Han Kim‡a,
Jung Hee Moona and
Youngjo Kim*a
aDepartment of Chemistry, BK21+ Program Research Team, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: ykim@chungbuk.ac.kr
bDepartment of Chemistry, Institute for Molecular Science and Fusion Technology, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea
First published on 13th October 2017
Abstract
The solid-state structures of dimeric alumatranes with three five-membered rings chelated by [(OCMe2CH2)nN(CH2CH2O)3−n]3− (n = 1, L1; n = 2, L2; n = 3, L3), which vary by the number of CMe2 groups adjacent to the OH functionality [1 (L1H3), 2 (L2H3), and 3 (L3H3)], were determined by single-crystal X-ray diffraction. The X-ray structures revealed that the aluminum geometries were slightly distorted trigonal bipyramids. The obtained aluminum complexes are the first structurally characterized dimeric alumatranes with tricyclic five-membered rings. Quite unexpectedly, the sterically bulky side arms with dimethyl substituents were always located in the bridging sites, as determined by density functional theory calculations. Their solution-state structures were analyzed by 1H, 13C, and 27Al NMR techniques, and their gas-phase structures were determined by mass spectrometry. Unlike Al(OCH2CH2)3N, complexes 1–3 were all dimeric in the solid state, solution phase, and gas phase. In addition, they were found to promote the reaction of aryl, heteroaryl, and alkyl aldehydes with trimethylsilylcyanide to provide the corresponding products in excellent yields under mild conditions of room temperature, a short reaction time of 1 h, and a very low catalyst loading of 0.5 mol%.
Introduction
Cyanohydrin trimethylsilylethers prepared via the trimethylsilylcyanation of aldehydes are versatile and critical intermediates for α-hydroxy aldehydes and β-amino alcohols.1,2 Various catalysts for the reaction of trimethylsilylcyanide (TMSCN) with aldehydes and ketones have been reported.3–5 Among them, aluminum-based compounds may be one of the most attractive groups of catalysts because aluminum is the most abundant metal in the Earth's crust, and its complexes have relatively low toxicity and high Lewis acidity, rendering them suitable for use as catalysts. Some examples of aluminum-based catalysts for the efficient transformation of this reaction have been reported.6–18 However, high Al catalyst loadings (5–25 mol%) and prolonged reaction times (3–72 h) are required for the completion of this reaction.6–17 Interestingly, Raders and Verkade have demonstrated that a 0.5 mol% catalyst loading of alumatranes with three six-membered rings could complete the trimethylsilylcyanation of aldehydes at room temperature in only 9–12 h.18 Here, the interesting point is that they used a dimeric alumatrane as the starting material. Because five-coordinate aluminum complexes are generally believed to be important intermediates in aluminum-catalyzed reactions, five-coordinate alumatranes could function as Lewis acid catalysts for the trimethylsilylcyanation reaction. Although their alumatranes are potentially excellent Lewis acid catalysts with a loading of only 0.5 mol%,18 the reaction time of 9–12 h should be shortened to achieve practical application.As shown in Chart 1, alumatranes with tricyclic five-19–33 and six-membered17,18,34–38 rings are well known. Like other atranes,39–41 all alumatranes with tricyclic five- or six-membered ring systems also have a transannular N → Al interaction from the bridgehead N atom in the tetradentate ligand to the Al atom. Although alumatranes with tricyclic six-membered rings (Chart 1(b)) are monomeric or dimeric in solution and in the solid state, Al(OCH2CH2)3N (Chart 1(a)) has been described as a dimer28 in the gas phase, a hexamer29 and octamer30 in solution, and a tetramer31 in the solid state.
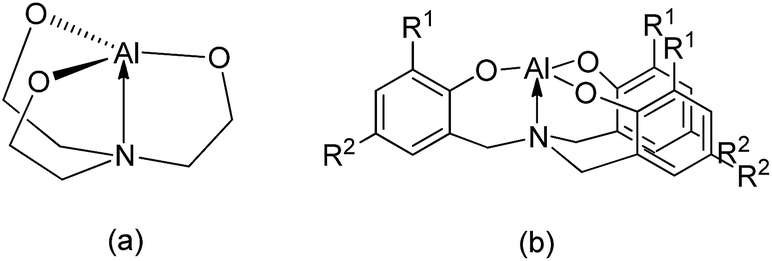 | ||
| Chart 1 Types of alumatranes with (a) five- and (b) six-membered rings. | ||
Even though some examples of structurally characterized alumatranes with tricyclic six-membered rings have been reported,17,34–38 few examples of five-membered alumatranes and their derivatives20,31–33 have been identified.
Thus, to the best our knowledge, no studies on a structurally characterized dimeric alumatrane with a tricyclic five-membered ring have appeared in the literature. In addition, examples of five-membered alumatranes having the same structure in the gas phase, solution phase, and solid state have, to our knowledge, not been reported. Herein, we report the logical synthesis, characterization, X-ray structures, theoretical density functional theory (DFT) studies, and catalytic application of dimeric alumatranes.
Results and discussion
Synthesis of new alumatrane complexes 1–3
As shown in Chart 2, the R1 groups of the tetradentate tris(2-oxy-3,5-dialkylbenzyl)amine ligand in alumatranes with tricyclic six-member rings could act as “picket fences” to prevent oligomer formation. A similar approach was applied to make dimeric alumatranes with tricyclic five-membered rings. Thus, the introduction of steric congestion in the vicinity of the hydroxyl groups of the triethanolamine ligands could inhibit oligomer formation. Using this idea, we have successfully prepared monomeric boratranes,42 monomeric germatranes,43 and monomeric or dimeric titanatranes.44–46 This idea further prompted us to prepare alumatranes with tricyclic five-membered rings, which may be dimeric in the gas phase, solution phase, and solid state.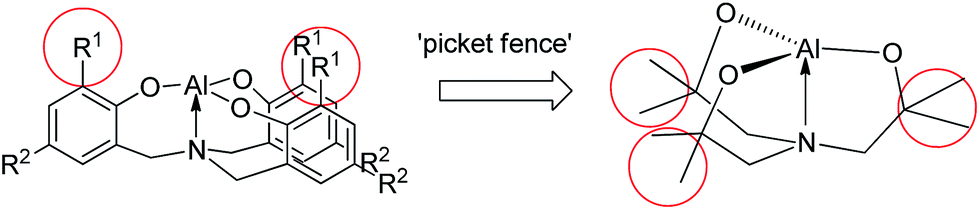 | ||
| Chart 2 Logical design of dimeric alumatranes with tricyclic five-membered rings. | ||
The alcoholysis of AlMe3 has proven to be a useful synthetic route for alumatranes.17–38 As shown in Scheme 1, the addition of AlMe3 to a solution of (HOCMe2CH2)nN(CH2CH2OH)3−n (n = 1, L1H3; n = 2, L2H3; n = 3, L3H3) in toluene gave novel alumatranes 1–3 as colorless crystals after workup. These reactions proceeded readily at ambient temperature, resulting in good isolated yields of 62–79%. The crude compounds were purified by washing with n-hexane and were recrystallized in toluene. Importantly, in contrast to aluminum complexes with Al–Me bonds,47 complexes 1–3 are very stable at room temperature for more than 1 week, even in chloroform-d1 and benzene-d6 solutions. They are soluble in various solvents, including toluene, chloroform, methanol, and acetone.
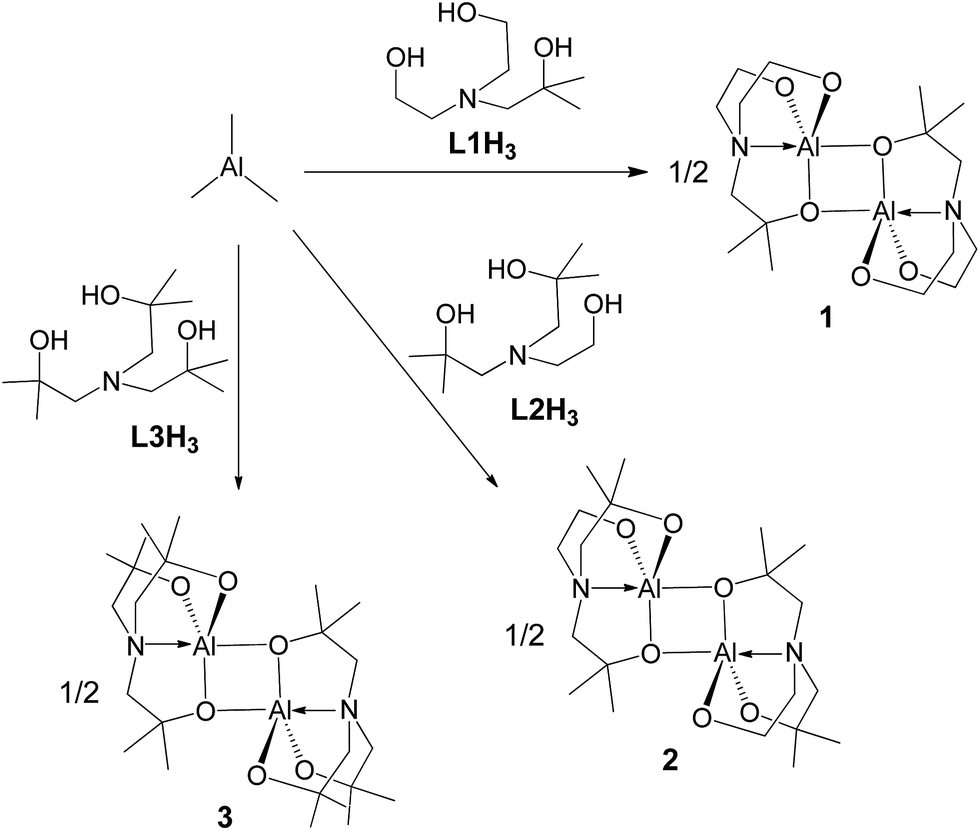 | ||
| Scheme 1 Synthetic routes for dimeric alumatranes 1–3. | ||
Solid-state structures of alumatranes 1–3
Complexes 1–3 were subjected to X-ray diffraction analysis to determine the geometry around the central Al atoms and binding modes of the ligands. X-ray-quality single crystals were obtained from toluene solutions maintained at −20 °C in a refrigerator for a few days. The molecular structures and their selected bond lengths and angles are shown in Fig. 1 and Table 1, respectively.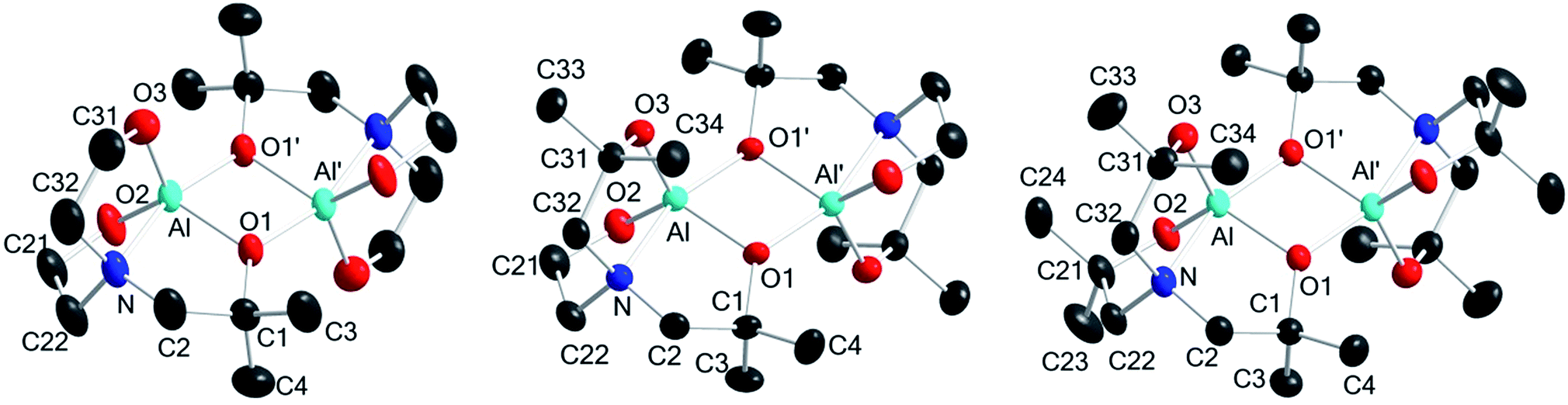 | ||
| Fig. 1 X-ray crystal structures of 1 (left), 2 (center), and 3 (right) (50% thermal ellipsoids). All H atoms and toluene molecules (for 1 and 2) are omitted for clarity. | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| Al–O1 | 1.841(2) | 1.8540(10) | 1.8593(11) |
| Al–O1′ | 1.848(2) | 1.8565(10) | 1.8515(11) |
| Al–O2 | 1.753(3) | 1.7604(11) | 1.7436(11) |
| Al–O3 | 1.755(3) | 1.7539(11) | 1.7601(12) |
| Al–N | 2.074(3) | 2.1055(12) | 2.1158(14) |
| O1–Al–O2 | 121.41(14) | 120.52(6) | 116.65(6) |
| O1–Al–O3 | 117.23(14) | 120.08(5) | 120.85(5) |
| O2–Al–O3 | 119.88(15) | 117.56(6) | 120.75(6) |
| O1–Al–O1′ | 78.18(11) | 77.85(4) | 78.54(5) |
| O2–Al–O1′ | 101.37(12) | 102.72(5) | 101.46(5) |
| O3–Al–O1′ | 102.11(13) | 102.94(5) | 102.45(5) |
| O1–Al–N | 83.53(11) | 82.68(5) | 82.71(5) |
| O2–Al–N | 86.95(12) | 86.90(5) | 87.30(5) |
| O3–Al–N | 87.39(13) | 86.99(5) | 86.75(5) |
| O1′–Al–N | 161.66(12) | 160.53(5) | 161.25(5) |
| Al–O1–Al′ | 101.81(11) | 102.14(4) | 101.46(5) |
In the solid state, complexes 1–3 exist as dimers with pseudo-Ci symmetry. Each is composed of two alumatrane units with a four-membered Al2O2 ring linked by two Al–O bonds. To our knowledge, compounds 1–3 represent the only structurally characterized examples of an alumatrane dimer with all five-membered rings reported thus far. The aluminum atoms in 1–3 adopt a slightly distorted trigonal bipyramid geometry with Oax–Al–Nax angles (∠O1′–Al–N in Table 1) of 161.66(12)° in 1, 160.53(5)° in 2, and 161.25(5)° in 3. Trigonal bipyramidal and square pyramidal are two possible coordination geometries around the metal center in five-coordinate systems. They could also be determined by the trigonality parameter τ (τ = [α − β]/60, where α and β are the largest and next-largest interligand bond angles, respectively).48,49 The largest and next-largest interligand bond angles are ∠O1′–Al–N [161.66(12)° in 1, 160.53(5)° in 2, and 161.25(5)° in 3] and ∠O2–Al–O3 [119.88(15)° in 1, 117.56(6)° in 2, and 120.75(6)° in 3], respectively. Thus, the τ values of 0.71 for 1, 0.72 for 2, and 0.68 for 3 means that complexes 1–3 have distorted trigonal bipyramidal structures; the trigonality parameter τ for regular trigonal bipyramidal complexes is 1.0, and τ for perfect square pyramidal complexes is zero.
All Al–O bond distances of dimeric compounds 1–3 were observed as ca. 1.74–1.86 Å, which are similar to those found in typical pentacoordinate aluminum complexes.50,51 Moreover, two equatorial bonds, Al–O2 and Al–O3 [1.753(3) and 1.755(3) Å in 1, 1.7604(11) and 1.7539(11) Å in 2, and 1.7436(11) and 1.7601(12) Å in 3], are substantially shorter than those between the aluminum and bridging oxygen atoms in the other equatorial Al–O1 bond and in one axial Al–O1′ bond [1.841(2) and 1.848(2) Å in 1, 1.8540(10) and 1.8565(10) Å in 2, and 1.8593(11) and 1.8515(11) Å in 3]. The longer bridging Al–O1 and Al–O1′ distances are also observed in dimeric alumatranes with tricyclic six-membered rings.17,33
The transannular Al–Nax interaction distances of 2.074(3) Å in 1, 2.1055(12) Å in 2, and 2.1158(14) Å in 3 are slightly longer than the sum of the ionic radii of Al3+ and N3− (2.00 Å)52 and that of the covalent radii of Al and N (2.05 Å).53 This means that all dative N → Al coordinating bonds in 1–3 have a substantial degree of single bond character. All Al–N bond distances in 1–3 are among the longest of those observed for other structurally characterized alumatranes with tricyclic five-membered (2.003–2.094 Å)20,31–33 or six-membered rings (2.026–2.083 Å).17,34–38
The sum of the Oeq–Al–Oeq angles (∠O1–Al–O2 + ∠O1–Al–O3 + ∠O2–Al–O3) are 358.52° in 1, 358.16° in 2, and 358.25° in 3. Interestingly, the obtuse Oeq–Al–Oax angle [av = 93.89° for 1, 94.50° for 2, and 94.15° for 3; (∠O1–Al–O1′ + ∠O2–Al–O1′ + ∠O3–Al–O1′)/3] and acute Oeq–Al–Nax angle [av = 85.96° for 1, 85.52° for 2, and 85.59° for 3; (∠O1–Al–N + ∠O2–Al–N + ∠O3–Al–N)/3] reflect a displacement of the aluminum atoms toward the bridging oxygen atoms. No direct Al–Al′ interactions occur in 1–3.
Even though boron and aluminum, which are in the same group of the periodic table, have similar chemical properties, boratranes42 and alumatranes chelated by L1–L3 are monomeric and dimeric, respectively. In addition, titanatranes showed dramatic dimer-to-monomer structural changes induced by an increase of steric hindrance in the side arm of the tetradentate ligands.45 However, alumatranes are insensitive to the effects of ligand change. Unexpectedly, the sterically bulky side arm with dimethyl substituents in dimeric 1–3 is always located in the bridging sites; however, the less hindered side arms with no substituents in the dimeric titanatranes45 are placed in the same positions.
Theoretical calculations for the structures of alumatranes 1–3
Unlike compound 3, compounds 1 and 2 may have five additional geometric isomers, i.e., 1a–1e and 2a–2e, which can be classified by how many dimethyl-substituted bridging arms exist (see Chart 3). Thus, dimeric alumatranes chelated by L1 could have six isomers such as 1 (with two dimethyl substituents at the bridging side arm), 1a and 1d (with one dimethyl substituent at the bridge), and 1b, 1c, and 1e (with no dimethyl substituents at the bridge). The same trends for 2 could be applied. Among these structures, 1a, 1b, 2a, and 2b is the enantiomer of 1d, 1e, 2d, and 2e, respectively. Thus, enantiomeric pairs of 1a/1d, 1b/1e, 2a/2d, and 2b/2e have the nonsuperimposable mirror image.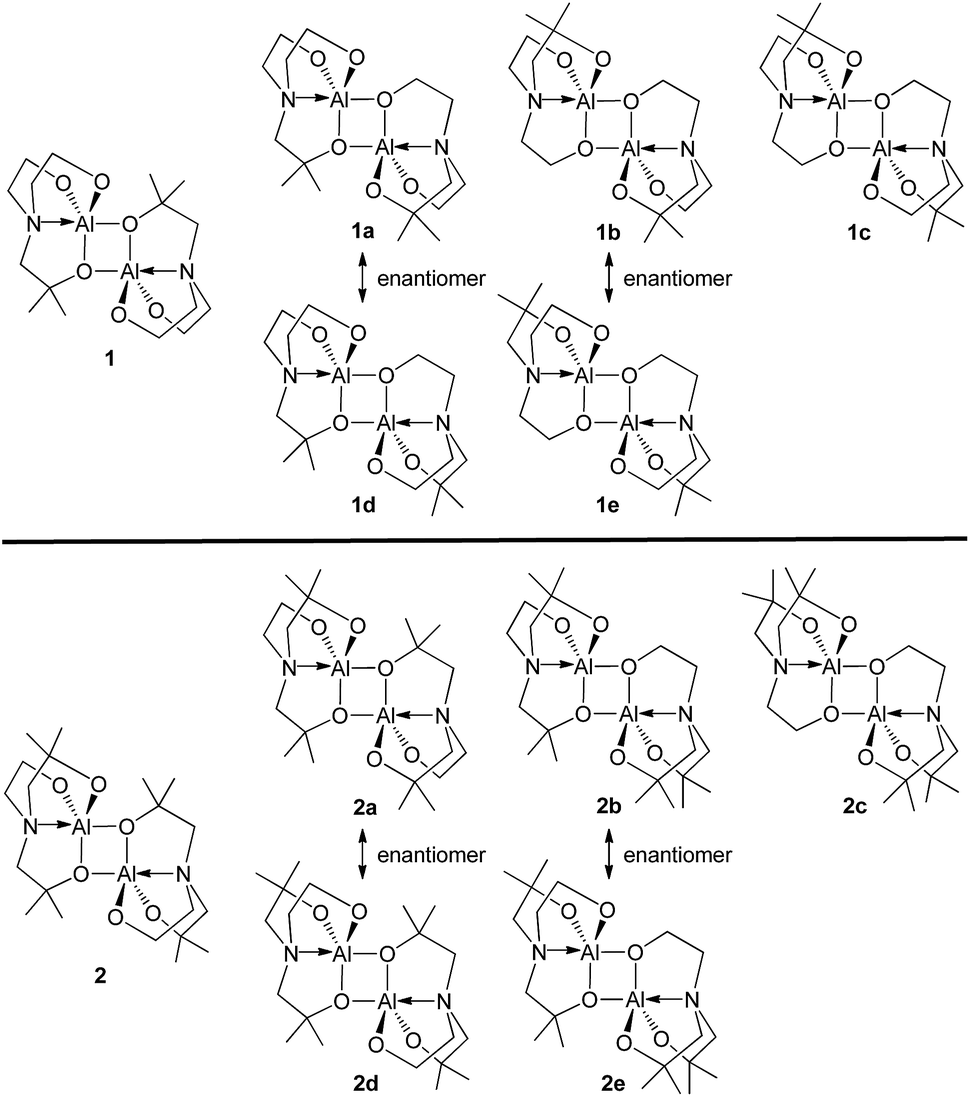 | ||
| Chart 3 All possible structural isomers for 1 and 2. | ||
The presence of dimethyl-substituted bridging arms in alumatrane isomers could play a significant role in determining their thermodynamic stability. To obtain the thermodynamic stabilities for each isomers shown in Chart 3, the relative free energy (ΔG/kcal mol−1) of the ground-state optimized structures in the gas phase was calculated using the B3LYP functional and 6-31G(d) basis set. The structural geometries used for the calculations were optimized on the basis of the X-ray structures of 1 and 2, and the energy states of the isomers were given relative to 1 or 2 because 1 and 2 were assigned to zero ΔG (kcal mol−1). Computed free energy diagram for 1, 2, and their possible isomers is shown in Fig. 2. Since enantiomers exhibit identical thermodynamic stabilities, data for only one enantiomer was given.
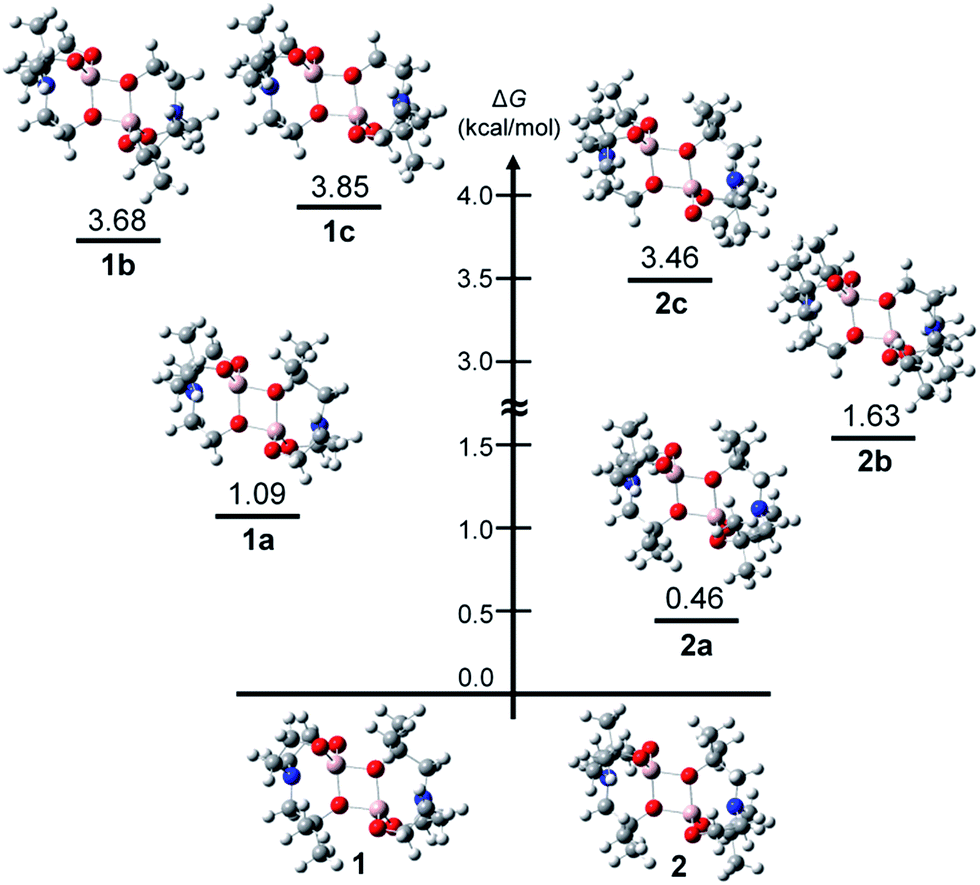 | ||
| Fig. 2 Computed free energy diagram for 1, 2, and their theoretical isomers. | ||
According to Fig. 2, the ΔG values of 1a–1c were 1.09, 3.68 and 3.85 kcal mol−1, respectively, higher than that of 1 (Fig. 2, left), distinctly indicating that the structure of 1 is the most thermodynamically stable isomer among six possible ones. The thermodynamic stability was proportional to the number of dimethyl-substituted bridging arms in the order of 1 > a pair of enantiomers 1a/1d > a pair of enantiomers 1b/1e > 1c. The ΔG values of 2a–2c were calculated to be 0.46, 1.63 and 3.46 kcal mol−1 higher than that of 2. Like 1 and its isomers, the similar stability order of 2 > a pair of enantiomers 2a/2d > a pair of enantiomers 2b/2e > 2c was also observed. In particular, the energy states for 1b and 1c (>3.6 kcal mol−1), which have no dimethyl-substituted bridging arms, are conspicuously enhanced compared to those for 1 and 1a. Whereas the ΔG values of 2 and 2a, bearing two dimethyl-substituted bridging arms each, differ slightly (by 0.46 kcal mol−1), those of 2b and 2c, which having one and no dimethyl-substituted bridging arms, respectively, linearly increase.
A key factor of the association between the structural features and thermodynamic stabilities of the isomers was found from the angle (φN) between the two unbridged arms of each structure optimized by theoretical calculations (Table S1 in ESI†). The φN is defined as the angle of CH2–N–CH2 (CH2 from the unbridged arm, see the inset figure of Table S1†), indicating the angle strain between the two unbridged arms centered at the N atom. These dimeric structures have two φN values (φN1 and φN2), and each value is dictated by the number of CH3 pairs substituted onto the unbridged arms. Whereas the φN between the two unbridged arms without CH3 substituents was observed to be ∼114° (φN1 and φN2 for 1 and φN1 for 1a, Table S1†), this angle between the unbridged arm with CH3 substituents and the unbridged arm without CH3 substituents was increased to ∼116° (φN2 for 1a and φN1 − φN2 for 1b–c, Table S1†). These φN values are the same for 2 and 2a. Furthermore, the φN between both unbridged arms with CH3 substituents (in 2b or 2c) was observed as >118°. These results distinctly indicate that the CH3 substituents on the unbridged arms increase the φN values and that CH3 substituents evoke angle strain centered at the N atom in the structure of each isomer. Consequently, we found that 1 and 2 are the most thermodynamically stable structures in comparison to the other isomers because these structures have the smallest angle strain among the isomer structures.
Unlike dimeric alumatranes 1 and 2, we recently found that the corresponding titanatranes chelated by L1 and L2 always had the less hindered side arms with no substituents in the bridging sites.45 Even though titanatranes have different site preference, such the correlation between the angle strains and thermodynamic stabilities were exhibited once again from the calculation results of dimeric titanatrane complexes45 (see Ti1–Ti3 in Table S2 in ESI†). The more increasing the substituted methyl groups on unbridged arms, the larger φN values are (Ti1: ∼106°, Ti2: ∼105° and Ti3: ∼104.6° in Table S2†) and simultaneously, these complexes become thermodynamically unstable (ΔG of Ti1: 0.48 kcal mol−1 and ΔG of Ti2: 0.22 kcal mol−1 in comparison of ΔG of Ti3, Table S2†). These results distinctly indicate that the angle strain between unbridged arms in dimeric complexes can evoke those thermodynamical unstability.
Solution- and gas-phase structures of alumatranes 1–3
Structurally characterized compounds 1–3 were also investigated by 1H, 13C, and 27Al NMR spectroscopies, elemental analysis, and electron ionization mass spectrometry (EI-MS) to determine their solution- and gas-phase structures. All chemical shifts of the protons and carbons for 1–3 were within their expected ranges. Compounds 1 and 2 may exist as isomers, as shown in Chart 1; however, their 1H NMR spectra display well-defined, sharp resonances with expected integrations. In compound 1, the 1H NMR spectrum shows two triplets and one singlet for the methylene protons in an integration ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (Fig. S1 and S4 in ESI†). In addition, the 1H NMR for compound 2 shows four singlets for the methyl protons with the integration ratio of 1:1:1:1 (Fig. S10 and S13 in ESI†). These data support only structures 1 and 2 among all possible structural isomers shown in Chart 3. The 1H and 13C NMR spectra support dimeric structures in solution; NMR separations for the bridging and terminal side arms were observed.
:2:1 (Fig. S1 and S4 in ESI†). In addition, the 1H NMR for compound 2 shows four singlets for the methyl protons with the integration ratio of 1:1:1:1 (Fig. S10 and S13 in ESI†). These data support only structures 1 and 2 among all possible structural isomers shown in Chart 3. The 1H and 13C NMR spectra support dimeric structures in solution; NMR separations for the bridging and terminal side arms were observed.
The coordination number and geometry around aluminum correlate well with the 27Al NMR chemical shift. The 27Al NMR spectra of 1–3 were collected with the samples dissolved in CDCl3, and two broad peaks at approximately 6 ppm and 65 ppm were observed (Fig. S7, S16 and S28 in ESI†).54 Low-field signals in their 27Al NMR spectra could be definitely assigned to the aluminosilicate peak of the NMR tube. The both pentacoordinate alumatranes N(C6H4O)3Al–NH2CH2Ph19 and Al(OCH2CH2)3N21 showed similar signals at 66 ppm in their 27Al NMR spectra. Although the 27Al NMR signal for the pentacoordinate six-membered system34 was shifted downfield to 37.2 ppm, which is within the expected region from 33 to 61 ppm for monomeric five-coordinate aluminum alkoxides,55 the 27Al NMR signal at approximately 6 ppm for 1–3 is very similar to those for other reported pentacoordinate alumatranes with tricyclic five-membered rings.19,21 Thus, 1H, 13C, and 27Al NMR data support that alumatranes 1–3 in the solution phase exist as pentacoordinate dimeric structures. Especially, 2D NMR (COSY, HSQC, and HMBC) spectra for compounds 2 and 3 made the proper assignment of NMR peaks (see ESI†).
The electron impact mass spectra (70 eV) of 1–3 show that the molecular peaks of compounds 1–3 appeared at 402, 458, and 514 m/z, respectively (Fig. S8, S20 and S32 in ESI†). In addition, the absence of other peaks between 500 and 1000 m/z excludes the existence of oligomeric species other than dimers. Similar data have been reported in the literature,28 and the mass spectra data indicate that 1–3 exist as dimeric structures in the gas phase.
Catalytic activities
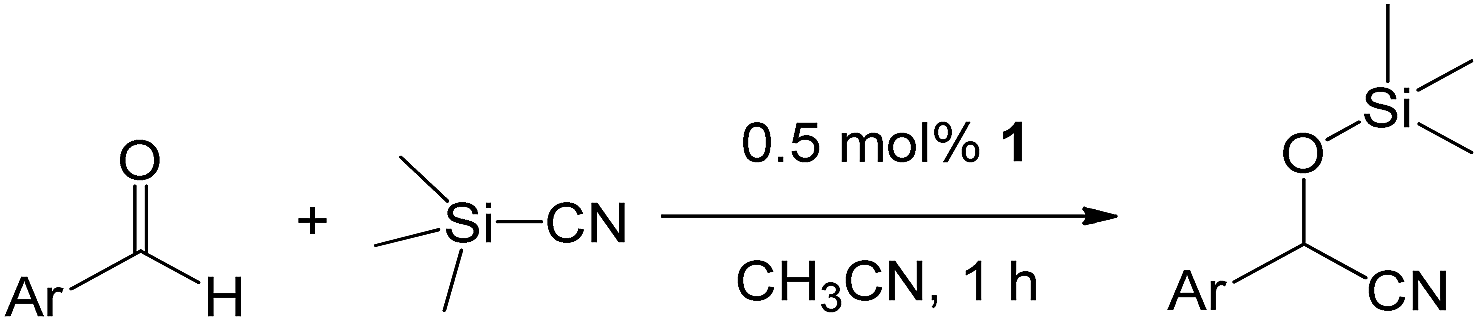
As shown in Table 2, to optimize the conditions for the trimethylsilylcyanation reaction, we used TMSCN and benzaldehyde as model substrates in four catalytic systems: compounds 1–3 and the previously reported alumatrane with a tricyclic six-membered ring,18 which was used as a trimethylsilylcyanation catalyst to synthesize 2-phenyl-2-trimethylsilyloxyacetonitrile in 92% isolated yield at room temperature in 9 h. Initially, we reduced the reaction time from 9 h to 1 h for the comparison of catalytic activity. Under the same reaction conditions, catalyst 1 showed the highest catalytic activity (entries 1–4). As expected, no catalytic activity was observed when no catalyst was used (entry 5). As the amount of catalyst was decreased from 0.5 mol% to 0.1 mol%, the reaction time required to achieve similar catalytic activity for all catalysts increased from 1 h to 6 h (entries 6–9). The order of the catalytic activity did not change between catalysts; thus, catalyst 1 was determined to be the best catalytic system among the four. Even at a low catalyst loading of 0.05 mol%, catalyst 1 showed an isolated yield of 39% for a reaction time of 6 h at room temperature (entries 10–13). Polar solvent such as MeCN is better than non-polar solvent of toluene under the same reaction condition (entries 6–8 and 14–16).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Solvent | t (h) | Yieldb,c (%) |
| a Reaction conditions: 2 mmol benzaldehyde, 3.5 mmol TMSCN, and 5 mL solvent.b Isolated yields after silica-gel column chromatography based on benzaldehyde.c Average of two runs.d [AlL]2 = [Al(OC6H2-2,4-Me2-6-CH2)3N]2. | |||||
| 1 | 1 | 0.5 | CH3CN | 1 | 95 |
| 2 | 2 | 0.5 | CH3CN | 1 | 90 |
| 3 | 3 | 0.5 | CH3CN | 1 | 84 |
| 4 | [AlL]2d | 0.5 | CH3CN | 1 | 15 |
| 5 | — | — | CH3CN | 1 | 0 |
| 6 | 1 | 0.1 | CH3CN | 6 | 93 |
| 7 | 2 | 0.1 | CH3CN | 6 | 87 |
| 8 | 3 | 0.1 | CH3CN | 6 | 83 |
| 9 | [AlL]2d | 0.1 | CH3CN | 6 | 13 |
| 10 | 1 | 0.05 | CH3CN | 6 | 39 |
| 11 | 2 | 0.05 | CH3CN | 6 | 31 |
| 12 | 3 | 0.05 | CH3CN | 6 | 26 |
| 13 | [AlL]2d | 0.05 | CH3CN | 6 | 5 |
| 14 | 1 | 0.1 | Toluene | 6 | 67 |
| 15 | 2 | 0.1 | Toluene | 6 | 59 |
| 16 | 3 | 0.1 | Toluene | 6 | 54 |
Such an order of the catalytic activities for 1–3 is significantly correlated with the dissociation free energy barrier of each bond between Al and bridged O atom. In order to exhibit efficiently catalytic behaviors of these alumatrane complexes, the dissociation of Al–O bond have to be especially well occurred for the insertion of substrates to Al center. These energy barriers could be calculated as the thermal stabilities between before and after dissociation of Al–O bonds in gas phase. The values of 1–3 were estimated as 1740 kcal mol−1 for 1, 1766 kcal mol−1 for 2 and 1780 kcal mol−1 for 3, indicating that the more decreasing the dissociation energy barrier of Al–O bond are, the more increasing the catalytic activities show. Consequently, the reason why the high catalytic activity in 1 could be shown compared to 2 or 3 is the weakest bond strength between Al and bridged O atom in 1 among those of alumatrane complexes. For the trimethylsilylcyanation reaction, a polar solvent such as acetonitrile is normally used to attain a high yield. As expected, changing the solvent from polar acetonitrile to nonpolar toluene caused a decrease of catalytic activity (entries 14–16). Since our systems 1–3 have higher solubility in CH3CN and toluene than [AlL]2,18 they showed higher catalytic activity than [AlL]2. However, the maintained homogeneity after completion of the reaction prevents the recycling of catalysts 1–3 for this reaction.
With the optimized conditions of 0.5 mol% 1, 5 mL CH3CN, rt and 1 h in hand, we examined the effect of various aryl aldehydes (instead of benzaldehyde) on the trimethylsilylcyanation reaction (Table 3). The coupling of benzaldehyde (entry 1) with TMSCN efficiently generated the desired product in 95% isolated yield. Electron-donating aldehydes such as p-tolualdehyde (entry 2) and p-anisaldehyde (entry 3), electron-withdrawing aldehydes such as α,α,α-trifluorotolualdehyde (entry 4), 4-nitrobenzaldehyde (entry 5), 4-cyanobenzaldehyde (entry 6), methyl 4-formylbenzoate (entry 7), and 4-chlorobenzaldehyde (entry 8) and electron-neutral aldehydes such as 1-naphthaldehyde (entry 9) and 2-naphthaldehde (entry 10) were also effective in the trimethylsilylcyanation reaction. When 0.5 mol% 1 was employed in this reaction, three aryl aldehydes—the highly electron-withdrawing α,α,α-trifluorotolualdehyde (entry 4) and 4-chlorobenzaldehyde (entry 8) and the sterically hindered electron-neutral 1-naphthaldehyde (entry 9)—resulted in only 83–85% isolated yields. Other aryl aldehydes showed activities similar to that of benzaldehyde.
|
||
|---|---|---|
| Entry | ArC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H O)H |
Yieldb,c (%) |
| a Reaction conditions: 2 mmol ArC(O)H, 3.5 mmol TMSCN, 5 mL CH3CN, 0.5 mol% 1, rt, and 1 h.b Isolated yields after silica-gel column chromatography based on aryl aldehyde.c Average of two runs. |
||
| 1 | Benzaldehyde | 95 |
| 2 | p-Tolualdehyde | 90 |
| 3 | p-Anisaldehyde | 94 |
| 4 | α,α,α-Trifluoro-p-tolualdehyde | 85 |
| 5 | 4-Nitrobenzaldehyde | 91 |
| 6 | 4-Cyanobenzaldehyde | 92 |
| 7 | Methyl 4-formylbenzoate | 90 |
| 8 | 4-Chlorobenzaldehyde | 83 |
| 9 | 1-Naphthaldehyde | 85 |
| 10 | 2-Naphthaldehde | 91 |
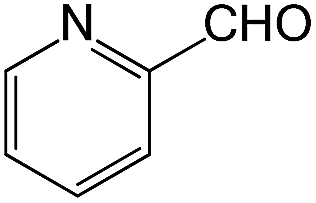
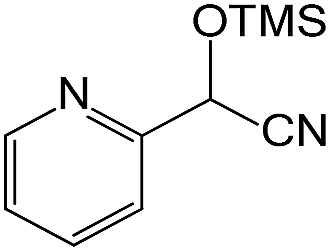
We also screened various heterocyclic and alkyl aldehydes using the optimized trimethylsilylcyanation conditions of 0.5 mol% 1, 5 mL CH3CN, rt and 1 h (Table 4). With 2- and 3-pyridine-carboxaldehyde and 6-methyl-2-pyridine-carboxaldehyde, good product yields of 91%, 86%, and 93%, respectively, were obtained (entries 1–3). As shown in Table 4 (entries 4–6), 6,2- and 3-thiopenecarboxaldehyde and 4-methyl-2-thiazolecarboxaldehyde also worked well in this reaction, giving 90%, 83%, and 91% yields of the corresponding products, respectively. Straight-chain alkyl aldehydes (entries 7 and 8), a conjugated aldehyde (entry 9), and an aldehyde with a sterically hindered cyclic ring (entry 10) also provided the corresponding products in reasonably good yields of 80–87%.
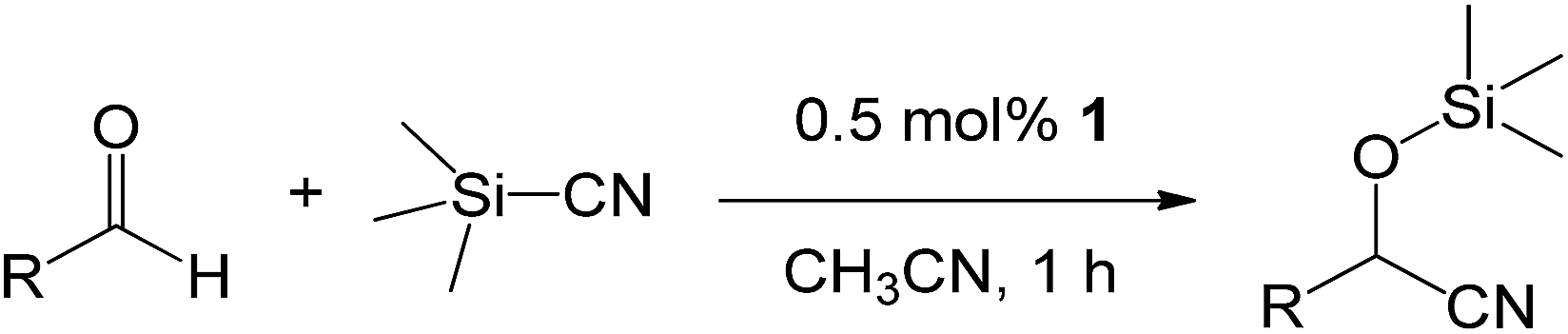
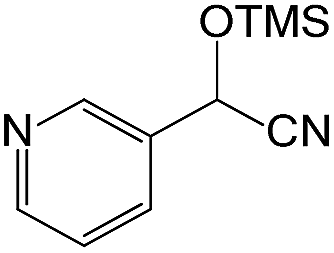
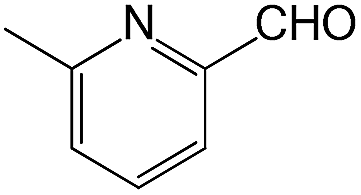
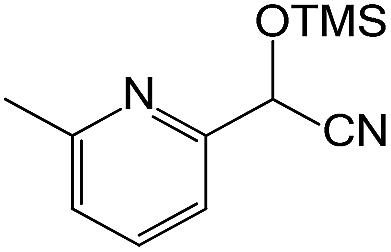
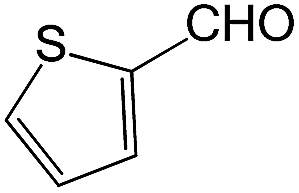
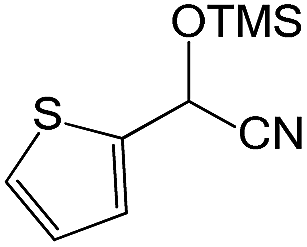
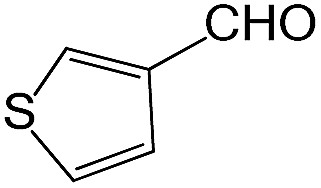
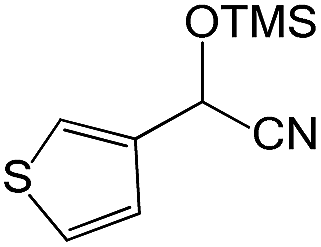
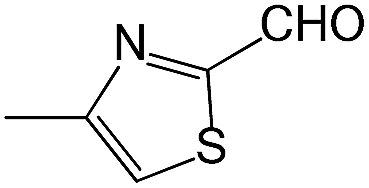
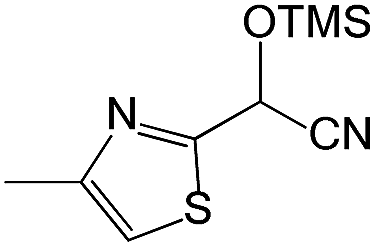
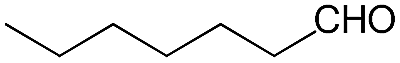
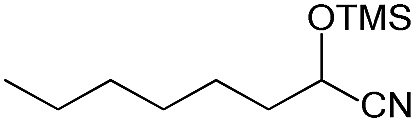
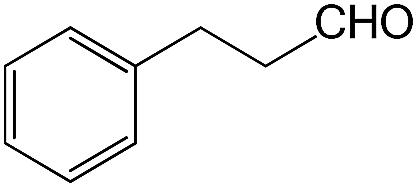
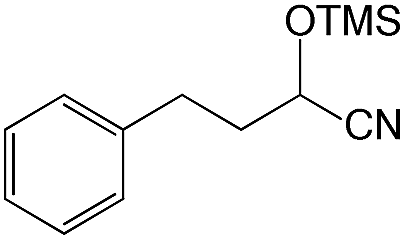
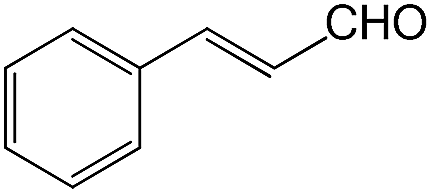
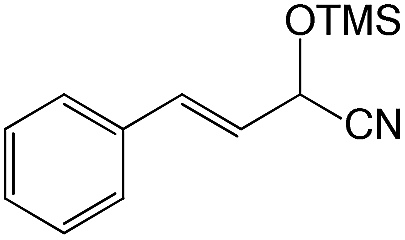
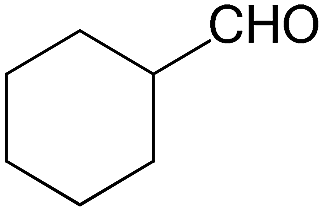
A possible mechanism for alumatrane catalyzed trimethylsilylcyanation reaction is proposed in Scheme 2. The catalytic cycle starts with the coordination of aldehyde to the Lewis acidic Al center in alumatrane 1, generating neutral monomeric alumatrane 1′. Generally, dimeric alumatranes in the presence of aldehyde could be easily converted to monomeric specie, which was supported by structurally characterized monomeric alumatrane adduct with benzaldehyde obtained from the reaction between dimeric alumatrane and benzaldehyde.17 In addition, we also calculated the dissociation free energy barrier of Al–O bond as 1740 kcal mol−1 for 1, 1766 kcal mol−1 for 2 and 1780 kcal mol−1 for 3, indicating that monomeric alumatrane containing aldehyde could be easily generated. Then, a cyanide anion as a nucleophile attacks the electron-deficient carbonyl carbon on the activated aldehyde to make new ionic species 1′′. Finally, the production of the desired product facilitates the regeneration of the catalyst 1.
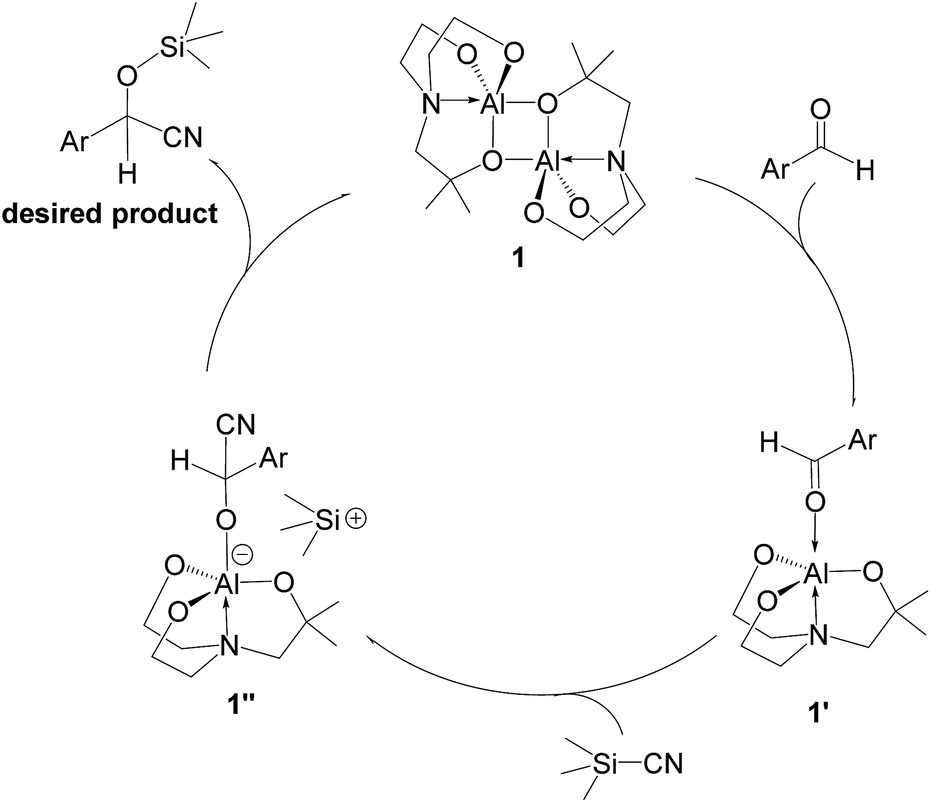 | ||
| Scheme 2 Plausible mechanism for trimethylsilylcyanation of aldehydes using catalyst 1. | ||
Experimental
General considerations
All reactions of air- and moisture-sensitive materials were carried out under dinitrogen using standard Schlenk-type glassware on a dual manifold Schlenk line in a glove box.56 Dinitrogen was deoxygenated using an activated Cu catalyst and dried with drierite.57 All chemicals were purchased from Aldrich and used as supplied unless otherwise indicated. Toluene, THF, diethylether, and n-hexane were dried with sodium diphenylketyl and stored over activated 3 Å molecular sieves. All deuterated solvents such as CDCl3 and C6D6 (Cambridge Isotope Laboratories) were used after drying over activated molecular sieves (5 Å).Measurements
1H and 13C NMR spectra were recorded at ambient temperature on a 400 MHz NMR spectrometer using standard parameters. All chemical shifts are reported in δ units with reference to the peaks of residual CDCl3 (δ 7.24, 1H NMR; δ 77.0, 13C NMR) or C6D6 (δ 7.16, 1H NMR; δ 128.0, 13C NMR). 27Al NMR spectroscopy was carried out at the Korea Basic Science Institute. EI-MS was performed on a VG Auto Spec. Elemental analyses were performed using an EA 1110-FISONS analyzer (CE Instruments).Synthesis
(HOCH2CH2)2N(CH2CMe2OH) (L1H3), (HOCH2CH2)N(CH2CMe2OH)2 (L2H3), and N(CH2CMe2OH)3 (L3H3) were synthesized by literature procedures.44Representative procedures for the trimethylsilylcyanation reaction
In a glove box, a 10 mL vial was charged with 1–3 (0.01 mmol, 0.5 mol% relative to aldehyde); the vial was removed from the glove box, and 5 mL of freshly distilled acetonitrile was added via a syringe, resulting in a clear solution. The corresponding aldehyde (2 mmol) and TMSCN (3.5 mmol) were added sequentially under nitrogen, and the reaction mixture was stirred at room temperature. After 1 h, the solvent and excess TMSCN were evaporated at reduced pressure on a Schlenk line at 70 °C, and then 10 mL of hexanes was added. The precipitated catalyst was filtered, and the crude product was purified by column chromatography (5% ethyl acetate in hexane).X-ray structural determination for 1–3
The crystallographic measurements were performed at 296(2) K for all complexes 1–3 using a Bruker APEX II diffractometer with Mo Kα (λ = 0.71073 Å) radiation. Specimens of suitable quality and size were selected, mounted, and centered in the X-ray beam using a video camera. The structures were solved by direct methods and refined by full-matrix least-squares methods using the SHELXTL58 program package with anisotropic thermal parameters for all non-hydrogen atoms, resulting in the X-ray crystallographic data of 1–3 in CIF formats (CCDC 1494116–1494118). Final refinement based on the reflections (I > 2σ(I)) converged at R1 = 0.0903, wR2 = 0.2667, and GOF = 1.082 for 1, at R1 = 0.0579, wR2 = 0.1526, and GOF = 1.017 for 2, and at R1 = 0.0513, wR2 = 0.1350, and GOF = 1.060 for 3. Further details are listed in Table 5.| 1 | 2 | 3 | |
|---|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |||
| Chemical formula | C15H24AlNO3 | C68H112Al4N4O12 | C12H24AlNO3 |
| Formula weight | 293.33 | 1285.53 | 257.30 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | P21/n |
| a (Å) | 10.3390(5) | 13.6533(6) | 9.4843(5) |
| b (Å) | 9.2969(4) | 14.1939(6) | 10.5629(6) |
| c (Å) | 17.3082(8) | 18.8530(8) | 14.1175(7) |
| α (deg) | 90.00 | 90.00 | 90.00 |
| β (deg) | 107.191(3) | 98.997(2) | 91.340(3) |
| γ (deg) | 90.00 | 90.00 | 90.00 |
| V (Å3) | 1589.35(13) | 3608.6(3) | 1413.93(13) |
| Z | 4 | 2 | 4 |
| dcalcd (g cm−3) | 1.226 | 1.183 | 1.209 |
| F(000) | 632 | 1392 | 560 |
| Reflections collected | 19264 |
79871 |
24117 |
| # of independent reflections | 3343 [R(int) = 0.0340] | 13281 [R(int) = 0.0668] |
4377 [R(int) = 0.0571] |
| # of parameters | 184 | 407 | 160 |
| R1 (I > 2σ(I))a | 0.0903 | 0.0579 | 0.0513 |
| wR2 (I > 2σ(I))b | 0.2667 | 0.1526 | 0.1350 |
| GOF (I > 2σ(I)) | 1.082 | 1.017 | 1.060 |
Computational details for 1, 2 and their isomers
The geometry optimization for the ground-state (S0) structures of 1, 2 and their isomers based on the X-ray structures of 1 and 2 were performed at the B3LYP/6-31+G(d) level of theory. Imaginary frequencies for the optimized structures were not observed. All the calculations were performed for gas-phase molecules and were carried out using the Gaussian 09 software package.59 The dissociation energy barriers between Al and bridged O atom of 1–3 could be calculated as the thermal stabilities between before and after dissociation of Al–O bonds in gas phase.Conclusions
We designed and explored novel dimeric alumatranes with tricyclic five-membered rings. The obtained alumatranes were all dimeric in the solid state, solution phase, and the gas phase. According to single-crystal X-ray analysis, the first structurally characterized dimeric alumatranes abnormally had their sterically bulky side arms with dimethyl substituents in the bridging sites of the tetradentate ligand, which was also determined by DFT calculations. The new alumatranes were used as catalysts for the trimethylsilylcyanation reaction of aldehydes under extremely mild conditions of room temperature, less than 0.5 mol% catalyst loading, and a short reaction time of 1 h. The new catalytic systems showed high catalytic activities regardless of the aldehyde type, which included electron-rich, neutral, and deficient aryl aldehydes, heterocyclic aldehydes, and alkyl aldehydes. Further explorations of the synthesis and application of chiral alumatranes are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), the Korean Ministry of Education (MOE) through Basic Science Research Program (grant number 2015R1D1A1A01061043).References
- R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS PubMed.
- J.-M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed.
- F.-X. Chen and X. Feng, Synlett, 2005, 892–899 CAS.
- T. R. J. Achard, L. A. Clutterbuck and M. North, Synlett, 2005, 1828–1847 CAS.
- J. Gawronski, N. Wascinska and J. Gajewy, Chem. Rev., 2008, 108, 5227–5252 CrossRef CAS PubMed.
- Y. Izumi and M. Onaka, J. Mol. Catal., 1992, 74, 35–42 CrossRef CAS.
- Y. Hamashima, D. Sawada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 2641–2642 CrossRef CAS.
- H. Deng, M. P. Isler, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 2002, 41, 1009–1012 CrossRef CAS PubMed.
- Y. Qin, L. Liu and L. Pu, Org. Lett., 2005, 7, 2381–2383 CrossRef CAS PubMed.
- G. Rajagopal, S. S. Kim and J. M. Kwak, Bull. Korean Chem. Soc., 2006, 27, 1907–1909 CrossRef CAS.
- K. Iwanami, J.-C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 1002–1004 RSC.
- J. Niemeyer, J. Cloppenburg, R. Fröhlich, G. Kehr and G. Erker, J. Organomet. Chem., 2010, 695, 1801–1812 CrossRef CAS.
- J.-J. Cao, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4976–4980 CrossRef CAS PubMed.
- M. North, M. Omedes-Pujol and C. Williamson, Chem.–Eur. J., 2010, 16, 11367–11375 CrossRef CAS PubMed.
- J. Ternel, F. Agbossou-Niedercorn and R. M. Gauvin, Dalton Trans., 2014, 43, 4530–4536 RSC.
- X.-P. Zeng, Z.-Y. Cao, X. Wang, L. Chen, F. Zhou, F. Zhu, C.-H. Wang and J. Zhou, J. Am. Chem. Soc., 2016, 138, 416–425 CrossRef CAS PubMed.
- W. Su, Y. Kim, A. Ellern, I. A. Guzei and J. G. Verkade, J. Am. Chem. Soc., 2006, 128, 13727–13735 CrossRef CAS PubMed.
- S. M. Raders and J. G. Verkade, Tetrahedron Lett., 2009, 50, 5317–5321 CrossRef CAS.
- M. A. Paz-Sandoval, C. Fernandez-Vincent, G. Uribe and R. Contreras, Polyhedron, 1988, 7, 679–684 CrossRef CAS.
- M. D. Healy and A. R. Barron, J. Am. Chem. Soc., 1989, 111, 398–399 CrossRef CAS.
- J. Pinkas and J. G. Verkade, Inorg. Chem., 1993, 32, 2711–2716 CrossRef CAS.
- J. Pinkas, B. Gaul and J. G. Verkade, J. Am. Chem. Soc., 1993, 115, 3925–3931 CrossRef CAS.
- R. Narayanan and R. M. Laine, Appl. Organomet. Chem., 1997, 11, 919–927 CrossRef CAS.
- M.-A. Munoz-Hernandez, P. Wei, S. Liu and D. A. Atwood, Coord. Chem. Rev., 2000, 210, 1–10 CrossRef CAS.
- Y. Opornsawad, B. Ksapabutr, S. Wongkasemjit and R. M. Laine, Eur. Polym. J., 2001, 37, 1877–1885 CrossRef CAS.
- A. Singh and R. C. Mehrotra, Coord. Chem. Rev., 2004, 248, 101–118 CrossRef CAS.
- L. Fernandez, P. Viruela-Martin, J. Latorre, C. Guillem, A. Beltrán and P. Amorós, J. Mol. Struct.: THEOCHEM, 2008, 850, 94–104 CrossRef CAS.
- M. J. Lacey and C. G. MacDonald, Aust. J. Chem., 1976, 29, 1119–1121 CrossRef CAS.
- R. C. Mehrotra and R. K. Mehrotra, J. Indian Chem. Soc., 1962, 39, 677–682 CAS.
- F. Hein and P. W. Albert, Z. Anorg. Allg. Chem., 1952, 269, 67–75 CrossRef CAS.
- V. E. Shklover, Y. T. Struchkov, M. G. Voronkov, Z. A. Ovchinnikova and V. P. Baryshok, Dokl. Akad. Nauk SSSR, 1984, 277, 1185–1189 CAS.
- B. Lee, F. Moise, W. T. Pennington and G. H. Robinson, J. Coord. Chem., 1992, 26, 187–197 CrossRef CAS.
- E. Müller and H.-B. Bürgi, Helv. Chim. Acta, 1987, 70, 520–533 CrossRef.
- Y. Kim and J. G. Verkade, Inorg. Chem., 2003, 42, 4804–4806 CrossRef CAS PubMed.
- W. Su, J. Kobayashi, A. Ellern, T. Kawashima and J. G. Verkade, Inorg. Chem., 2007, 46, 7953–7959 CrossRef CAS PubMed.
- G. Licini, M. Mba and C. Zonta, Dalton Trans., 2009, 5265–5277 RSC.
- A. L. Johnson, M. G. Davidson, Y. Pérez, M. D. Jones, N. Merle, P. R. Raithby and S. P. Richards, Dalton Trans., 2009, 5551–5558 RSC.
- J. Zhang, A. Liu, X. Pan, L. Yao, L. Wang, J. Fang and J. Wu, Inorg. Chem., 2011, 50, 9564–9570 CrossRef CAS PubMed.
- M. G. Voronkov and V. P. Baryshok, J. Organomet. Chem., 1982, 239, 199–249 CrossRef CAS.
- J. G. Verkade, Acc. Chem. Res., 1993, 26, 483–489 CrossRef CAS.
- J. G. Verkade, Coord. Chem. Rev., 1994, 137, 233–295 CrossRef CAS.
- D. J. Kim, S.-d. Mun, S. Yoon, C. H. Oh, H.-R. Park, T.-S. You, J. Lee and Y. Kim, Polyhedron, 2011, 30, 1076–1079 CrossRef CAS.
- J. H. Moon, S. H. Kim, K. M. Lee, T.-S. You, Y. Do and Y. Kim, Polyhedron, 2011, 30, 2333–2338 CrossRef CAS.
- S.-d. Mun, J. Lee, S. H. Kim, Y. Hong, Y.-h. Ko, Y. K. Shin, J. H. Lim, C. S. Hong, Y. Do and Y. Kim, J. Organomet. Chem., 2007, 692, 3519–3525 CrossRef CAS.
- S.-d. Mun, S. H. Kim, J. Lee, H.-J. Kim, Y. Do and Y. Kim, Polyhedron, 2010, 29, 379–383 CrossRef CAS.
- J. H. Kim, S. Yoon, S.-d. Mun, S. H. Kim, J. Lee, Y. Chung, S. H. Kwon, K. S. Lee, C. Lee and Y. Kim, J. Organomet. Chem., 2011, 696, 1729–1735 CrossRef CAS.
- J. Lewinski and A. E. H. Wheatley, Top. Organomet. Chem., 2013, 41, 1–58 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. H. Kim, S. Y. Han, J. H. Kim, Y. Y. Kang, J. Lee and Y. Kim, Eur. J. Inorg. Chem., 2015, 2323–2329 CrossRef CAS.
- J. Lee, S. H. Kim, K. M. Lee, K. Y. Hwang, H. Kim, J. O. Huh, D. J. Kim, Y. S. Lee, Y. Do and Y. Kim, Organometallics, 2010, 29, 347–353 CrossRef CAS.
- S. H. Kim, D. Ahn, M. J. Go, M. H. Park, M. Kim, J. Lee and Y. Kim, Organometallics, 2014, 33, 2770–2775 CrossRef CAS.
- G. Rayner-Canham and T. Overton, Descriptive Inorganic Chemistry, Freeman, W. H. and Company, New York, 5th edn, 2010, p. 95 Search PubMed.
- J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, Harper-Collins, New York, 4th edn, 1993, p. 292 Search PubMed.
- R. Benn, A. Rufinska, E. Janssen and H. Lehmkuhl, Organometallics, 1986, 5, 825–827 CrossRef CAS.
- O. Kriz, B. Casensky, A. Lycka, J. Fusek and S. Hermanek, J. Magn. Reson., 1984, 60, 375–381 CAS.
- D. F. Shriver and M. A. Drezdzon, The Manipulation of Air-Sensitive Compounds, John Wiley & Sons, New York, 2nd edn, 1986 Search PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier, New York, 6th edn, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, IR, EI mass spectra and computational details. CCDC 1494116–1494118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09851k |
| ‡ The first, second, and third authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |