
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Raman spectro-microscopic investigation of ETFE-based radiation-grafted anion-exchange membranes†
Wai Hin Lee,
Carol Crean ,
John R. Varcoe and
Rachida Bance-Soualhi*
,
John R. Varcoe and
Rachida Bance-Soualhi*
Department of Chemistry, The University of Surrey, Guildford, GU2 7XH, UK. E-mail: r.bance-soualhi@surrey.ac.uk
First published on 11th October 2017
Abstract
This study used Raman spectro-microscopy to investigate the synthesis and degradation of radiation-grafted anion-exchange membranes (RG-AEM) made using 50 μm thick poly(ethylene-co-tetrafluoroethylene) (ETFE) films, vinylbenzyl chloride (VBC) monomer, and 1-methylpyrrolidine (MPY) amination agent. The data obtained confirmed the operation of the grafting-front mechanism. VBC grafting times of 1 and 4 h led to low degrees of grafting homogeneity, while 72 h led to extreme levels of grafting that resulted in mechanically weak RG-AEMs due to the excessive H2O contents. A grafting time of 16 h was optimal yielding a RG-AEM with an ion-exchange capacity (IEC) of 2.06 ± 0.02 mmol g−1 (n = 3). An excess of grafting was detected at the surface of this RG-AEM (at least within the first few μm of the surface). This RG-AEM was then degraded in O2-purged aqueous KOH (1.0 mol dm−3) for 14 d at 80 °C. Degradation was detected throughout the RG-AEM cross-section, where the Raman data was quantitatively consistent with the loss of IEC. A slight excess of degradation was detected at the surface of the RG-AEM. Degradation involved the loss of whole benzyl-1-methypyrrolidinium grafted units as well as the direct attack on the pendent (cationic) pyrrolidinium groups by the hydroxide anions.
Introduction
Radiation-grafting is a useful technique for the production of functional membranes (and other polymeric materials).1 This technique has been commonly used to produce solid electrolytes, e.g. ion-exchange membranes (IEM), including proton-exchange membranes (PEM) for use in technologies such as proton-exchange membrane fuel cells (PEMFCs) and PEM-based water electrolysers.2 A more recent line of research is the development of anion-exchange membranes (AEM) for use in alkaline Anion-Exchange Membrane Fuel Cells (AEMFCs) and AEM-based Alkaline Water Electrolysers (AEM-AWE):3 this includes the use of radiation-grafted (RG) types.4 The use of high pH, hydroxide-conducting solid electrolytes allows for a broader range of electrocatalysts that do not involve scarce, expensive Pt.5 Scheme 1 outlines the steps involved in the synthesis of such a RG-AEM: pre-irradiation in air (peroxidation) of poly(ethylene-co-tetrafluoroethylene) (ETFE) precursor film, followed by grafting with vinylbenzyl chloride (VBC) monomer and amination with 1-methylpyrrolidine (MPY).6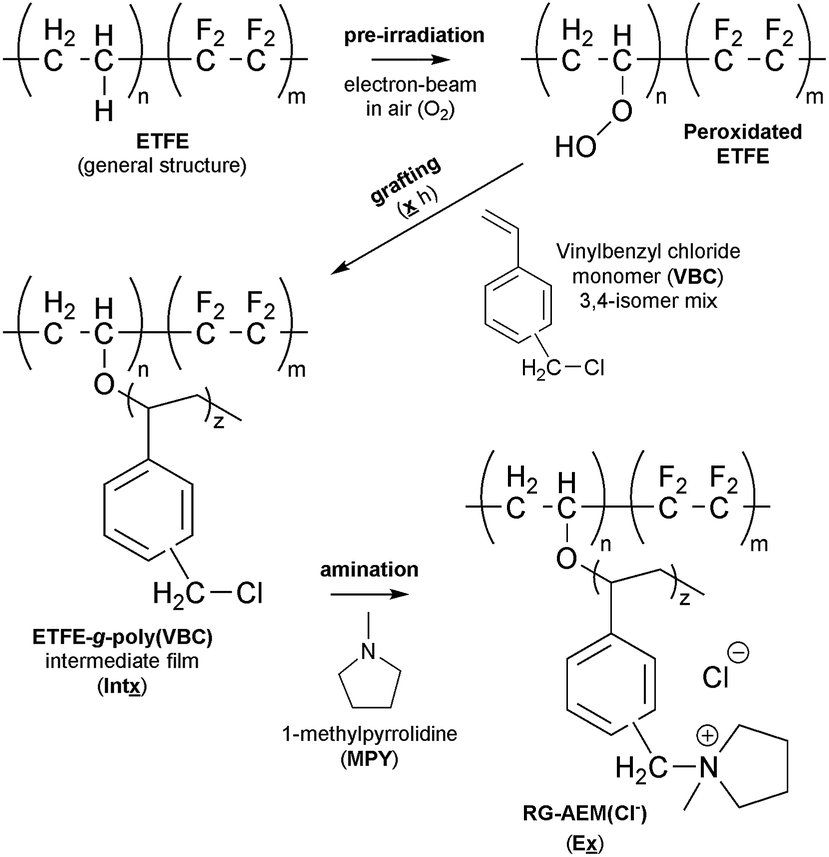 | ||
| Scheme 1 An outline of the synthesis of ETFE-based pyrrolidinium-type radiation-grafted anion-exchange membranes (RG-AEM) in the chloride-anion form. The precursor ETFE film used in this study was 50 μm thick. The main variable in this study was the grafting time (x). | ||
Incomplete grafting can lead to RG-IEMs with lower than ideal functionalisation, which are then too ionically resistive for application. Radiation-grafting often involves the grafting-front mechanism, especially with thicker precursor films:6–8 this is where grafting initiates on the surfaces of the precursor films with the gradual penetration of the grafts into the bulk of the films (Fig. 1). As radiation-grafting involves the modification of solid polymer films (up to 150 μm thickness in the literature),1 it is essential to assess the uniformity of the monomer grafting (and any subsequent functionalisation) throughout the core of the resulting RG-IEMs.9 Techniques that have been used to study the uniformity of grafting include Scanning Electron Microscopy coupled with Energy-Dispersive X-ray Spectroscopy (SEM-EDX)8,10 and Raman spectro-microscopy.6,9,11
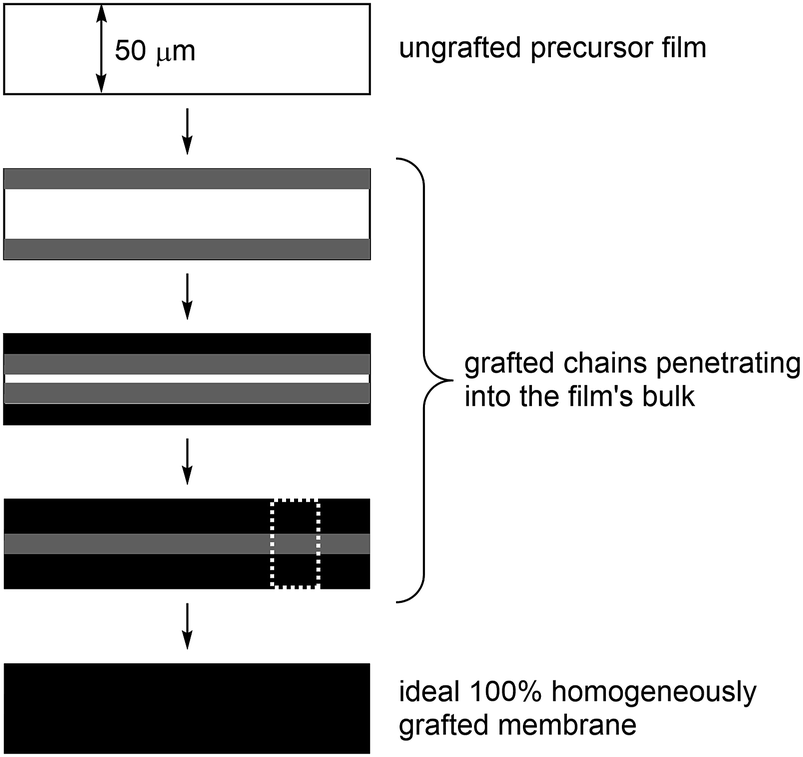 | ||
| Fig. 1 A diagram showing the grafting front mechanism that is commonly encountered in the synthesis of radiation-grafted membranes. The white dashed rectangle indicates a typical Raman spectro-microscopy cross-sectional mapping region (i.e. the 30 × 65 μm areas used in this study). | ||
Raman spectroscopy can achieve spectral resolutions down to ca. 0.3 cm−1 and is a powerful spectroscopy that both complements and supports other spectroscopies (vibrational, rotational, and electronic).12 As Raman spectrometers involve the use of lasers, the spectrometers are commonly coupled to (confocal) microscopic equipment that allows individual spectra to be recorded on different parts of a sample with spatial resolutions as low as ca. 1 μm diameter.13‡ As the cost of Raman instrumentation has generally decreased in the last 10 years§ and the familiarity with the technique has increased, the use of Raman spectro-microscopy is becoming more widespread and routine in the field of materials science. The use of Raman spectro-microscopy often has the advantage of requiring minimal levels of sample preparation. The primary disadvantage is that the quality of spectra is often degraded due to the presence of photoluminescence interferences such as fluorescence. If Raman equipment is available that possesses multiple laser wavelengths (λ), it is often possible to select a laser wavelength that minimises such interferences (but, obviously, this adds to the cost of the equipment).
Given the spatial resolution of Raman spectro-microscopy can be as low as 1 μm during routine measurements (and that previous RG-IEMs reported in the literature are typically up to ca. 150 μm in thickness),1 this technique can be a useful quality assurance tool for the study of the radiation-grafting homogeneity (especially through the membrane cross-section). Raman spectro-microscopic mapping has already been used to study RG-AEMs, but these experiments have typically taken 8–16 h per map (as Raman scattered light is of low intensity):6,9 this is non-ideal considering the life-times of expensive (£10k+) lasers is typically only a few thousand hours (of in use time).
Herein, the aim of this study is to investigate the optimum laser wavelength and spectral collection parameters that will allow for the more rapid and routine use of Raman spectro-microscopy in the study of each stage (grafting, amination) of the synthesis of RG-AEMs. This study will then use this knowledge to conduct an initial investigation into the homogeneity of the degradation of a RG-AEM when it is exposed to aqueous KOH (1.0 mol dm−3) at 80 °C for 14 d.
Experimental
Materials and chemicals
Commercial Nowoflon ET-6235Z ETFE film of 50 μm thickness was supplied by Nowofol Kunstoffprodukte GmbH (Germany) and was used as the precursor material. Vinylbenzyl chloride (VBC, 97%, mixture of 3- and 4-isomers, used without removal of 4-tert-butylcatechol and nitromethane inhibitors), 1-octy-2-pyrrolidone dispersant, and 1-methylpyrrolidine (MPY, 97%) were purchased from Sigma-Aldrich. Standardised analytical aqueous solutions of AgNO3 (20.00 ± 0.06 mmol dm−3) and aqueous HNO3 (2.0 mol dm−3), used for the IEC determinations, were supplied from Fluka. All other chemicals, including analytical grade aqueous solutions of NaCl (1.0 mol dm−3) and KOH (1.0 mol dm−3), toluene, and NaNO3(s) were used as received. Ultrapure water (UPW, resistivity = 18.2 MΩ cm) was used throughout this study.Preparation of the radiation-grafted anion-exchange membranes
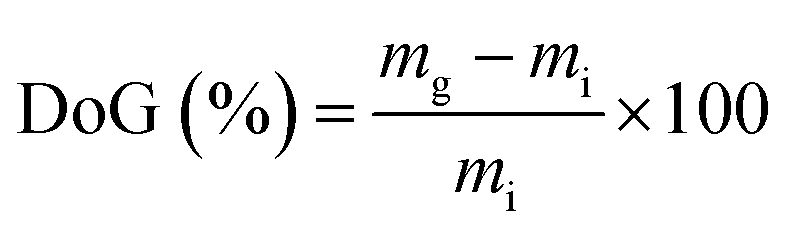 | (1) |
Membrane designations
The following nomenclature is used to unambiguously identify the different samples (see Scheme 1):Ex a final RG-AEM(Cl−) synthesised with x h grafting time;
Intx the pre-aminated ETFE-g-poly(VBC) membranes used to fabricate Ex;
AE16 the E16 RG-AEM(Cl−) that has been aged for 14 d at 80 °C in aqueous KOH (1.0 mol dm−3) solution that has been purged with O2 (see below).
Water uptakes (WU)
RG-AEM(Cl−) samples were removed from the storage water and excess surface water was removed by dabbing with filter paper. The hydrated masses (mhyd) were speedily recorded (to avoid dehydration on prolonged exposure to the atmosphere). The AEM samples were subsequently dried in a vacuum oven at 50 °C for 24 h before the dry masses (mdry) were speedily recorded (dry AEMs are generally hygroscopic). All measurements were repeated on n = 3 samples of each RG-AEM(Cl−). The WU value for each sample was then calculated:
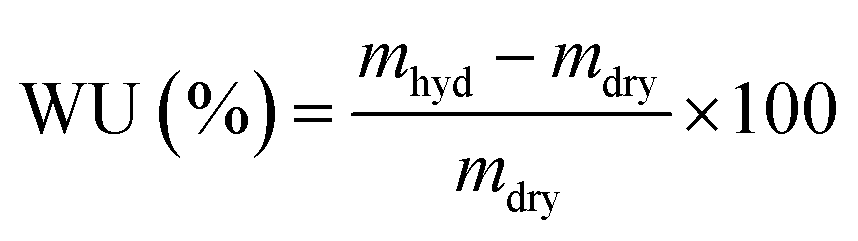 | (2) |
Ion-exchange capacities (IEC)
The dehydrated RG-AEM(Cl−)s samples (mdry/g) collected straight from the WU measurements above were individually immersed into excess aqueous NaNO3 (2.4 mol dm−3, 20 cm3) solutions for 24 h. The solutions, still containing the AEM samples, were then acidified with aqueous HNO3 (2.0 mol dm−3, 2.0 cm3) and titrated using the aqueous AgNO3 (20.00 ± 0.06 mmol dm−3) solution. A Metrohm 848 TitrinoPlus autotitrator equipped with a Ag-Titrode was used for the titrations. The IEC for each sample was calculated from the end-point (Ep):
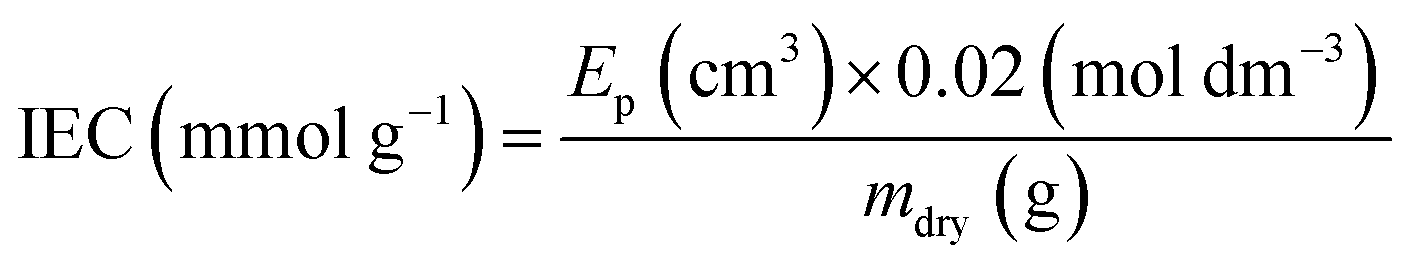 | (3) |
Alkali stability testing
A select RG-AEM(Cl−), synthesised with 16 h of grafting reaction duration (E16), was subjected to alkali aging tests. A sample of E16 (7 × 7 cm) was immersed in an excess of aqueous KOH (1.0 mol dm−3) solution that was then purged with O2 for 1 h. Previous studies have indicated that alkali degradation is more extreme in O2-containing aqueous alkali compared to inert-gas purged aqueous alkali.14 The polypropylene bottle was then immediately sealed (after purging) and placed in oven at 80 °C for 14 d. After this alkali ageing process, the sealed bottle was cooled to room temperature and the aged sample (designated AE16) was then re-exchanged back to the Cl−-anion form using ion-exchange with aqueous solution NaCl (1.0 mol dm−3) followed by thorough washing with UPW for 16 h (with at least 5 changes of fresh UPW during this period). As well as the Raman experiments, the IEC of AE16 was also determined.Raman spectro-microscopic procedures
Raman spectra and cross-sectional maps were recorded using an InVia Reflex Raman Microscope (Renishaw, UK) using 4 laser excitation wavelengths (Table 1).¶ The Raman microscope was fitted with a cooled charged coupled detector (CCD) along with holographic notch filters and gratings tailored for each laser wavelength. The attached Leica DMLM optical microscope was equipped with different objective lenses and a trinocular viewer that accommodates a video camera, allowing direct viewing of the sample. Daily calibration of the instrument was conducted by recording the Raman spectrum of silicon in static mode. If necessary, an offset correction was performed to ensure that the position of the silicon peak to be 520 ± 1 cm−1.| a Theoretical smallest laser spot diameter calculated (1.22 × λ/NA) assuming the use of a 50× (NA = 0.75) objective: the 785 nm line laser was used with the pinhole applied to expose the sample to a well-defined laser spot (rather than a laser line) but this reduces the power at the sample. | ||||
|---|---|---|---|---|
| Wavelength/nm | 785 | 633 | 532 | 457 |
| Max. power/mW | 300 | 20 | 50 | 20 |
| Colour | Near-IR | Red | Green | Blue |
| Type | Line | Spot | Spot | Spot |
| Grating/lines mm−1 | 1200 | 1800 | 2400 | 2400 |
| Laser spot sizea/μm | 1.28 | 1.03 | 0.87 | 0.74 |
| Filter cut-off/cm−1 | 50 | 100 | 100 | 100 |
In this study, the laser beam was focused on the sample using a 50× (NA = 0.75) objective to collect the backscattered light so the resulting laser spot diameter was ca. 1 μm (see Table 1). Other spectral collection parameters are presented in the figure captions (as appropriate). The raw data was processed using Renishaw's Wire 4.3 software: processing included baseline correction, smoothing, normalization, interpolation, and curve fitting of the peaks with the Gaussian–Lorentzian function to obtain accurate peak areas (where appropriate).
For the cross-sectional (through-plane) mapping experiments (Fig. 1), the membrane samples were held and pressed between two metal holders and fresh edges were obtained by sectioning using a scalpel. The membrane samples were mounted perpendicular to the sample-stage so that the exposed cross-sections are parallel to the stage. The stage was moved in the x- and y-direction under computer control. Cross-sectional maps were collected with point mapping using the instrument's static scan mode over the spectral range 600–1700 cm−1. Maps were recorded over 30 × 65 μm cross-sectional areas (all the same so that the experiments could be timed).
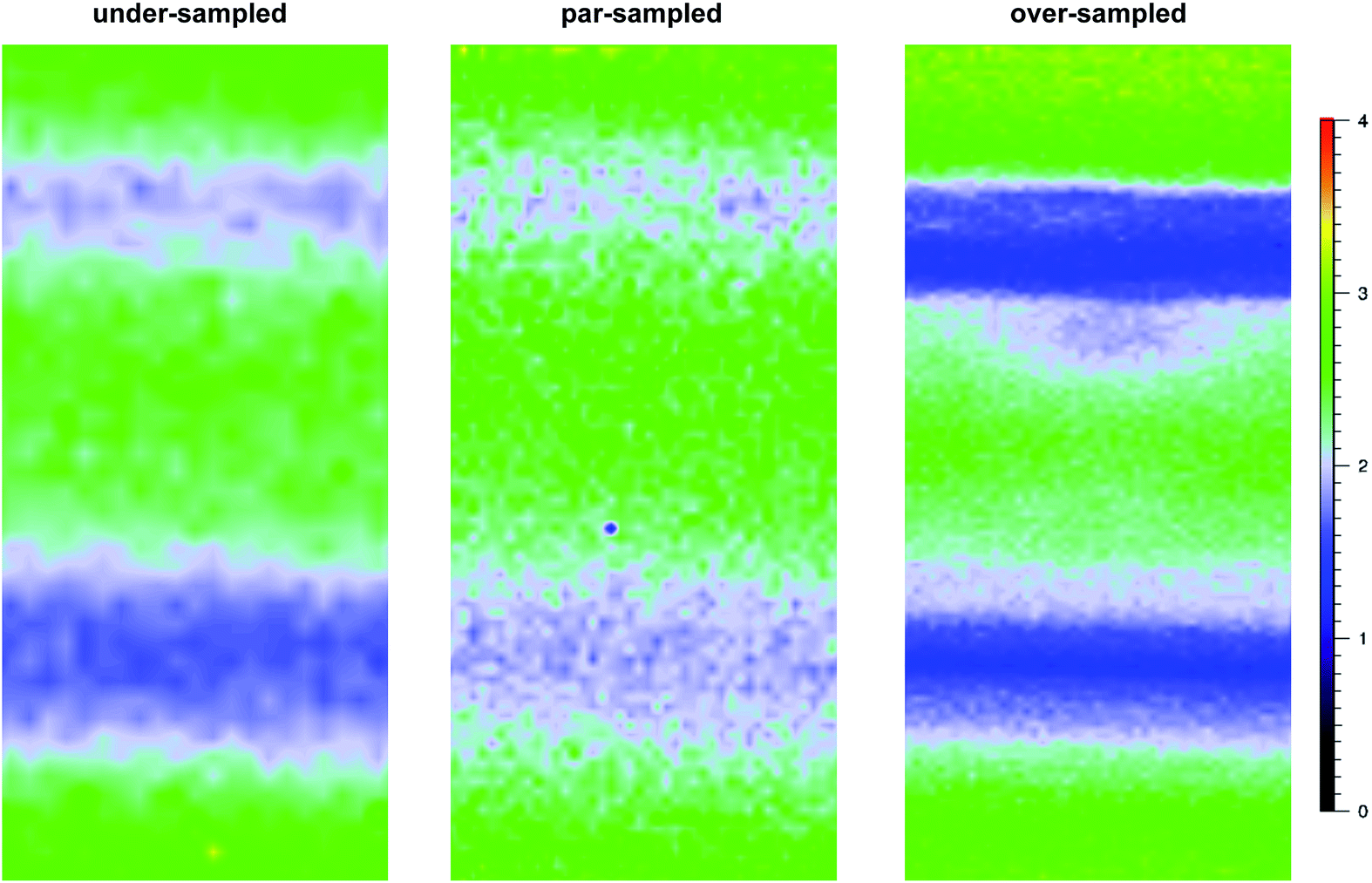
For the pre-aminated ETFE-g-poly(VBC) membranes, the optimised mapping parameters used are presented in Table 2 (for par-sampling where sample step size = theoretical min. laser spot diameter). Maps with different sample step sizes were also performed on a sample of Int4 to determine an appropriate step size to be used for mapping experiments (Fig. 2): both under-sampling (step size > theoretical min. laser spot diameter) and over-sampling (step size < theoretical min. laser spot diameter) were studied.
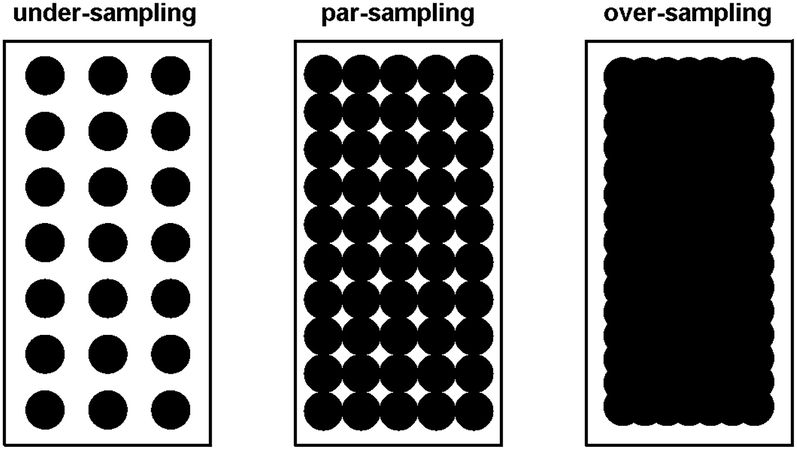 | ||
| Fig. 2 A diagram summarising the different sampling modes used in the Raman mapping experiments. | ||
For the final E16 and the alkali-aged AE16 samples, cross-sectional maps were collected using the 785 nm line laser (pinhole applied, 300 mW power) with a step size of 1.28 μm (par-sampling) in both the x- and y-directions. These spectral maps were collected using a 50× objective, 5 s exposure times, and averaging over 2 accumulations (mapping time of 220 min).
Results and discussion
Membrane characterisation
A series of pyrrolidinium-based RG-AEM(Cl−)s with various degrees of grafting (DoG) were prepared via the peroxidation radiation-grafting of VBC monomer onto ETFE films followed by amination with 1-methylpyrrolidine (MPY) (Scheme 1). The IECs and WUs of the AEMs are presented in Table 3 (Fig. S2 in the ESI† shows the variation in IEC vs. DoG for the RG-AEM(Cl−)s). The IECs of E16 and E72 were 80% of the theoretical IECs (calculated from DoG) due to side-reactions as explained previously (e.g. conversion of –CH2Cl to –CH2OH).9 The WU is related to the number of ion-exchange sites. As expected, the WUs of the AEM(Cl−)s increased with IEC (Fig. S2 in the ESI†), due to the higher hydrophilic content levels. The IEC and WU values obtained for RG-AEM(Cl−)s are bulk averages and do not give any information on the spatial distribution of the grafted quaternary ammonium groups. This is why techniques, such as Raman microscopy, are required to evaluate such micro-phase segregation in IEMs, e.g. the RG-AEMs in this study.||| a Number of H2O molecules per Cl− anion [not to be confused with laser wavelength λ], calculated as: λwater = WU (%)/(100 × 18.02 × IEC), where IEC is in mol g−1. | ||||
|---|---|---|---|---|
| RG-AEM(Cl−)s | E1 | E4 | E16 | E72 |
| Grafting time/h | 1 | 4 | 16 | 72 |
| DoG (%) | 18 | 44 | 99 | 155 |
| IEC (Cl− anion)/mmol g−1 | 0.91 ± 0.05 | 1.70 ± 0.06 | 2.06 ± 0.02 | 2.36 ± 0.07 |
| WU (%) | 8 ± 3 | 79 ± 5 | 104 ± 7 | 145 ± 6 |
| λwatera | 5 | 26 | 28 | 34 |
Raman spectroscopy to elucidate optimal laser wavelength
A vital consideration for Raman spectro-microscopy is the selection of laser excitation wavelength (λ).15 This choice is not always straightforward and must balance the following requirements:• Maximised intensity of the Raman scattered radiation to allow for more rapid collection of spectral data: this is dictated (at each wavelength) by the Raman efficiency and the highest level of laser power that can be applied that does not cause sample damage;
• Minimised level of fluorescence to facilitate spectral data processing and to ensure all relevant peaks are observed;
• Maximised spectral resolution to ensure adequate resolution of overlapping peaks;
• Ability to obtain the spatial resolution required, which is partially controlled by the laser spot diameter obtainable (Table 1): the minimum Airy disk diameter = 1.22 × λ/NA (where NA is the aperture of the objective being used).13
The intensity of Raman scattering is proportional to λ−4,16 hence, a shorter wavelength will yield a stronger Raman signal: the intensity of the scattered radiation with a 633 nm laser is only 27% of the intensity obtained with a 457 nm laser. For the higher wavelength laser, this intrinsic lower intensity can often be offset by increasing the laser power as long as sample damage does not occur (higher wavelength radiation yields less J of energy per photon). Fluorescence is commonly a decisive factor and arises from the coincidence of the excitation energy with electronic transitions. A way of avoiding this problem is to choose a longer wavelength (e.g. near-IR) with the compromise of lower spatial resolutions, or to move to UV wavelengths with the compromise of lower spectral resolutions and with the increased risk of sample damage occurring. Finally, the spatial resolution is especially important for point mapping, where a small laser spot diameter is often required.
Fig. 3 displays the Raman spectra of the Int16 membrane at the four-different wavelengths both with and without baseline correction and normalisation. We selected Int16 for this initial laser λ study as this pre-aminated membrane visually showed evidence (increase in opacity) of a high level of grafting. The use of 457 and 532 nm lasers led to moderate fluorescence, whereas the 633 and 785 nm lasers yielded spectra with minimal fluorescence. After baseline correction and normalisation, a number of Raman peaks were obtained with enhanced intensities with the 457 nm laser (especially the aromatic peak at 1612 cm−1) indicating an intriguing but small Raman enhancement effect. Therefore, the 457 and 633 nm spot lasers were down-selected for the characterisation of pre-aminated ETFE-g-poly(VBC) membranes using Raman cross-sectional mapping. The 457 nm laser could be used up to 10% maximum power before sample damage occurs, while the 633 nm laser could be used at maximum power.
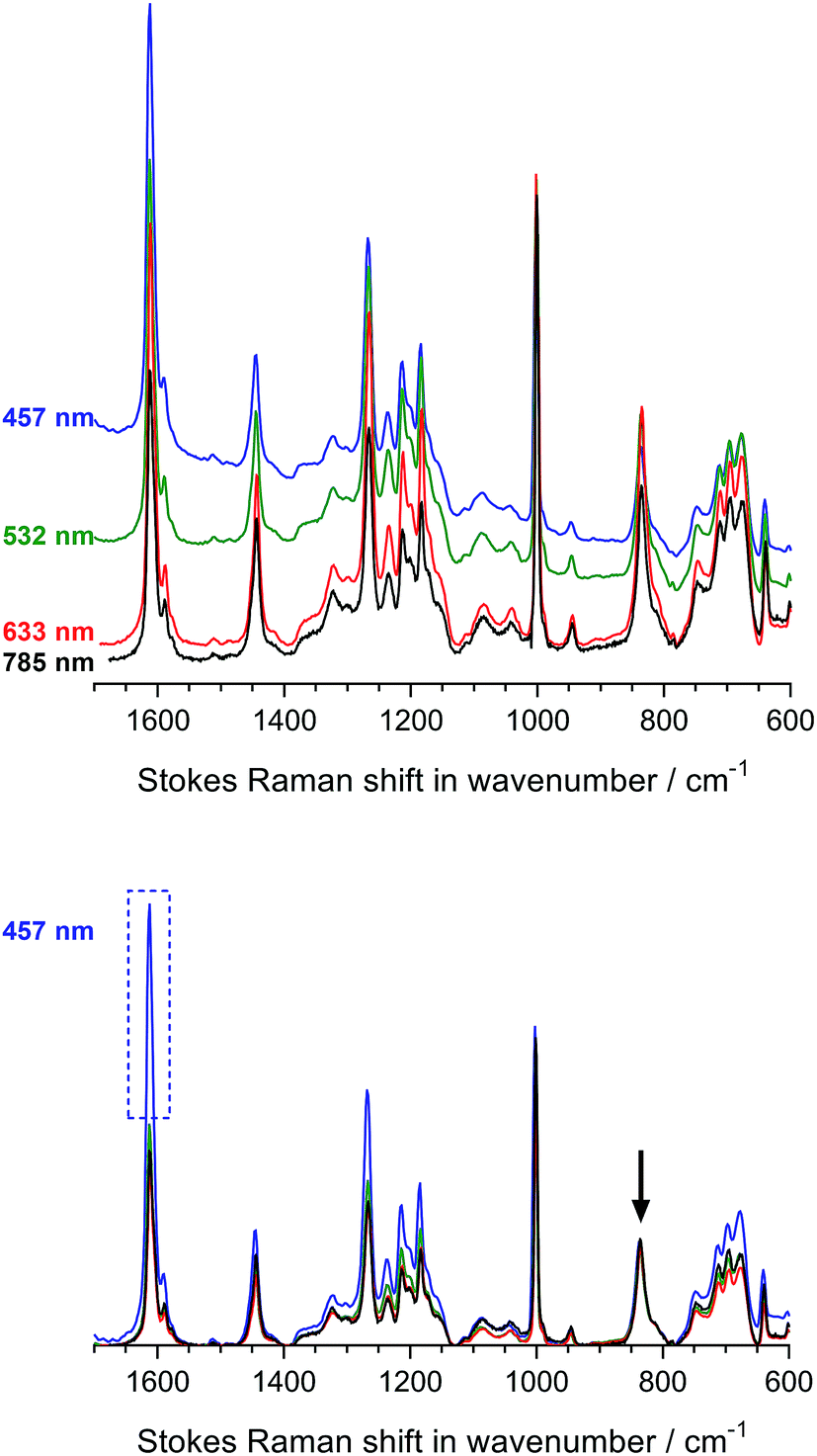 | ||
| Fig. 3 The Raman spectra of Int16 both with (bottom graph) and without (top graph) baseline correction and normalisation. The baseline-corrected spectra were normalised to the intensity of the CF2 stretch peak at 835 cm−1 peak to aid visual comparison (the peak indicated by arrow). The enhancement in the 1612 cm−1 peak when using the 457 nm laser is shown by the dashed box. | ||
Regarding the (orange-brown coloured) RG-AEM(Cl−)s, the Raman spectra recorded using 457 and 532 nm lasers showed excessively strong fluorescence backgrounds that could not be reliably subtracted (see Fig. S3 in the ESI†). In contrast, the 785 nm laser yielded spectra with lower levels of baseline noise. Furthermore, it was impossible to record adequate spectra with the dark brown AE16 sample with all but the 785 nm laser due to the very high levels of fluorescence observed. The 785 nm laser proved to be the only available option that was suitable for recording spectral maps of both the pre- and post-alkali-treated RG-AEMs. On the downside, the use of this longer wavelength laser (with pinhole applied to make it a spot laser) gave weak signals and so longer acquisition times were required (even when using maximum power).
Chemical characterisation using Raman spectroscopy
The chemical structures of the pristine ETFE along with Int16, E1, E4, E16, and E72 were evaluated using Raman spectroscopy on the surfaces of the membranes (Fig. 4). The ETFE spectrum is dominated by the presence of two sharp peaks: CF2 stretches at 835 cm−1 (ref. 17) and CH2 bends at 1444 cm−1. The grafting of the VBC onto the peroxidated ETFE film introduced new peaks at 1612, 1266, and 1000 cm−1,** which correspond to the aromatic ring breathing, CH2Cl deformations,6,9 and meta-disubstituted benzene rings,18 respectively. The spectra of RG-AEM(Cl−)s reveals that the CH2Cl peak at 1266 cm−1 (* in Fig. 4) disappears indicating successful amination with the MPY. In addition, a new band at 900 cm−1 was observed, which derives from the presence of cationic pyrrolidinium groups. All this data corroborates the Raman data previously reported for ETFE-MPY-based RG-AEMs.6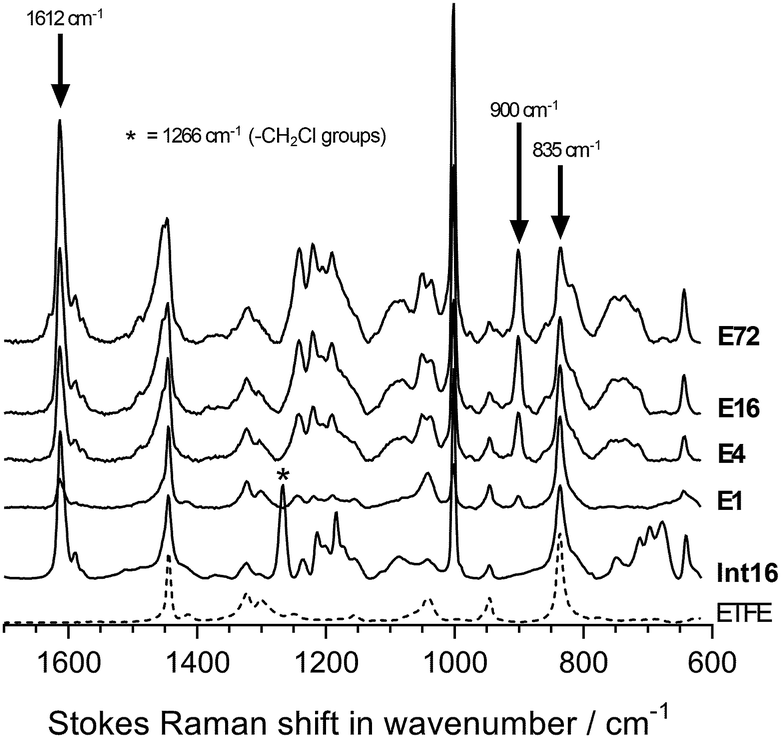 | ||
| Fig. 4 The Raman spectra of (from bottom to top): the pristine ETFE, Int16, E1, E4, E16 and E72. Spectra were recorded with a 785 nm laser at 300 mW power (the only laser that could be used for all samples). The spectra were normalised to the intensity of ETFE peak at 835 cm−1 to aid visual comparison. The arrows indicate the key peaks used for the Raman cross-section mapping experiments. | ||
Raman mapping of the pre-aminated ETFE-g-poly(VBC) membranes
Raman mapping was first performed on cross-sectional samples of the pre-aminated ETFE-g-poly(VBC) membranes to measure the through-plane distributions of the poly(VBC) grafts (Fig. 1). This was assessed using the peak area ratio A1612/A835: this is the area of the 1612 cm−1 aromatic peak (deriving from the grafted poly(VBC) chains) vs. the area of the 835 cm−1 peak (deriving from the backbone of the ETFE films). Peak areas were calculated by curve fitting using the Gaussian–Lorentzian function (after the spectra had been baseline corrected).Fig. 5 shows the interpolated Raman maps obtained using the 457 nm laser for cross-sections of Int4 with the following stage (x–y) step sizes: 0.5 μm (over-sampling), 0.74 μm (par-sampling) and 1.5 μm (under-sampling). Recall, Fig. 2 schematically highlights the difference between these sampling modes. Note, these maps were produced from different cross-sections of Int4, and so the grafting distributions show a natural level of variation. For Int4, the distribution of poly(VBC) grafts shows more intensity at the surface and the middle of the membranes with bands of slightly lower intensity separating these regions. It is hypothesised that this is an effect of the diffusion of monomer. The monomer fronts diffuse from both surfaces of the ETFE film to finally meet in the middle, which increases the concentration of monomer at this location: this leads to more rapid grafting for a period of time in the centre of the membrane. This effect was previously seen in a study that used propan-2-ol in the grafting medium,6 rather than the water used in this current study.
 | ||
| Fig. 5 Raman cross-sectional interpolated maps of Int4 recorded using the 457 nm laser (2 mW power at sample) and sample-stage step-sizes of: (left) under-sampled 1.5 μm; (middle) par-sampled 0.74 μm; and (right) over-sampled 0.5 μm. The colour scale represents the peak area ratio = A1612/A835 (content of benzene rings vs. ETFE) where Ay is the area under the peak located at wavenumber = y cm−1. Each map is 30 × 65 μm in size (y-axis is the through-plane direction). | ||
The data recorded using over-sampling demonstrated a higher fidelity Raman image, at the expense of experiment duration: map time was 220 min (compared to 30 min for the under-sampled image). A step size of 0.74 μm (par-sampling) was chosen for this study as it balances image fidelity with measurement time (map time of 110 min). Despite this, under-sampling may still be useful if a future necessity arose that required regularly conducted quality assurance measurements on a large number of samples (e.g. routine checks for homogeneous grafting of multiple batches of RG-AEM) or if measurements on larger cross-sectional areas of individual samples were desired: the fidelity of the under-sampled maps is still adequate for such tasks.
The Raman maps collected with the 633 nm laser are presented in Fig. S5 in the ESI:† with this laser, faster mapping times of 20 min, 40 min, and 76 min were obtained with under- (1.50 μm step-size), par- (1.03 μm), and over-sampling (0.74 μm), respectively. These mapping times were quicker as the 633 nm laser could be applied at higher powers (at the sample): 20 mW compared to the maximum 2 mW that could be used with the 457 nm laser (due to sample damage at higher powers). Par-sampling (stage step size of 1.03 μm) was again down-selected to map the distribution of the grafted poly(VBC) chains with the 633 nm laser.
Fig. 6 shows cross-sectional maps (457 nm, par-sampled) obtained with the pre-aminated ETFE-g-poly(VBC) membranes synthesised using increasing grafting reaction times (Int1, Int4, Int16, and Int72). Fig. S4 in the ESI† gives the box and whisker plots that summarises this data for all the pre-aminated ETFE-g-poly(VBC) membranes studied. For additional clarity, Fig. 7 presents line-map data (457 nm) taken from the centres of the cross-sectional maps.
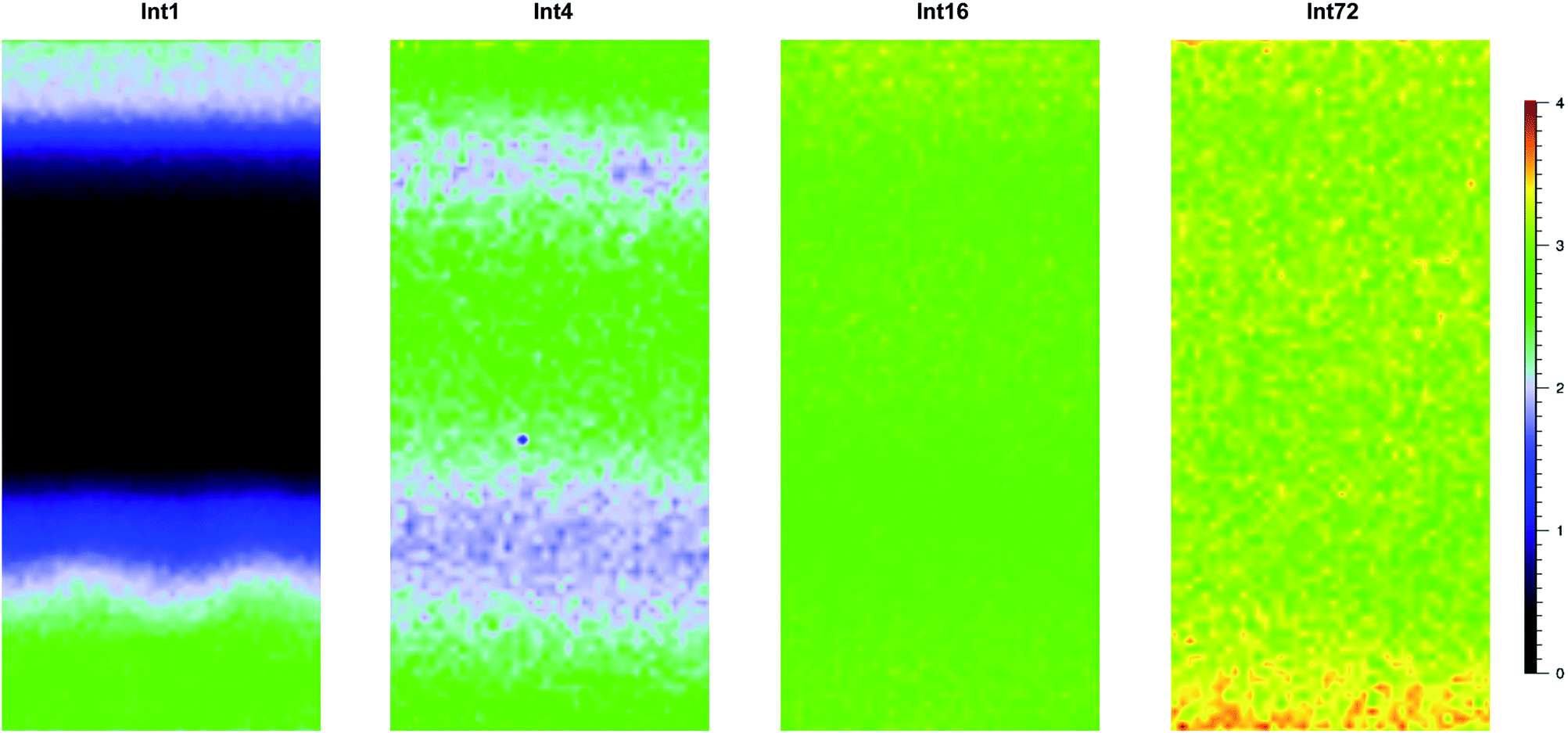 | ||
| Fig. 6 Raman cross-sectional interpolated maps recorded using the 457 nm laser (2 mW power at sample) with par-sampling of (from left to right): Int1, Int4, Int16, and Int72. The colour scale represents the peak area ratio = A1612/A835 (content of benzene rings vs. ETFE) where Ay is the area under the peak located at wavenumber = y cm−1. Each map is 30 × 65 μm in size (y-axis is the through-plane direction). | ||
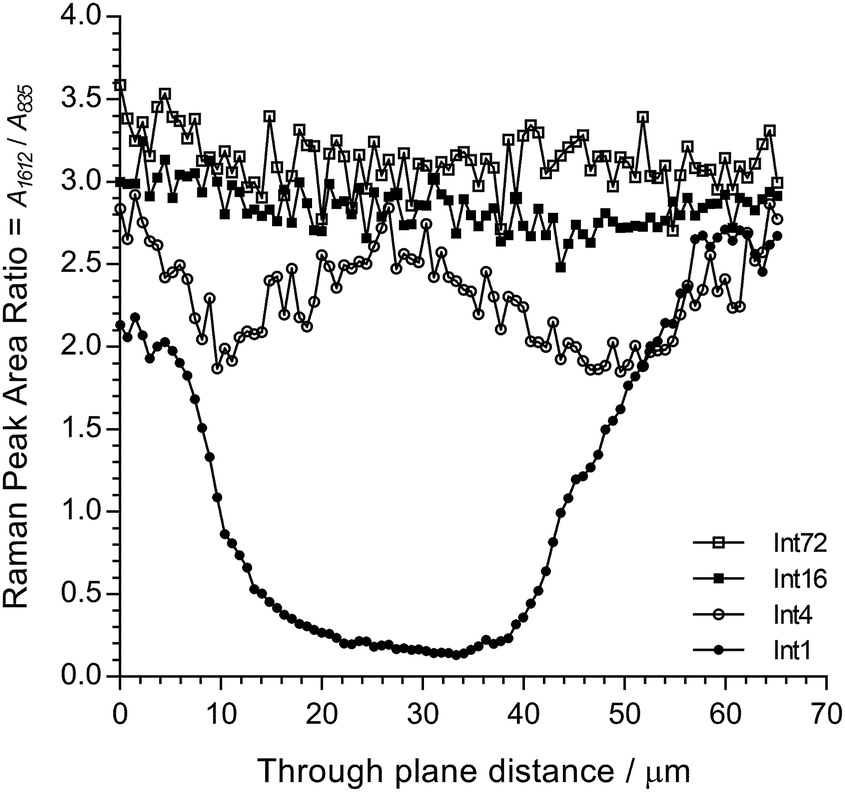 | ||
| Fig. 7 Line-map data for Int1, Int4, Int16, and Int72 taken from the centres of the cross-sectional maps presented in Fig. 6 (in the through-plane y-axis direction). | ||
From this Raman data, it was observed that Int1 (with the lowest DoG = 18%) contained poly(VBC) grafted chains at the surface and a relatively ungrafted core. Increasing the grafting time to 4 h resulted in a wider through-plane distribution of grafting in the resulting membrane (Int4), but the grafting distribution was still not fully homogeneous (discussed previously). As the DoG approaches a value of 99% (Int16), a more homogeneous grafting of the poly(VBC) chains was achieved. The 4-probe (in-plane) chloride-anion conductivities of fully hydrated E1, E4, and E16 (measured using the method described in ref. 9) were 14 ± 3, 39 ± 2, and 49 ± 2 mS cm−1 (n = 3), respectively, which is consistent with this Raman data.
Prolonging the grafting time to 72 h (Int72, DoG = 155%) led to higher grafting levels but this has detrimental consequences: e.g. E72 was observed to possess an excessive water content with λwater > 30 (Table 3), where λwater is the average number of H2O molecules per anion-exchange site. Prior studies have shown that not all of the H2O molecules are directly associated with the ionic groups: many H2O molecules are “non-useful” in that they form domains of water aggregates that lead to excessive RG-AEM swelling, which results in a weakening of the mechanical properties.19 Table 4 gives the results of a statistical analysis comparing the poly(VBC) grafting levels between Int16 and Int72 using all of the A1612/A835 peak area ratios recorded in the production of their Raman maps in Fig. 6. Int72 has a (statistically significant) higher amount of grafting than Int16.
| a Test for normality.b Non-parametric two-tailed Mann–Whitney U test. | ||
|---|---|---|
| ETFE-g-poly(VBC) | Int16 | Int72 |
| Number of measurements | 3738 | 3738 |
| Mean | 2.83 | 3.13 |
| Standard deviation | 0.13 | 0.16 |
| Relative standard deviation RSD (%) | 4.6 | 5.1 |
| Confidence intervals | ±0.005 | ±0.005 |
| D'Agostino–Pearson omnibus test:a p= | <0.0001 | <0.0001 |
| Passed normality test (α = 0.05)? | No | No |
| Mann–Whitney U test:b p= | <0.0001 | |
| Means significantly different? | Yes | |
All of the above indicates that the grafting front mechanism is operating when VBC is grafted onto 50 μm thick ETFE films. Additional maps, acquired using the 633 nm laser on different cross-sections of the pre-aminated ETFE-g-poly(VBC) membranes (Fig. S6 in the ESI†), confirm these observations. This Raman data is consistent with the bulk IEC values recorded for the final RG-AEM(Cl−)s that were made by the amination of Int1, Int4, Int16, and Int72: IECs increase in the order E1 < E4 < E16 < E72. However, this Raman microscopy data provides additional evidence that the homogeneity of grafting is poor at low grafting reaction times.
Raman studies on a RG-AEM(Cl−): pre- and post-alkali-aged E16
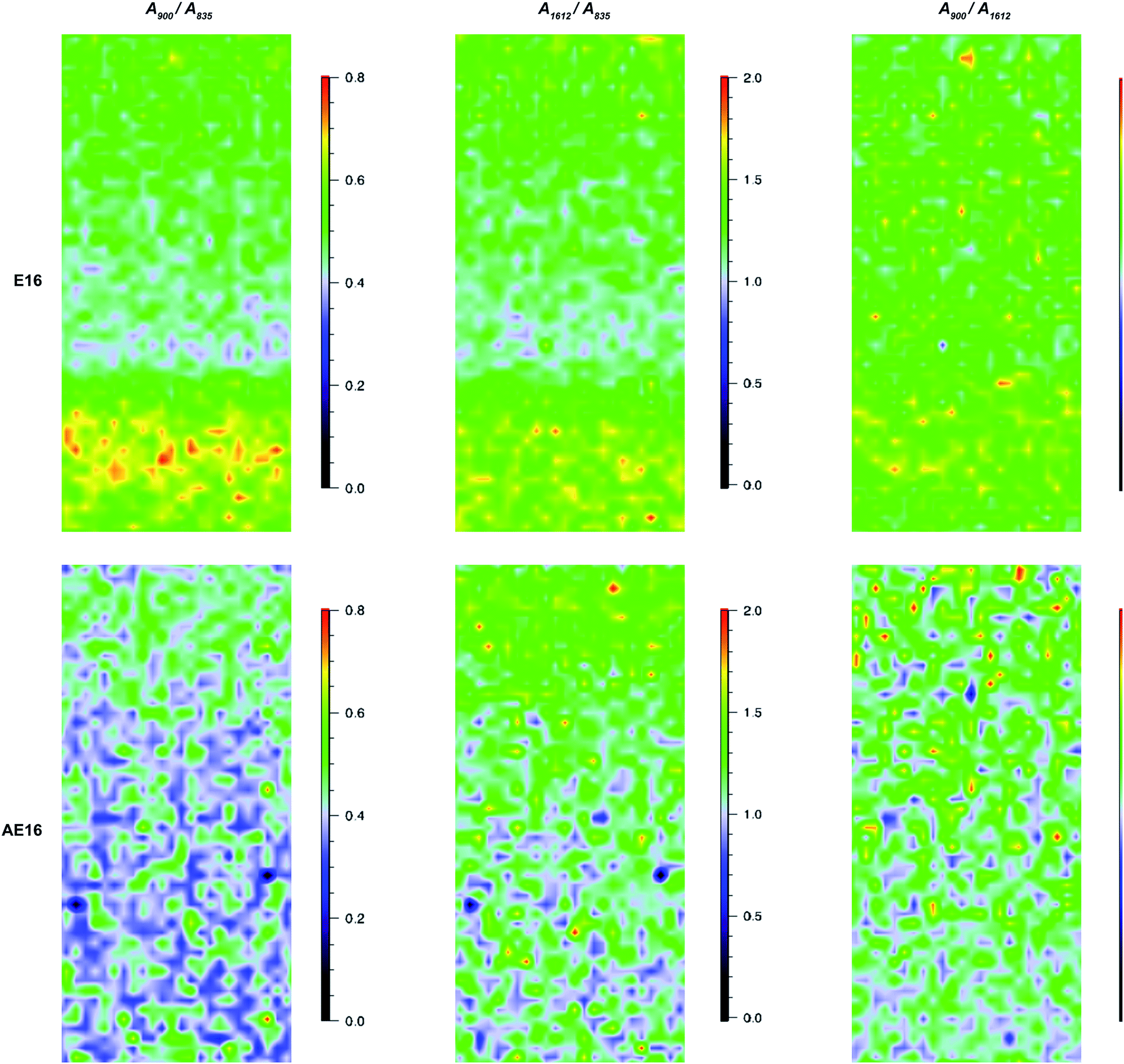 | ||
| Fig. 8 Raman cross-sectional interpolated maps of E16 (top row) and AE16 (bottom row) recorded using the 785 nm laser (300 mW power, pinhole applied) with par-sampling. The colour scale represents (from left to right columns): A900/A835 (pyrrolidinium vs. ETFE); A1612/A835 (benzene vs. ETFE); A900/A1612 (pyrrolidinium vs. benzene). Each map is 30 × 65 μm in size (y-axis is the through-plane direction). | ||
(1) Ratio = A900/A835: pyrrolidinium groups vs. ETFE;
(2) Ratio = A1612/A835: benzene rings vs. ETFE;
(3) Ratio = A900/A1612: pyrrolidinium groups vs. benzene rings;
(1) and (2) represent the homogeneity of grafting and (3) represents the homogeneity of amination of the benzene rings.
For the pre-alkali-aged E16, it was evident that functionalisation was less homogeneous compared to the grafting studies on Int16. This may be due to segregation of the hydrophilic and hydrophobic phases in the RG-AEM sample that occurs after the amination process.†† The obvious “health warning” must also be kept in mind in that the apparent lower homogeneity may be down to sampling: the specific cross-sectional sample of E16 that was mapped could have been a low homogeneity section of the RG-AEM. Fig. 9 gives the box and whisker plots for data extracted from the top row of Fig. 8.
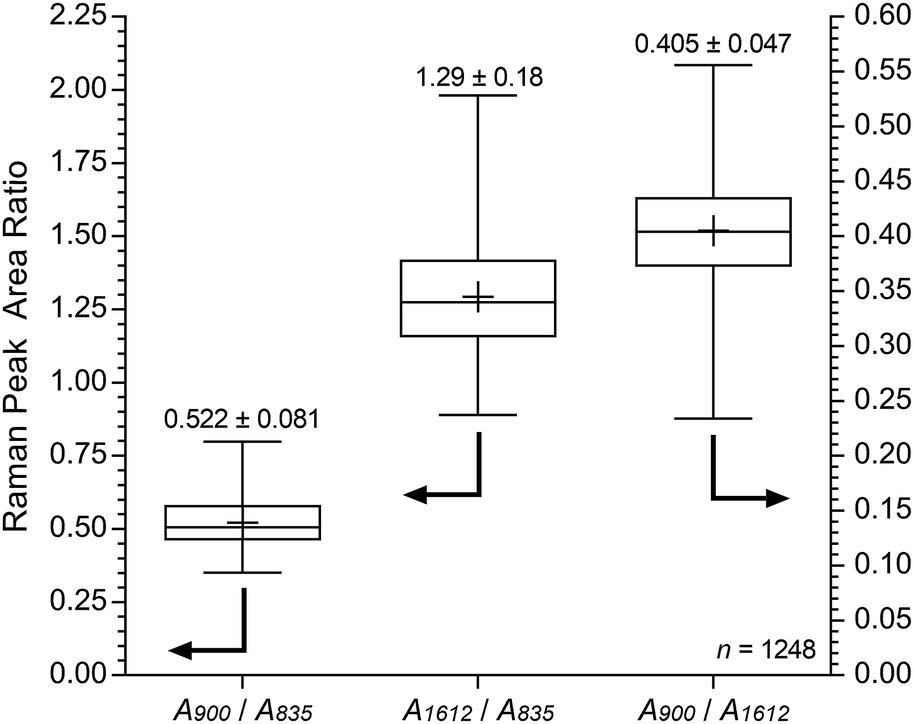 | ||
| Fig. 9 Box and whisker plot summarising the cross-sectional Raman spectral data (785 nm laser) for E16 (extracted from Fig. 8). The + symbols give the means, the middle horizontal lines give the medians, the boxes give the interquartile ranges, and the whiskers give the min and max values recorded. The numeric data presented gives the mean and sample standard deviations (n = 1248). | ||
The data on the alkali degraded AE16 (bottom row of Fig. 8) clearly shows that degradation is evident when the E16 was aged in O2-purged aqueous KOH (1.0 mol dm−3) at 80 °C for 14 d. This data also shows that the degradation occurs throughout the cross-section of the RG-AEM. Fig. S7 in the ESI† presents the box and whisker plots comparing the peak area ratios for E16 and AE16 (extracted from the Raman cross-sectional map data presented in Fig. 8). The larger variances seen in the AE16 data is due to the difficulty in reliably fitting all of the spectral data due to the significant fluorescence backgrounds encountered. Statistical analysis of the peak area ratio A900/A835 data (the ratio expected to change the most on RG-AEM degradation) is summarised in Table 5. This analysis shows that this peak area ratio data is significantly different for AE16 compared to E16 due to alkali degradation. The degradation observed was due to both: (1) loss of the positively-charged pyrrolidinium functionality (Scheme 2: nucleophilic hydroxide attack positions A, B, and C and Hofmann elimination attack position E); (2) loss of grafted benzene rings from the RG-AEM, complete with pyrrolidinium groups (Scheme 2: hydroxide attack position D). This is consistent with the observations reported in recent studies on both ETFE- and LDPE-based RG-AEMs.6,14
| a Test for normality.b Non-parametric two-tailed Mann–Whitney U test. | ||
|---|---|---|
| E16 | AE16 | |
| Number of measurements | 1248 | 1248 |
| Mean | 0.522 | 0.415 |
| Standard deviation | 0.081 | 0.076 |
| RSD (%) | 16 | 18 |
| Confidence intervals | ±0.005 | ±0.004 |
| D'Agostino–Pearson omnibus test:a p= | <0.0001 | <0.0001 |
| Passed normality test (α = 0.05)? | No | No |
| Mann–Whitney U test:b p= | <0.0001 | |
| Means significantly different? | Yes | |
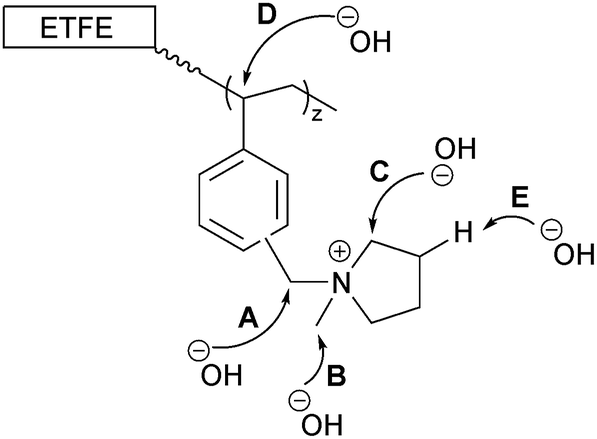 | ||
| Scheme 2 The possible sites that nucleophilic OH− anions can attack the grafted chains in RG-AEMs.6 | ||
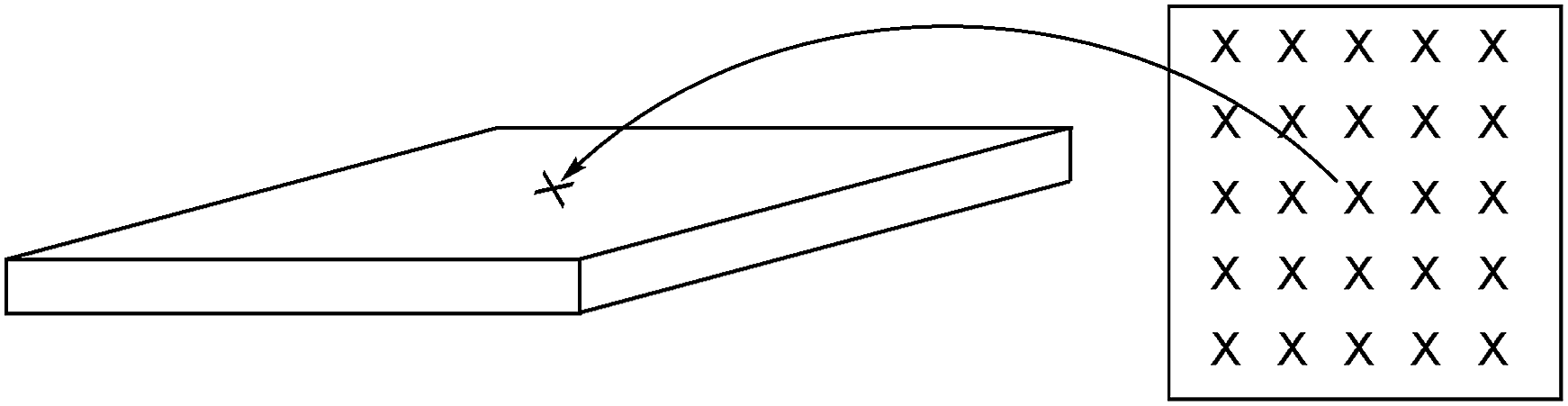 | ||
| Fig. 10 A diagram showing the sampling of the Raman spectra recorded on the surfaces of E16 and AE16 (ca. 1 cm2 samples in this study). | ||
Fig. 11 shows the results recorded on the surface of a sample of E16. This data shows good homogeneity with RSDs < 10% for all three peak area ratios. However, this Raman data shows that the surface analysis of E16 yields larger area ratios A900/A835 and A1612/A835, that are related to the level of grafting, compared to the cross-sectional analysis (Fig. 9). This suggests there is an excess of grafting directly at the surface of E16 (where the laser only penetrates a couple of μm into the membrane when focused on the surface). Therefore, for RG-AEMs, we strongly recommend that surface data should always be collected alongside cross-sectional mapping data (to obtain the fullest possible picture of the distribution of the grafted chains).
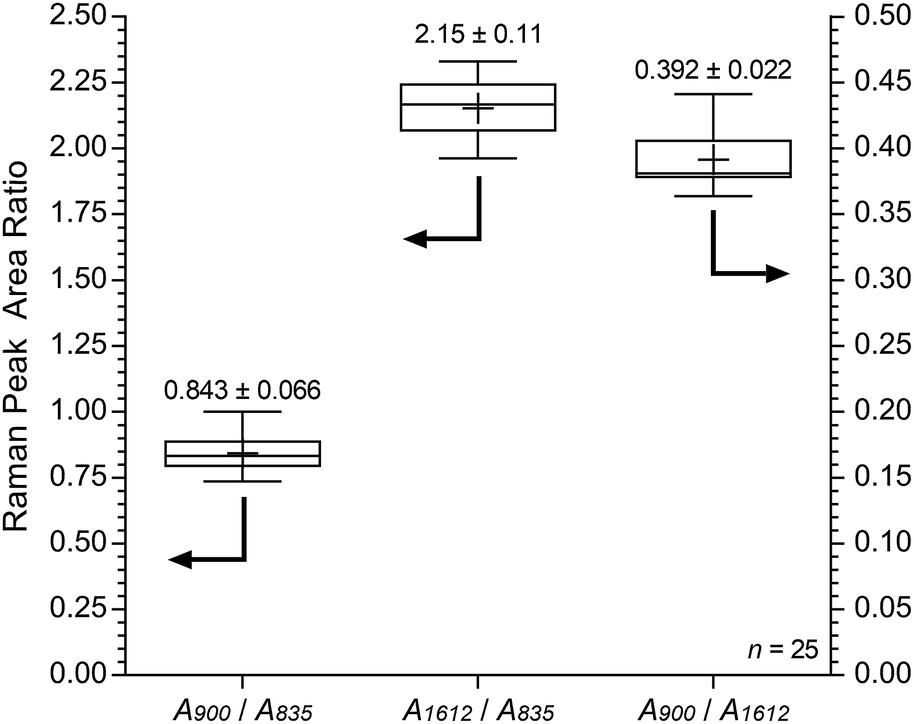 | ||
| Fig. 11 Box and whisker plot summarising the surface Raman spectral data (785 nm laser) for E16. The numeric data presented gives the mean and sample standard deviations (n = 25). | ||
Fig. S8 in the ESI† presents the box and whisker plots comparing the peak area ratios from the surface analyses of E16 and AE16. Statistical data for the peak area ratio A900/A835 is summarised in Table 6. This analysis again shows that this diagnostic peak area ratio is significantly different for AE16 compared to E16 due to alkali degradation. There was a 25% loss of this peak area ratio on surface degradation, while only a 21% loss was observed in the analysis of the cross-sectional data. It is not unexpected that degradation would be more severe at the surface of RG-AEMs. AE16 possessed an IEC of 1.65 ± 0.17 mmol g−1 (n = 3), which was 20% lower than the 2.06 ± 0.02 mmol g−1 recorded for the pre-alkali treated E16. This (bulk) IEC data directly correlates with the loss of the peak area ratio A900/A835 in the Raman cross-sectional data.
| a Test for normality.b Un-paired two-tailed t test with Welch's-correction (due to non-equal variances). | ||
|---|---|---|
| E16 | AE16 | |
| Number of measurements | 25 | 25 |
| Mean | 0.843 | 0.63 |
| Standard deviation | 0.066 | 0.15 |
| RSD (%) | 7.9 | 24 |
| Confidence intervals | ±0.027 | ±0.063 |
| D'Agostino–Pearson omnibus test:a p= | 0.512 | 0.407 |
| Passed normality test (α = 0.05)? | Yes | Yes |
| F-test: p= | 0.0001 | |
| Variances statistically different? | Yes | |
| t-test:b p= | <0.0001 | |
| Means statistically different? | Yes | |
Conclusions
This study highlights the usefulness of using Raman spectro-microscopy in the study of radiation-grafted membranes (anion-exchange membranes in this case). This technique is especially helpful for the study of the homogeneity of the cross-sectional grafting (on the micro-scale). This technique has the potential to be used as a quality assurance tool regarding the grafting process (with cross-sectional mapping times of <1 h being possible with under-sampling). This technique can also be useful for the study of the homogeneity of any post-grafting functionalisation process (e.g. amination) as well as for the (post-mortem) investigation of membrane degradation processes. For radiation-grafted membranes, the analysis of the surfaces must always be conducted in addition to any cross-sectional analysis, in order to obtain the whole picture regarding the distribution of membrane functionality (and membrane degradation).For the radiation-grafted anion-exchange membranes (RG-AEM), made from 50 μm thick ETFE films in this investigation, a vinylbenzyl grafting time of 16 h appears to be optimal (with the specific synthesis conditions used). The Raman data is consistent with the operation of the grafting front mechanism. Amination with 1-methylpyrrolidine was homogeneous and yielded RG-AEMs with ion-exchange capacities (IEC) of >2.0 mmol g−1. There was an excess of grafting at the surfaces of the RG-AEMs (at least in the first few μm). The level of degradation, measured using Raman spectro-microscopy of the RG-AEM cross-sections after ageing in hot aqueous alkali, matched the loss of IEC; however, the high levels of photoluminescence that occurs with the (dark-brown-coloured) alkali degraded RG-AEMs led to more problematic spectral analysis. Degradation occurred throughout the RG-AEM cross-sections but appeared to be more severe at the surfaces. The 457 and 633 nm laser wavelengths were the most useful for the study of the pre-aminated grafted membranes, while the 785 nm laser wavelength was the only option that could be used for the study of the pre- and post-alkali degraded RG-AEMs.
Author contributions
Dr Bance-Soualhi supervised the student and conducted a number of the experiments (surface maps and degradation data), as well as making a major contribution to authoring of this article. Dr Crean and Prof Varcoe are the principal investigators of the EPSRC grants who contributed to the formulation of this article and scientific interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the UK's Engineering and Physical Sciences Research Council (EPSRC) with resources provided by grants EP/M005933/1, EP/M014371/1, and EP/M022749/1 (the latter providing the capital funding that allowed for the purchase of the Renishaw Raman microscope system). Renishaw are thanked for their technical assistance related to our familiarisation with the instrument. Wai Hin Lee thanks the University of Surrey for providing additional funding: the bulk of the results in this paper was from his undergraduate final year project. All data is freely available with no restrictions: details on how to access this data can be found at DOI: 10.15126/surreydata.00842458.References
- T. R. Dargaville, G. A. George, D. J. T. Hill and A. K. Whittaker, Prog. Polym. Sci., 2003, 28, 1355 CrossRef CAS; M. M. Nasef, S. A. Gürsel, D. Karabelli and O. Güven, Prog. Polym. Sci., 2016, 63, 1 CrossRef.
- M. M. Nasef, Chem. Rev., 2014, 114, 12278 CrossRef CAS PubMed; L. Gubler, Adv. Energy Mater., 2014, 4, 201300827 Search PubMed; A. Albert, T. Lochner, T. J. Schmidt and L. Gubler, ACS Appl. Mater. Interfaces, 2016, 8, 15297 Search PubMed.
- J. Pan, C. Chen, L. Zhuang and J. Lu, Acc. Chem. Res., 2012, 45, 473 CrossRef CAS PubMed; J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 Search PubMed; K. D. Kreuer, Chem. Mater., 2014, 26, 361 CrossRef; M. A. Hickner, Interface, 2017, 26, 69 Search PubMed; T. J. Omasta, L. Wang, X. Peng, C. A. Lewis, J. R. Varcoe and W. E. Mustain, J. Power Sources, 2017 DOI:10.1016/j.jpowsour.2017.05.006.
- N. Walsby, M. Paronen, J. Juhanoja and F. Sundholm, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1512 CrossRef CAS; J. R. Varcoe, R. C. T. Slade, E. L. H. Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperley, Chem. Mater., 2007, 19, 2686 CrossRef; O. I. Deavin, S. Murphy, A. L. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584 Search PubMed; B.-M. Ko, J.-Y. Sohn and J. Shin, Polymer, 2012, 53, 4652 CrossRef; M. Mamlouk, J. A. Horsfall, C. Williams and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 11912 CrossRef; T. Zhou, R. Shao, S. Chen, X. He, J. Qiao and J. Zhang, J. Power Sources, 2015, 293, 946 CrossRef; R. Espiritu, M. Mamlouk and K. Scott, Int. J. Hydrogen Energy, 2016, 41, 1120 CrossRef; Y. Zhao, K. Yoshimura, H. Shishitani, S. Yamaguchi, H. Tanaka, S. Koizumi, N. Szekely, A. Radulescu, D. Richtere and Y. Maekawa, Soft Matter, 2016, 12, 1567 RSC.
- J. R. Varcoe, R. C. T. Slade, G. L. Wright and Y. Chen, J. Phys. Chem. B, 2006, 110, 21041 CrossRef CAS PubMed; S. F. Lu, J. Pan, A. B. Huang, L. Zhuang and J. T. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef; C. A. Hancock, A. Ong, P. R. Slater and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 3047 Search PubMed; H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. Di Benedetto, F. D. I. Acapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55, 6004 CrossRef PubMed; H. A. Miller, F. Vizza, M. Marelli, A. Zadick, L. Dubau, M. Chatenet, S. Geiger, S. Cherevko, H. Doan, R. K. Pavlicek, S. Mukerjee and D. R. Dekel, Nano Energy, 2017, 33, 293–305 CrossRef; H. Wang and H. Abruña, J. Am. Chem. Soc., 2017, 139, 6807 CrossRef PubMed; L. Wang, J. J. Brink, Y. Liu, A. M. Herring, J. Ponce-González, D. K. Whelligan and J. R. Varcoe, Energy Environ. Sci., 2017 10.1039/C7EE02053H.
- J. Ponce-González, D. K. Whelligan, L. Q. Wang, R. Bance-Soualhi, Y. Wang, Y. Q. Peng, H. Q. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724 Search PubMed.
- H.-P. Brack, H. G. Bührer, L. Bonorand and G. G. Scherer, J. Mater. Chem., 2000, 10, 1795 RSC; M. M. Nasef and E.-S. A. Hegazy, Prog. Polym. Sci., 2004, 29, 499 CrossRef CAS; Y. Buchmüller, A. Wokaun and L. Gubler, Fuel cells, 2013, 13, 1177 CrossRef.
- V. Sproll, T. J. Schmidt and L. Gubler, Polym. Int., 2016, 65, 174 CrossRef CAS.
- L. Q. Wang, E. Magliocca, E. L. Cunningham, W. E. Mustain, S. D. Poynton, R. Escudero-Cid, M. M. Nasef, J. Ponce-González, R. Bance-Soualhi, R. C. T. Slade, D. K. Whelligan and J. R. Varcoe, Green Chem., 2017, 19, 831 RSC.
- S. Holmberg, J. H. Näsman and F. Sundholm, Polym. Adv. Technol., 1998, 9, 121 CrossRef CAS; L. I. Şanli and S. A. Gürsel, J. Appl. Polym. Sci., 2011, 120, 2313 CrossRef.
- F. Cardona, G. A. George, D. J. T. Hill, F. Rousoul and J. Maeji, Macromolecules, 2002, 35, 355 CrossRef CAS; F. Cardona, G. A. George, D. J. T. Hill and S. Perera, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3191 CrossRef; H. Herman, R. C. T. Slade and J. R. Varcoe, J. Membr. Sci., 2003, 218, 147 CrossRef.
- J. E. Katon, Vib. Spectrosc., 1994, 7, 201 CrossRef CAS; A. Chaplin, I. Hamerton, H. Herman, A. K. Mudhar and S. J. Shaw, Polymer, 2000, 41, 3945 CrossRef; E. Widjaja and M. Garland, J. Raman Spectrosc., 2012, 43, 828 CrossRef; R. Zeng, J. Handsel, S. D. Poynton, A. J. Roberts, R. C. T. Slade, H. Herman, D. C. Apperley and J. R. Varcoe, Energy Environ. Sci., 2011, 4, 4925 Search PubMed; D. M. Hillman, S. H. Stephens, S. D. Poynton, S. Murphy, A. Ong and J. R. Varcoe, J. Mater. Chem. A, 2013, 1, 1018 Search PubMed; R. Zeis, Beilstein J. Nanotechnol., 2015, 6, 68 CrossRef PubMed; S. Mukherjee and A. Gowen, Anal. Chim. Acta, 2015, 895, 12 CrossRef PubMed; C. Cardell and I. Guerra, Trends Anal. Chem., 2016, 77, 156 CrossRef; T. R. Pozegic, J. V. Anguita, I. Hamerton, K. D. G. I. Jayawardena, J.-S. Chen, V. Stolojan, P. Ballocchi, R. Walsh and S. R. P. Silva, Sci. Rep., 2016, 6, 37334 CrossRef PubMed.
- A. Zoubir, Raman Imaging: Techniques and Applications, Springer, Heidelberg NY (USA), 2012 Search PubMed.
- R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Mater. Chem. A, 2017, 5, 1248 CAS; R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Power Sources, 2017 DOI:10.1016/j.jpowsour.2017.07.074.
- E. Smith and G. Dent, Modern Raman Spectroscopy: A practical Approach, Wiley, Hoboken NJ (USA), 2005 Search PubMed.
- R. L. McCreery, Raman Spectroscopy for Chemical Analysis, John Wiley & Sons, New York (USA), 2005 Search PubMed.
- G. Galleja, A. Houdayer, S. Etienne-calas, D. Bourgogne, V. Flaud, G. Silly, S. Shibahara, A. Takahara, A. Jourdan, A. Hamwi and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1517 CrossRef.
- P. J. Larkin, Infrared and Raman spectroscopy: Principles and spectral interpretation, Elsevier, Amsterdam, 2011 Search PubMed.
- M. A. Vandiver, B. R. Caire, J. R. Carver, K. Waldrop, M. R. Hibbs, J. R. Varcoe, A. M. Herring and M. W. Liberatore, J. Electrochem. Soc., 2014, 161, H677 CrossRef CAS; T. P. Pandey, A. M. Maes, H. N. Sarode, B. D Peters, S. Lavinia, K. Vezzu, Y. Yang, S. Poynton, J. R. Varcoe, S. Seifert, M. Liberatore, V. Di Noto and A. Herring, Phys. Chem. Chem. Phys., 2015, 17, 4367 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional data and Raman spectra/maps in support of the main article. See DOI: 10.1039/c7ra09650j |
| ‡ Sub-micron resolution can be less routinely obtained with the use of high cost equipment and low wavelength lasers, especially with the use of tip-enhanced Raman spectroscopy (TERS), which couples atomic force microscopes with Raman instrumentation. |
| § Raman microscopes are still generally more expensive than for IR instrumentation (>£100k vs. <£50k). |
| ¶ The Renishaw Raman instrument used contains a 5th laser (244 nm deep UV) but this is not appropriate for this study: the laser damages the polymer samples, even at the lowest power setting. |
| || If determination of nano-scale phase segregations of hydrophilic vs. hydrophobic components is required, techniques such as small angle neutron scattering (SANS) and transmission electron microscopy (TEM) are required. Such data is beyond the scope of this current study, which is focussed on the homogeneity of the grafting and degradation processes (where we show that micro-scale characterisation is sufficient). |
| ** This peak is only present with 1,3-disubstituted benzene rings (in the 3-VBC monomer and 3-VBC grafted chains) and is not observed for the grafted 4-VBC chains (recall that mixed 3- and 4-VBC monomers are being grafted). |
| †† Note: with the use of the IR (785 nm) laser, the scale-bar for the A1612/A835 peak area ratio (middle column in Fig. 8) is compressed to the range 0–2, compared to the range 0–4 in Fig. 6, that was required to accommodate the higher ratios observed for Int72 with the use of the blue (457 nm) laser. |
| This journal is © The Royal Society of Chemistry 2017 |