DOI:
10.1039/C7RA09711E
(Paper)
RSC Adv., 2017,
7, 49701-49709
Asymmetry-unit-dominated double slow-relaxation modes of 2,6-dimethyl-3,5-heptanedione dysprosium SMMs†
Received
1st September 2017
, Accepted 11th October 2017
First published on 25th October 2017
Abstract
A series of Hdmh mononuclear dysprosium complexes, namely [Dy(dmh)3(MeOH)] (1), [Dy(dmh)3(2,2′-bpy)]2 (2), and [Dy(dmh)3(phen)] (3), and analogue complexes of complex 2 [Lu(dmh)3(2,2′-bpy)]2 (4) (Hdmh = 2,6-dimethyl-3,5-heptanedione, 2,2′-bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline), have been isolated by reaction of Hdmh and LnCl3·6H2O with 2,2′-bpy/phen auxiliary ligands. X-ray crystallographic analysis reveals that all complexes 1–4 are mononuclear complexes. Complex 1 is seven-coordinated by three dmh ligands and one methanol. Complexes 2, 3 and 4 are all eight-coordinated by three dmh ligands and one 2,2′-bpy or phen. Magnetic studies indicate that complexes 2 and 3 exhibit slow relaxation at zero field but complex 1 does not. Strikingly, complex 2 exhibits mainly single slow relaxation under zero dc field and double slow relaxation under optimized dc field. In contrast, complex 3 exhibits double slow relaxation under zero dc field and single slow relaxation under optimized dc field. The origins of the double slow relaxation modes of complex 2 have been verified by dilution sample of complex 4.
Introduction
β-Diketone Dy-based single-molecule magnets (SMMs) have received extensive attention in recent years since β-diketone lanthanide complexes usually are of a simple mononuclear structure which enables the study of structure–magnetism relationship. Previously, the simplest β-diketone acetylacetonate (acac) dysprosium complex [Dy(acac)3(H2O)2] has been reported with an energy barrier of 66.1 K.1 Subsequently, several acac dysprosium complexes [Dy(acac)3(phen)], [Dy(dpq)(acac)3] and [Dy(dppz)-(acac)3]·CH3OH (phen = 1,10-phenanthroline, dpq = dipyrido-[3,2-d:20,30-f]-quinoxaline and dppz = dipyrido-[3,2-a:20,30-c]-phenazine) have been reported by changing the different capping nitrogen-containing auxiliary ligands.2,3 It is suggested that the larger capping auxiliary ligands may increase the energy barrier of these complexes. However, a SMM of [Dy(acac)3(dppn)]·C2H5OH exhibiting relatively small energy barrier (dppn = benzo[i]dipyrido-[3, 2-a:2′,3′-c]phenazine) containing even larger capping auxiliary ligands was reported in 2014.4 The authors thus claim that the energy barrier is not solely dominated by the size of the auxiliary ligand but also by the coordination symmetry around the Dy(III) ion. Thereafter, the study of magnetism–structure relationships has been focused on coordination symmetry around the Dy(III) ion in terms of α, φ angle and dihedral angle in the structure, e.g. complexes Dy(DBM)3(bpy), Dy(DBM)3(bpy) and Dy(TFI)3(bpy) etc. (DBM = dibenzoylmethane, TFI = 2-(2,2,2-trifluoroethyl)-1-indon).5–8 Recently, theoretical calculation, e.g. magnetic easy axis,9 has been employed in the explanation of the magnetism–structure relationship for the complex Dy(tBu-acac)3bpy (tBu-acac = 2,2,6,6-tetramethylheptane-3,5-dionate).10 Although a host of β-diketone dysprosium SMMs have been studied, there is still no complete theory for understanding of the magnetism–structure relationship for Dy(III)-based SMMs due to the complicated origin of the magnetism. To further explore the magnetism–structure relationship of β-diketone dysprosium SMMs with various auxiliary ligands, Hdmh and 2,2′-bpy/phen auxiliary ligands were employed in reactions with LnCl3·6H2O. As a result, a series of Hdmh mononuclear dysprosium complexes containing MeOH and 2,2′-bpy/phen auxiliary ligands have been isolated (Scheme S1, ESI†). Their crystal structures have been determined and their magnetism–structure relationship has been investigated.
Experimental
Materials and measurements
All chemicals except LnCl3·6H2O were obtained from commercial sources and used without further purification. LnCl3·6H2O was prepared by the reaction of Ln2O3 and HCl in aqueous solution. Elemental (C, H, O and N) analyses were performed using a PerkinElmer 2400 analyzer. FT-IR spectra were recorded using a PerkinElmer 100 spectrophotometer by using KBr pellets in the range of 4000–450 cm−1. UV spectra were recorded in solution using a PerkinElmer Lambda 35 spectrometer. Powder X-ray diffraction (PXRD) data were recorded using a Rigaku D/Max-3B X-ray diffractometer with Cu Kα as the radiation source (λ = 0.15406 nm) in the angular range θ = 5–50° at room temperature. Thermal analyses were carried out using an STA-6000 analyzer with a heating rate of 15 °C min−1 in the range of 30–800 °C under an O2 atmosphere. The magnetic susceptibilities of complexes 1–4 were investigated using a Quantum Design VSM superconducting quantum interference device (SQUID) magnetometer. The magnetic corrections were made using Pascal's constants.
Synthesis of [Dy(dmh)3(MeOH)] (1). NaOH (1.5 mmol, 60.0 mg) and Hdmh (1.5 mmol, 234.3 mg) in methanol (5 ml) were allowed to stir for 1.5 h. Then, DyCl3·6H2O (0.5 mmol, 189.8 g) was added to the solution and the mixture was allowed to stir for 24 h at room temperature and then was filtered. Colorless block crystals suitable for single-crystal X-ray diffraction were obtained in about one week by slowly evaporating the filtrate at 4 °C. Yield: 0.60 g (60%). Anal. calcd for C28H48DyO7: C 51.02 and H 7.34. Found (%): C 50.95 and H 7.35. IR (KBr, cm−1): 3457(m), 2966(m), 2429(w), 1584(s), 1427(s), 1159(s), 1074(w), 949(m), 866(m) and 552(s). UV-visible (CH3OH, λmax/nm): 292.
Synthesis of [Dy(dmh)3(2,2′-bpy)] (2). NaOH (1.5 mmol, 60.0 mg) and Hdmh (1.5 mmol, 234.3 mg) in methanol (5 ml) were allowed to stir for 1.5 h. Then DyCl3·6H2O (0.5 mmol, 189.8 mg) and bpy (0.5 mmol, 78.0 mg) were added to the solution. The mixture was allowed to stir 24 h at room temperature and then was filtered. Colorless block crystals suitable for single-crystal X-ray diffraction were obtained in about one week by slowly evaporating the filtrate at 4 °C. Yield: 0.75 g (64%). Anal. calcd for C37H47DyN2O6: C 57.10, H 6.09 and N 3.60. Found (%): C 57.02, H 6.10 and N 3.62. IR (KBr, cm−1): 2963(s), 1594(s), 1537(m), 1499(m), 1447(m), 1307(w), 1155(m), 1091(m), 913(w) and 760(m). UV-visible (CH3OH, λmax/nm): 235 and 287.
Synthesis of [Dy(dmh)3(phen)] (3). NaOH (1.5 mmol, 60.0 mg) and Hdmh (1.5 mmol, 234.3 mg) in methanol (5 ml) were allowed to stir for 1.5 h. Then, DyCl3·6H2O (0.5 mmol, 189.8 mg) and phen (0.5 mmol, 99.2 mg) were added to the solution. The mixture was allowed to stir for 24 h at room temperature and then was filtered. Colorless block crystals suitable for single-crystal X-ray diffraction were obtained in about one week by slowly evaporating the filtrate at 4 °C. Yield: 0.91 g (75%). Anal. calcd for C39H53DyN2O6: C 57.95, H 6.61 and N 3.47. Found (%): C 57.8, H 6.65 and N 3.4. IR (KBr, cm−1): 3453(m), 2963(m), 2434(w), 1594(s), 1454(m), 1155(s), 1066(s), 948(m), 852(s) and 543(m). UV-visible (CH3OH, λmax/nm): 229, 267 and 289.
Synthesis of [Lu(dmh)3(2,2′-bpy)] (4). NaOH (1.5 mmol, 60.0 mg) and Hdmh (1.5 mmol, 234.3 mg) in methanol (5 ml) were allowed to stir for 1.5 h. Then LuCl3·6H2O (0.5 mmol, 194.8 mg) and bpy (0.5 mmol, 78.0 mg) were added to the solution. The mixture was allowed to stir 24 h at room temperature and then was filtered. Colorless block crystals suitable for single-crystal X-ray diffraction were obtained in about one week by slowly evaporating the filtrate at 4 °C. Yield: 0.75 g (64%). Anal. calcd for C37H53LuN2O6: C 55.77, H 6.70 and N 3.52. Found (%): C 55.68, H 6.82 and N 3.49. IR (KBr, cm−1): 2965(s), 1596(s), 1538(m), 1500(m), 1445(m), 1308(w), 1154(m), 1093(m), 911(w) and 762(m). UV-visible (CH3OH, λmax/nm): 235 and 287.
X-ray crystallography
The single-crystal X-ray data of complexes 1–4 were collected using an Oxford Xcalibur Gemini Ultra diffractometer with graphite-monochromated Mo Kα (λ = 0.71073 Å) at room temperature. Empirical absorption corrections based on equivalent reflections were applied. The structures of complexes 1–4 were solved by direct methods and refined using the full matrix least-squares method on F2 using the SHELXS-97 crystallographic software package.11 All non-hydrogen atoms were anisotropically refined. The crystal data and structure refinement details for complexes 1–4 are summarized in Table 1. The selected bond lengths and angles for complexes 1–3 are given in Table S1.† CCDC no. 1515986, 1515987, 1515850 and 1563201 contain the supplementary crystallographic data for complexes 1–4, respectively.†
Table 1 Crystal data and structure refinement for complexes 1–4
| R1 = ∑||Fo| − |Fc||/|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Complex |
1 |
2 |
3 |
4 |
| Empirical formula |
C28H48DyO7 |
C37H47DyN2O6 |
C39H53DyN2O6 |
C37H53LuN2O6 |
| Formula weight |
659.16 |
778.27 |
808.33 |
796.78 |
| Color |
Colorless |
Colorless |
Colorless |
Colorless |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
| a (Å) |
16.643(5) |
13.266(5) |
12.878(5) |
12.910(5) |
| b (Å) |
12.983(5) |
16.803(5) |
24.058(5) |
16.376(5) |
| c (Å) |
18.688(4) |
18.056(5) |
16.567(5) |
17.851(4) |
| α (deg) |
90 |
92.267(5) |
90.000 |
90.622(5) |
| β (deg) |
119.996(18) |
94.493(5) |
128.376(19) |
94.243(5) |
| γ (deg) |
90 |
92.591(5) |
90.000 |
90.447(5) |
| V (Å3) |
3497(2) |
4005(2) |
4024(2) |
3763(2) |
| Z |
4 |
4 |
4 |
4 |
| ρ (g cm−3) |
1.252 |
1.291 |
1.334 |
1.406 |
| μ (mm−1) |
2.171 |
1.907 |
1.900 |
2.667 |
| F (000) |
1352.0 |
1588.0 |
1600.0 |
1630.0 |
| R1a, [I > 2σ(I)] |
0.0667 |
0.0393 |
0.0346 |
0.0297 |
| wR2b, [I > 2σ(I)] |
0.1504 |
0.1002 |
0.0752 |
0.0616 |
| R1a, (all data) |
0.1075 |
0.0605 |
0.0484 |
0.0398 |
| wR2b, (all data) |
0.1791 |
0.1098 |
0.0839 |
0.0672 |
| GOF on F2 |
0.995 |
1.094 |
1.097 |
1.070 |
Results and discussion
Spectral analysis of complexes 1–3
Complexes 1–3 were synthesized as shown in Scheme S1.† The IR spectrum of complex 1 exhibits the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the donors (1615 cm−1) shifted to higher wavenumber around 1640 cm−1. For complexes 2 and 3, two peaks appear around 1510 cm−1 and 1540 cm−1 which reveal the involvement of the auxiliary nitrogen ligands in the complexes (Fig. S1†). The UV-visible spectra show obvious absorption bands around 273 nm for pure Hdmh and 293 nm for coordinated Hdmh in complexes 1–3 (Fig. S2†). The 20 nm red-shift results from the singlet–singlet n–π* enol absorption of the β-diketonate.
O stretching of the donors (1615 cm−1) shifted to higher wavenumber around 1640 cm−1. For complexes 2 and 3, two peaks appear around 1510 cm−1 and 1540 cm−1 which reveal the involvement of the auxiliary nitrogen ligands in the complexes (Fig. S1†). The UV-visible spectra show obvious absorption bands around 273 nm for pure Hdmh and 293 nm for coordinated Hdmh in complexes 1–3 (Fig. S2†). The 20 nm red-shift results from the singlet–singlet n–π* enol absorption of the β-diketonate.
TG-DSC analysis of complexes 1–3
TG-DSC analysis for complexes 1–3 exhibits no step in the temperature range of 30–200 °C (Fig. S3†), supporting that there are no solvents in complexes 1–3.
PXRD analysis of complexes 1–3
PXRD patterns of complexes 1–3 are in agreement with the simulated patterns (Fig. S4†). It is worth noting that the PXRD patterns of dilute samples of complex 2 and complex 4 are in agreement with that of complex 2, suggesting that they are isomorphic. PXRD analysis demonstrates that the crystal structures of complexes 1–4 are truly representative of the bulk materials.
Structural descriptions of complexes 1–3
Crystal structural analysis suggests that complex 1 is mononuclear crystallizing in the monoclinic space group P2/c. Each Dy(III) ion is seven-coordinated by six oxygen atoms from three dmh ligands and one oxygen atom from one methanol molecule (Fig. 1a). The average bond length of Dy–O is 2.293 Å. Crystal structural analysis suggests that complex 2 is mononuclear crystallizing in the triclinic space group P. Notably, there are two crystallographically nonequivalent molecules of Dy(dmh)3(2,2′-bpy) in one asymmetry unit with different coordination symmetry in terms of 0.551 and 0.573 calculated by SHAPE 2.1 software, respectively. Each Dy(III) ion is eight-coordinated by six oxygen atoms from three dmh ligands and two nitrogen atoms from one bpy (Fig. 1b). Crystal structural analysis suggests that complex 3 is mononuclear crystallizing in the monoclinic space group P21/c. Each Dy(III) ion is eight-coordinated by six oxygen atoms from three dmh ligands and two nitrogen atoms from one phen (Fig. 1c). Crystal structure of complex 4 is isomorphic to complex 2. The coordination geometry of Dy(III) was calculated utilizing SHAPE 2.1 software on the basis of the crystal data.12 The corresponding parameters of representative coordination polyhedron are shown in Table S2 and S3.† On the basis of the calculation, the coordination geometry of the Dy(III) ion for complex 1 can be defined as a capped trigonal prism. In contrast, the geometry for complexes 2 and 3 can be defined as distorted square antiprism (Fig. 1d–f).
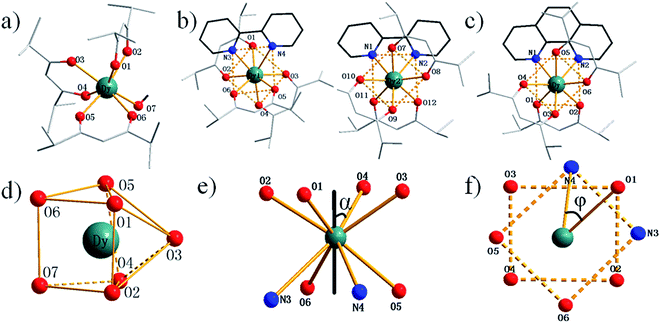 |
| | Fig. 1 Molecular structures of 1 (a), 2 (b) and 3 (c). Local coordination geometries of Dy(III) ions of complex 1 (d). Square-antiprismatic environment with skew angle α between the diagonals of the two squares (e). Square-antiprismatic environment showing φ angle between the S8 axis and a Dy–L vector (f). | |
Magnetic analysis
Direct-current (dc) magnetic analysis
Dc magnetic susceptibility for complexes 1–3 has been investigated at 1000 Oe in the temperature range from 1.8 K to 300 K (Fig. 2). The molar magnetic susceptibilities (χmT) at 300 K are 13.94, 13.92 and 14.17 cm3 K mol−1 for 1, 2 and 3, respectively, which are close to the expected value of 14.18 cm3 K mol−1 for an isolated Dy(III) ion (6H15/2, S = 5/2, L = 5, g = 4/3, C = 14.18 cm3 K mol−1). The M–H curves measured in the range of 0 to 65 kOe dc field at 1.8 K show that the maximum magnetizations are 5.03, 5.14 and 4.01 μB for complexes 1, 2 and 3, respectively (inset of Fig. 2), which are lower than the expected saturation value for one uncorrelated Dy(III) (gJ × J = 4/3 × 15/2 = 10 μB) but close to the usual value of 5.28 μB. The maximum magnetization of 4.01 μB for complex 3 is obviously lower than that for complex 2. This may result from the magnetic anisotropy and crystal field effects in the dysprosium center of complex 3 that dispel the 16-fold degeneration of the 6H15/2 ground state.13,14 Non-superposition curves of M versus H obtained in different magnetic fields (Fig. S5†) suggest the presence of a significant magnetic anisotropy and/or low-lying excited states in complexes 1–3.
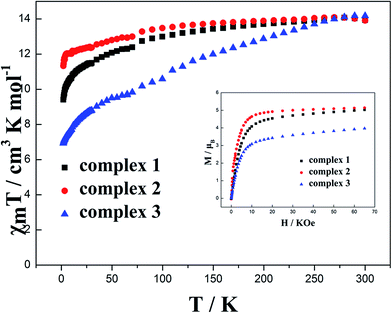 |
| | Fig. 2 Temperature dependence of χmT at 100 Oe for complexes 1–3 in the range 1.8–300 K. Inset: M vs. H/T data for complexes 1–3 at 1.8 K. | |
The hysteresis loops for complexes 2 and 3 were obtained in the temperature range of 1.8 K to 4.5 K with a sweep rate of 50 Oe s−1 (Fig. 3). Butterfly-shaped loops were clearly observed although the loop openings obviously change along with the temperature change. Upon raising the temperature, the openings of hysteresis loops become narrow until the magnetization is blocked at 4.5 K and 3 K for complexes 2 and 3, respectively. Noticeably, the blocking temperature of 4.5 K for complex 2 is higher than that for complex 3 and most previously reported β-diketone dysprosium complexes.5,6,8,15 This is attributed to the relatively weak quantum tunneling of magnetization (QTM) at zero dc field in comparison to that for complex 3 and other previously reported β-diketone dysprosium complexes.
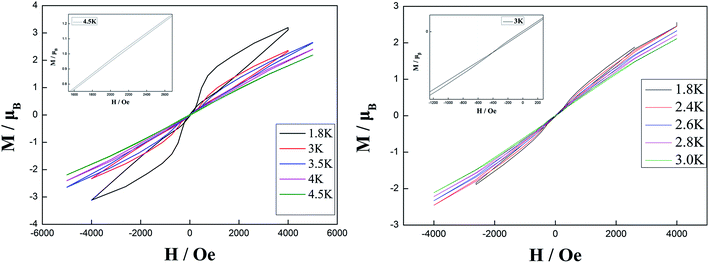 |
| | Fig. 3 Hysteresis loops for complex 2 with sweep rate of 50 Oe s−1 at 1.8–4.5 K (left) and for complex 3 with sweep rate of 50 Oe s−1 at 1.8–3 K (right). | |
Alternating-current (ac) magnetic analysis
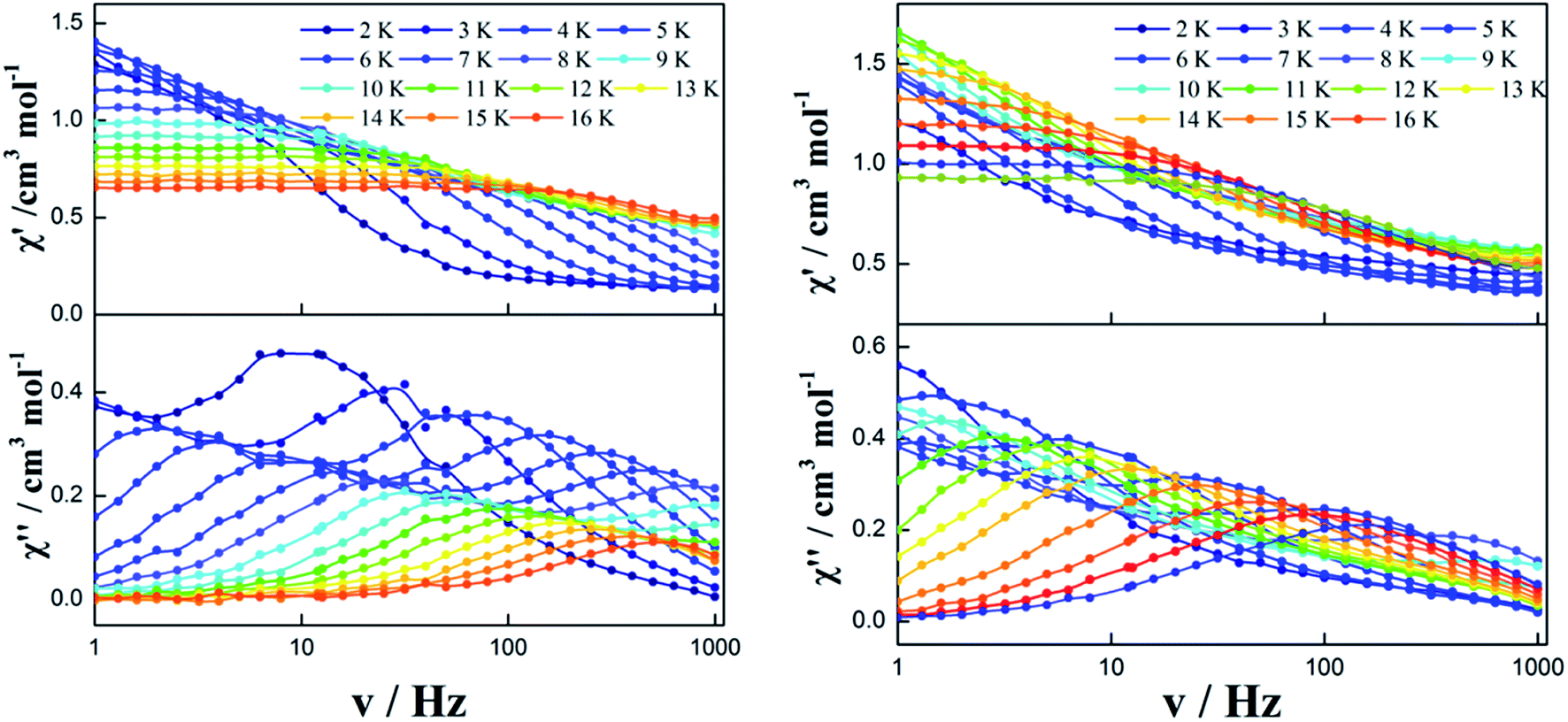
Zero dc field. To further investigate the dynamics of the magnetization, alternating-current (ac) magnetic susceptibilities for complexes 1–3 were investigated under zero dc field. The out-of-phase (χ′′) susceptibilities of complex 1 did not show a significant temperature dependence in the range of 1.8 K to 20 K (Fig. S6†), indicating the onset of pure QTM as often seen in other dysprosium SMMs.1–3,5–8,16–21 However, a temperature/frequency dependence was observed below 16 K for complexes 2 and 3, although the QTM still exists below 5 K (Fig. 4 and S7†). Noticeably, complex 2 exhibits a single relaxation process in both temperature- and frequency-dependent out-of-phase susceptibility (χ′′) curves (Fig. 4 left). In contrast, there is a double relaxation processes in the out-of-phase susceptibility (χ′′) curves for complex 3 (Fig. 4 right).
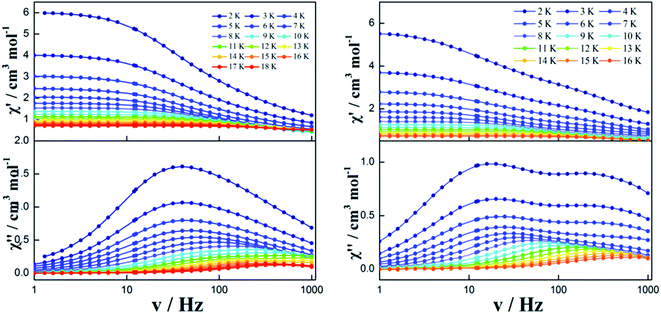 |
| | Fig. 4 Frequency dependence of the in-phase (χ′) and out-of-phase (χ′′) ac susceptibility of complexes 2 (left) and 3 (right) under 0 Oe. | |
The resulting Cole–Cole plots of complexes 1–3 can be fitted to the generalized Debye model at zero dc field (Fig. 5 and S8†). For complex 1, there are no semicircular curves observed confirming the existence of QTM. For complex 2, one semicircle is clearly observed in the Cole–Cole plots in the temperature range of 2.0–12 K. The values of α are in the narrow range of 0.268–0.315 indicating that a single relaxation mode is mainly involved at zero dc field (Table S4†). However, the plots and fitting of the Cole–Cole plots show a long flat semicircle in the high-temperature area (9–12 K) hinting at a double slow-relaxation mode. Thus, the single relaxation mode in the low-temperature area (2–8 K) is attributed to the low-temperature QTM effect. The real relaxation process is highly likely masked. For complex 3, however, two separate semicircles are clearly observed in the Cole–Cole plots in the low-temperature range of 2–5 K and one semicircle is observed in the Cole–Cole plots in the high-temperature range of 6–12 K. This indicates that there are two relaxation pathways in the low-temperature region and one relaxation pathway in the high-temperature region. This is consistent with the values of α in the range of 0–0.416 (Table S5†) implying two or multiple relaxation pathways.22 Notably, the two relaxation pathways in the low-temperature range of 2–5 K may result from the QTM effect.
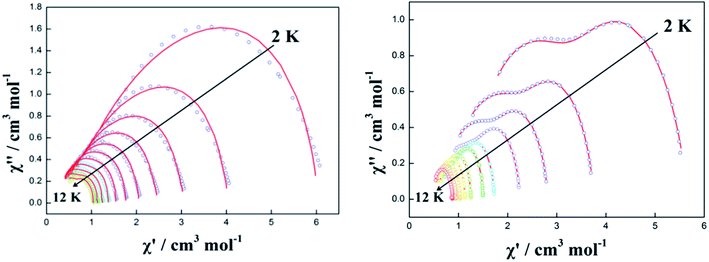 |
| | Fig. 5 Cole–Cole plots measured in the temperature range of 2.0–12.0 K under 0 Oe for complexes 2 (left) and 3 (right). The red solid lines correspond to the best fit obtained with a generalized Debye model. | |
Optimized dc field
In order to observe the real relaxation process and obtain an effective barrier, various dc fields in the range of 0–2000 Oe were applied at 8 K and 5 K for complexes 2 and 3, respectively23–25 (Fig. S9 and S10†). As a result, 2000 Oe and 1200 Oe were selected as optimized external dc fields on the basis of their lowest frequencies for complexes 2 and 3, respectively. In contrast to the temperature-/frequency-dependent curves for both in-phase and out-of-phase susceptibilities under zero dc field, the quantum tunneling effect was efficiently suppressed under the optimized external dc field (Fig. 6 and S11†). Strikingly, two thermally activated relaxation modes for complex 2 were highlighted under the optimized 2000 Oe field in contrast to a single peak in the temperature range of 10–20 K at zero dc field. Simultaneously, a single relaxation mode was observed for complex 3 in the plots of both temperature and frequency dependence. The Cole–Cole plots of complexes 2 and 3 can also be fitted to the generalized Debye model under optimized dc field (Fig. 7). For complex 2, two semicircles are clearly observed in the Cole–Cole plots in the temperature range of 7–16 K. The values of α in the range of 0–0.432 suggest two or multiple relaxation pathways (Table S6†). However, for complex 3, only one semicircle is clearly observed in the Cole–Cole plots in the temperature range of 2.0–12 K. The values of α in the range of 0.2–0.438 (Table S7†) suggest a single relaxation mode involved in the present relaxation pathway. Therefore, the Cole–Cole plots for both complexes 2 and 3 at the optimized dc field support that the QTM effect dominated the slow relaxation mode at zero dc field.
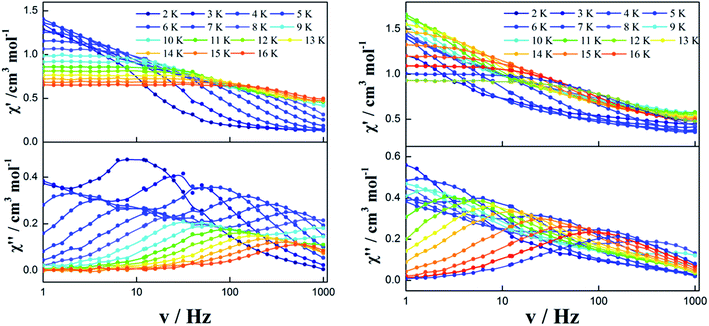 |
| | Fig. 6 Temperature/frequency dependence of the in-phase (χ′) and out-of-phase (χ′′) ac susceptibility of complex 2 under 2000 Oe (left) and complex 3 under 1200 Oe (right) in the temperature range 2.0–16 K. | |
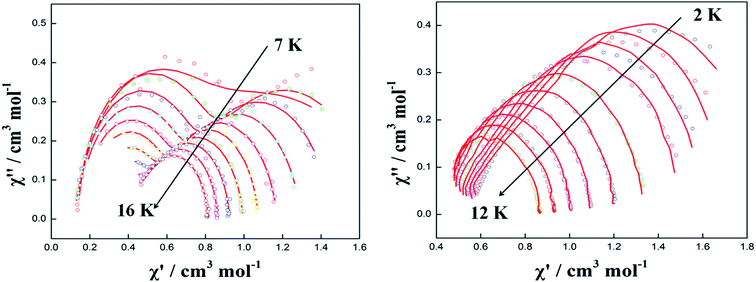 |
| | Fig. 7 Cole–Cole plots measured in the temperature range of 7–16 K for complex 2 (left) and 2–12 K for complex 3 (right) under optimal field. The red solid lines correspond to the best fit obtained with a generalized Debye model. | |
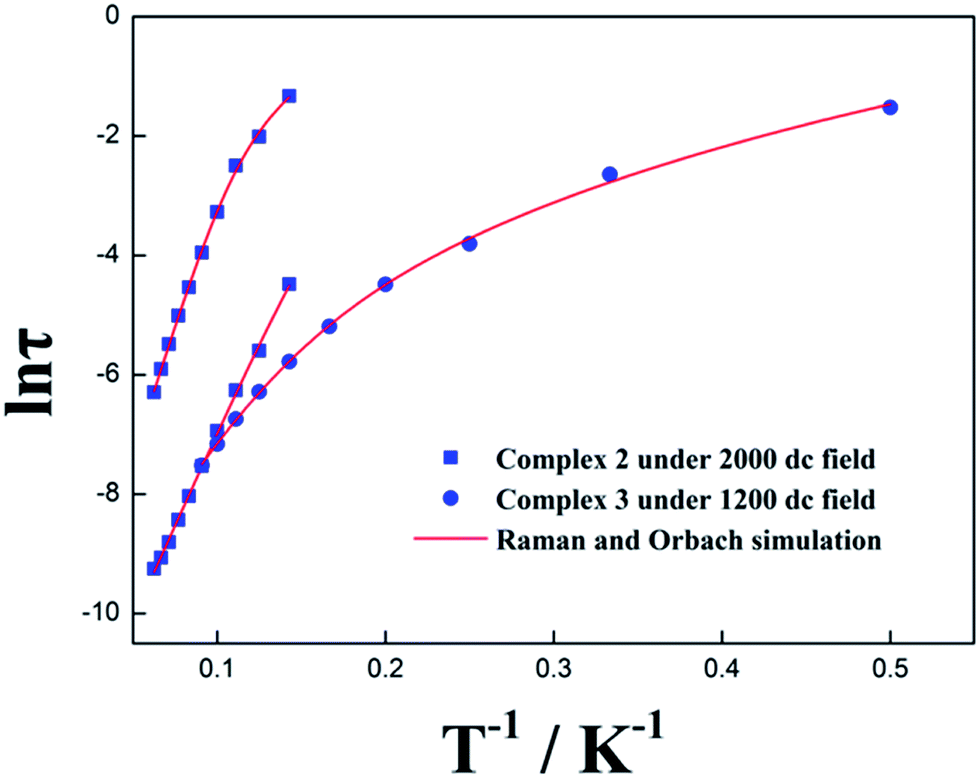
The anisotropic energy barrier Ueff can be obtained from the τ parameters of Cole–Cole plots on the basis of the optimized Arrhenius law 1/τ = CTn + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ueff/kBT) under optimum dc field (where C is the coefficient of Raman process, Ueff is the energy barrier to magnetization reversal and kB is the Boltzmann constant).26–29 As a result, Ueff/kB and τ0 were afforded as 62.84 K, 91.76 K and 1.87 × 10−6 s, 6.60 × 10−6 s for 2 as well as 73.69 K and τ0 = 2.49 × 10−6 s for 3, respectively, upon considering the spin-lattice relaxation of Raman and Orbach processes. It is also noteworthy that the plots of ln(τ) versus 1/T for complexes 2 and 3 under optimum dc field exhibit an obvious curvature, indicating that another relaxation process (Raman process) is also operative (Fig. 8 and Table S8†).
exp(−Ueff/kBT) under optimum dc field (where C is the coefficient of Raman process, Ueff is the energy barrier to magnetization reversal and kB is the Boltzmann constant).26–29 As a result, Ueff/kB and τ0 were afforded as 62.84 K, 91.76 K and 1.87 × 10−6 s, 6.60 × 10−6 s for 2 as well as 73.69 K and τ0 = 2.49 × 10−6 s for 3, respectively, upon considering the spin-lattice relaxation of Raman and Orbach processes. It is also noteworthy that the plots of ln(τ) versus 1/T for complexes 2 and 3 under optimum dc field exhibit an obvious curvature, indicating that another relaxation process (Raman process) is also operative (Fig. 8 and Table S8†).
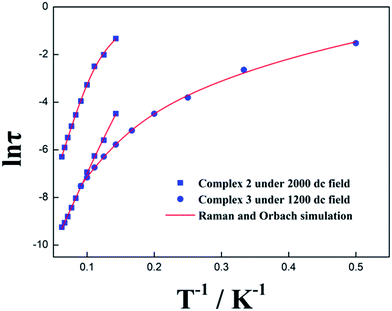 |
| | Fig. 8 The anisotropic energy barrier Ueff obtained from the τ parameters of Cole–Cole plots on the basis of the optimized Arrhenius law under zero dc field and optimized dc field for complexes 2 and 3, respectively. | |
In order to eliminate the interactions between the two asymmetry-nonequivalent Dy(III) in complex 2, a dilution sample was designed and synthesized by doping a large amount of Lu(III) ions (Lu:Dy = 0.957:0.043, verified by ICP). The temperature dependence of the out-of-phase (χ′′) ac susceptibility for diluted complex 2 reveals the double relaxation processes at optimized dc field (Fig. S12†), verifying that the double relaxation process results from the two different symmetric Dy(III) ions in the same asymmetric unit of complex 2. Namely, each Dy(III) ion from two different molecules in one asymmetric unit acts as a quasi-independent molecular object with its own intrinsic SMM behavior in which there is no coupling or interaction between two Dy(III) ions.30 Such a phenomenon is similar to that for previously reported β-diketone mononuclear SMMs of [Dy(hfac)3(boaDTDA)]217 (hfac = 1,1,1,5,5,5-hexafluoroacetylacetonato, boaDTDA = 4-(benzoxazol-2′-yl)-1,2,3,5-dithiadiazolyl) and other mononuclear SMMs31–34 exhibiting double relaxation modes with two crystallographically nonequivalent Dy(III) ions. Noticeably, sometimes only one relaxation process can be observed even though two crystallographically nonequivalent Dy(III) ions exist in one asymmetric unit of a complex depending on the symmetry difference in the same asymmetry unit of two Dy(III) ions and/or the probable existence of interaction/coupling between two Dy(III) ions.8
In general, the anisotropic energy barriers of eight-coordinated Dy-based SMMs are dominated by the coordination symmetry of the Dy(III) ion. However, the anisotropic energy barriers are not consistent with their symmetry for complexes 2 and 3. To investigate the correlation between the magnetism and structure for complexes 2 and 3, the magnetic anisotropy axis of Dy(III) ions in complexes 2 and 3 was calculated by Magellan software35–41 (Fig. 9). According to Long's work,42 complexes 2 and 3 involve oblate electron density of Dy(III) ions. The compression of the ligand field along the axis of Dy(III) ions will increase the magnetic anisotropy.43–45 Therefore, the shorter the Dy–O bonds, the greater the compression along the axis of Dy(III) ions and the higher the anisotropic energy barrier Ueff. On the basis of the average Dy–O bond lengths along the axis of Dy(III) ions in complex 2 (Dy1, 2.2792 Å) < complex 3 (2.3076 Å) < complex 2 (Dy2, 2.3269 Å), this corresponds to energy barriers of complex 2 (Dy1) (91.76 K) > complex 3 (73.69 K) > complex 2 (Dy2) (62.84 K), further verifying that the compression along the axis of Dy(III) ions can enhance energy barriers.
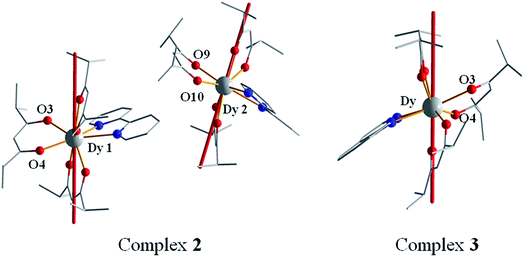 |
| | Fig. 9 The calculated easy axis (red line) of complexes 2 (left) and 3 (right) by Magellan software. Hydrogen atoms are omitted for clarity. | |
Conclusion
Isolation of a series of Hdmh dysprosium complexes demonstrates that dmh is able to react with Dy(III) ions and bpy/phen affording mononuclear Dy(III) complexes. Magnetic studies indicate that the ligand bpy/phen efficiently enhanced the magnetic energy barrier of complexes 2 and 3. QTM at 0 Oe not only blocks the thermally activated relaxation pathway for complex 2 but also causes the quantum relaxation pathway for complex 3. Optimized dc field is able to revive the thermally activated relaxation pathway for complex 2 and eliminate the quantum relaxation pathway for complex 3. Magnetic analysis of a dilution sample of complex 2 reveals that the double-relaxation mode results from the two types of independent Dy(III) ions in the same asymmetry unit. The magnetic easy axis analysis by Magellan software suggests that the Hdmh ligands along the axis of Dy(III) ion compression increase the magnetic anisotropy of Dy(III) ions for complex 2 in comparison with complex 3. This approach provided a facile way to afford a series of β-diketone mononuclear lanthanide complexes with high anisotropic energy barrier for further study of the magnetism–structure relationship.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21471051 and 21601132) and University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2015113).
References
- S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed.
- G. J. Chen, C. Y. Gao, J. L. Tian, J. Tang, W. Gu, X. Liu, S. P. Yan, D. Z. Liao and P. Cheng, Dalton Trans., 2011, 40, 5579–5583 RSC.
- G. J. Chen, Y. N. Guo, J. L. Tian, J. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem.–Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed.
- G. J. Chen, Y. Zhou, G. X. Jin and Y. B. Dong, Dalton Trans., 2014, 43, 16659–16665 RSC.
- J. Zhu, C. Wang, F. Luan, T. Liu, P. Yan and G. Li, Inorg. Chem., 2014, 53, 8895–8901 CrossRef CAS PubMed.
- Y. Dong, P. Yan, X. Zou and G. Li, Inorg. Chem. Front., 2015, 2, 827–836 RSC.
- Y. Dong, P. Yan, X. Zou, T. Liu and G. Li, J. Mater. Chem. C, 2015, 3, 4407–4415 RSC.
- Y. Dong, P. Yan, X. Zou, X. Yao, G. Hou and G. Li, Dalton Trans., 2016, 45, 9148–9157 RSC.
- Y. L. Wang, C. B. Han, Y. Q. Zhang, Q. Y. Liu, C. M. Liu and S.-G. Yin, Inorg. Chem., 2016, 55, 5578–5584 CrossRef CAS PubMed.
- K. Qian, J. J. Baldoví, S.-D. Jiang, A. Gaita-Ariño, Y.-Q. Zhang, J. Overgaard, B.-W. Wang, E. Coronado and S. Gao, Chem. Sci., 2015, 6, 4587–4593 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- D. C. M. Llunell, J. Cirera, P. Alemany and S. ALvarez, SHAPE, V2.1, University of Barcelona and The Hebrew University of Jerusalem, Barcelona and Jerusalem, 2013 Search PubMed.
- Y. Gao, G. F. Xu, L. Zhao, J. Tang and Z. Liu, Inorg. Chem., 2009, 48, 11495–11497 CrossRef CAS PubMed.
- P. P. Cen, S. Zhang, X. Y. Liu, W. M. Song, Y. Q. Zhang, G. Xie and S. P. Chen, Inorg. Chem., 2017, 56, 3644–3656 CrossRef CAS PubMed.
- Y. Bi, Y. N. Guo, L. Zhao, Y. Guo, S. Y. Lin, S. D. Jiang, J. Tang, B. W. Wang and S. Gao, Chem.–Eur. J., 2011, 17, 12476–12481 CrossRef CAS PubMed.
- D. P. Li, T. W. Wang, C. H. Li, D. S. Liu, Y. Z. Li and X. Z. You, Chem. Commun., 2010, 46, 2929–2931 RSC.
- E. M. Fatila, M. Rouzières, M. C. Jennings, A. J. Lough, R. Clérac and K. E. Preuss, J. Am. Chem. Soc., 2013, 135, 9596–9599 CrossRef CAS PubMed.
- B. Yao, B. Gu, M. Su, G. Li, Y. Ma, L. Li, Q. Wang, P. Cheng and X. Zhang, RSC Adv., 2017, 7, 2766–2772 RSC.
- X.-H. Lv, S.-L. Yang, Y.-X. Li, C.-X. Zhang and Q.-L. Wang, RSC Adv., 2017, 7, 38179–38186 RSC.
- M. Zhu, Y. Li, L. Jia, L. Zhang and W. Zhang, RSC Adv., 2017, 7, 36895–36901 RSC.
- Q. Y. Liu, Y. L. Li, W. L. Xiong, Y. L. Wang, F. Luo, C. M. Liu and L. L. Chen, CrystEngComm, 2014, 16, 585–590 RSC.
- F. Gao, L. Cui, Y. Song, Y. Z. Li and J. L. Zuo, Inorg. Chem., 2014, 53, 562–567 CrossRef CAS PubMed.
- K. L. M. Harriman, J. J. Le Roy, L. Ungur, R. J. Holmberg, I. Korobkov and M. Murugesu, Chem. Sci., 2017, 8, 231–240 RSC.
- R. X. Zhang, Y. X. Chang, H. Y. Shen, W. M. Wang, X. P. Zhou, N. N. Wang, J. Z. Cui and H. L. Gao, Dalton Trans., 2016, 45, 19117–19126 RSC.
- D. Zeng, M. Ren, S. S. Bao, Z.-S. Cai, C. Xu and L. M. Zheng, Inorg. Chem., 2016, 55, 5297–5304 CrossRef CAS PubMed.
- L. Zhang, Y. Q. Zhang, P. Zhang, L. Zhao, M. Guo and J. Tang, Inorg. Chem., 2017, 56, 7882–7889 CrossRef CAS PubMed.
- J. Wu, J. Jung, P. Zhang, H. Zhang, J. Tang and B. Le Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC.
- J. Li, C. Yuan, L. Yang, M. Kong, J. Zhang, J. Y. Ge, Y. Q. Zhang and Y. Song, Inorg. Chem., 2017, 56, 7835–7841 CrossRef CAS PubMed.
- J. Y. Ge, H. Y. Wang, J. Li, J. Z. Xie, Y. Song and J. L. Zuo, Dalton Trans., 2017, 46, 3353–3362 RSC.
- Y. Wang, X.-L. Li, T.-W. Wang, Y. Song and X.-Z. You, Inorg. Chem., 2010, 49, 969–976 CrossRef CAS PubMed.
- U. J. Williams, B. D. Mahoney, P. T. DeGregorio, P. J. Carroll, E. Nakamaru-Ogiso, J. M. Kikkawa and E. J. Schelter, Chem. Commun., 2012, 48, 5593–5595 RSC.
- C. M. Liu, D. Q. Zhang and D. B. Zhu, Inorg. Chem., 2013, 52, 8933–8940 CrossRef CAS PubMed.
- K. R. Meihaus, J. D. Rinehart and J. R. Long, Inorg. Chem., 2011, 50, 8484–8489 CrossRef CAS PubMed.
- Y. N. Guo, G. F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC.
- N. F. Chilton, D. Collison, E. J. McInnes, R. E. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 Search PubMed.
- M. Murugesu, Nat. Chem., 2012, 4, 347–348 CrossRef CAS PubMed.
- S. Zhang, H. Wu, L. Sun, H. Ke, S. Chen, B. Yin, Q. Wei, D. Yang and S. Gao, J. Mater. Chem. C, 2017, 5, 1369–1382 RSC.
- M. Vonci, M. J. Giansiracusa, W. Van den Heuvel, R. W. Gable, B. Moubaraki, K. S. Murray, D. Yu, R. A. Mole, A. Soncini and C. Boskovic, Inorg. Chem., 2017, 56, 378–394 CrossRef CAS PubMed.
- J. Li, R.-M. Wei, T.-C. Pu, F. Cao, L. Yang, Y. Han, Y.-Q. Zhang, J.-L. Zuo and Y. Song, Inorg. Chem. Front., 2017, 4, 114–122 RSC.
- T. Lacelle, G. Brunet, A. Pialat, R. J. Holmberg, Y. Lan, B. Gabidullin, I. Korobkov, W. Wernsdorfer and M. Murugesu, Dalton Trans., 2017, 46, 2471–2478 RSC.
- M. Guo, Y. Wang, J. Wu, L. Zhao and J. Tang, Dalton Trans., 2017, 46, 564–570 RSC.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- J. Tang and P. Zhang, Lanthanide Single Molecule Magnets, Springer, Berlin, Heidelberg, 2015 Search PubMed.
- E. C. Mazarakioti, J. Regier, L. Cunha-Silva, W. Wernsdorfer, M. Pilkington, J. Tang and T. C. Stamatatos, Inorg. Chem., 2017, 56, 3568–3578 CrossRef CAS PubMed.
- E. L. Gavey and M. Pilkington, Coord. Chem. Rev., 2015, 296, 125–152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra, UV spectra, additional magnetic measurements, powder X-ray diffraction, TG-DSC curves, selected bond lengths and calculation details of Cole–Cole plots, coordination geometry of Dy(III) and Arrhenius law for complexes 1–4. CCDC 1515986 1515987 1515850 and 1563201. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09711e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yanping Donga and
Guangming Li
a,
Yanping Donga and
Guangming Li