
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSPIONs/DOX loaded polymer nanoparticles for MRI detection and efficient cell targeting drug delivery
Le Shanga,
Qiu-yue Wanga,
Kang-long Chena,
Jing Qua,
Qing-han Zhou *a,
Jian-bin Luo*a and
Juan Lin*b
*a,
Jian-bin Luo*a and
Juan Lin*b
aCollege of Chemical and Environment Protection, Southwest Minzu University, First Ring Road, 4th Section No. 16, Chengdu, Sichuan 610041, China. E-mail: zhqinghan@swun.edu.cn; Fax: +86-28-85524382; Tel: +86-28-85522269
bSchool of Biomedical Sciences, Chengdu Medical College, 601 Tianhui Road, Chengdu, Sichuan 610083, China. E-mail: linjuan.scu@gmail.com
First published on 11th October 2017
Abstract
In this research, reducible polydopamine coated magnetic nanoparticles (SPIONs@PDA) were prepared and doxorubicin (DOX) was encapsulated for both magnetic resonance imaging (MRI) detection and cell targeting drug delivery. The SPIONs@PDA exhibited excellent water-solubility and colloidal stability, because the coating of the magnetic nanoparticle was a crosslinked reducible polydopamine synthesized with N,N-bis(acrylate) cystamine (BACy) as a crosslinker, dopamine methacrylamide (DMA) as an anchor to immobilize SPIONs, and polyethylene glycol (PEG) to provide colloidal stability. The SPIONs@PDA showed reduction-sensitivity due to the reducible disulfide bond in the polymer network. Subsequently, a sustained and high cumulative drug release in glutathione (GSH) was observed, rather than the low drug release in PBS (pH 7.4). Fluorescent imaging studies by confocal laser scanning microscopy indicated that the prepared SPIONs@PDA could specifically target tumor cells. The results of the cytotoxicity assays further demonstrated that the DOX-loaded SPIONs@PDA presented efficient antitumor activity to HeLa cells. These favorable properties suggested SPIONs@PDA as promising targeted magnetic nanoparticles for MRI detection and stimuli-triggered drug delivery in cancer theranostics.
1. Introduction
Recently, superparamagnetic iron oxide nanoparticles (SPIONs) have been extensively studied as a promising theranostic candidate for both magnetic resonance imaging (MRI) detection and cell targeting drug delivery.1–4 However, clinical applications have been disappointing because of the oxidation, acid erosion, severe aggregation, and poor water solubility of SPIONs.5–9 Increasing efforts are devoted to preparing biostable and biocompatible SPIONs in aqueous media with functional polymeric coatings to avoid these disadvantages.10–12 Conventionally, to stabilize magnetic nanoparticles for medical applications, SPIONs were incorporated into crosslinked polymers to form magnetic nanocomposites due to the excellent water-solubility and colloidal stability of crosslinked polymers,13–16 which processed a phase transfer reaction based on exchanging surface surfactants on SPIONs with amphiphilic molecules, such as PEG,17 dextran,18 or PEI.19 However, the drug release will be prevented due to an excessively stabilized or nondegradable crosslinked polymer structure at aimed tissue or cell, and the nanoparticles will accumulate in the normal cells causing a long-term toxicity, thus reducing the theranostic efficiency.20–23 To solve this problem SPIONs can be incorporated into a biodegradable crosslinked polymer, which can provide SPIONs the biodegradability, colloidal stability, high drug delivery efficacy, and enhanced contrast effect for MRI detection.24,25 However, so far, only a few examples for biodegradable crosslinked polymer based magnetic nanoparticle were developed as both MRI contrast agent and stimuli-responsive drug delivery system.Because the concentration of glutathione (GSH) is about 1000 times in the various subcellular organelles in cytoplasm than the extracellular environment, and at least 4-fold higher concentrations in the tumor cells over normal cells, biodegradable nanoparticles with reduction stimulus sensitivity demonstrated effective drug release behavior.26,27 Nanoparticles containing reducible disulfide bonds can be degraded easily by thiol/disulfide exchange with GSH, and achieve the intracellular delivery of drug molecules,28 DNA,29 and siRNA for cancer therapy.30 Therefore, biodegradable nanoparticles based on disulfide are of particular interest for efficiency and targeted intracellular drug delivery.
On the other hand, due to the weak interaction between polymer and SPIONs, magnetic nanoparticle with polymer coating often shows only short-term stability, which can be easily disassociated by slight disruptions in different chemical environment of human body.31–33 It has been demonstrated that catechol-containing molecules present the ability to adhere to almost any material of either organic or inorganic origin. For example, 3,4-dihydroxy-L-phenylalanine (DOPA) can form strong bonds with various inorganic/organic surfaces by coordination of metal ions, formation of π-electrons, or hydrogen bond interactions.34–36 However, to the best of our knowledge, there was no report on crosslinked reducible polydopamine coated magnetic nanoparticles in theranostic system, although the dopamine moiety has been widely utilized for surface modification of inorganic nanoparticles.37–42
Herein, in this article our object is to design and fabricate the biostable, biocompatible, and reducible polydopamine coated magnetic nanoparticles (SPIONs@PDA) that can achieve both MRI diagnosis and anticancer drug delivery. The shell of the magnetic nanoparticle was a crosslinked reducible polydopamine (PDA) synthesized with N,N-bis(acrylate) cystamine (BACy), dopamine methacrylamide (DMA), and polyethylene glycol (PEG) methyl ether methacrylate, where BACy served as crosslinker, PEG provided colloidal stability, and DOPA moiety as anchor to immobilize SPIONs. The experimental results showed that the theranostic magnetic nanoparticles showed good stability in physiological condition with high DOX loading efficacy, and have effective T2 relaxivity and good negative contrast effect for MRI detection, and achieve high efficiency of drug delivery by the quick degradation in reductive environment due to the cleavage of the disulfide linkages. The cytotoxicity assays and cellular uptake demonstrated that the blank nanoparticles were nontoxic and the drug-loaded nanoparticles enabled specific targeting to HeLa cells for drug delivery through the CCK8 assay and confocal laser scanning microscopy images. Fig. 1 illustrates the preparation and reduction-sensitive DOX release process of the magnetic nanoparticles in the GSH condition.
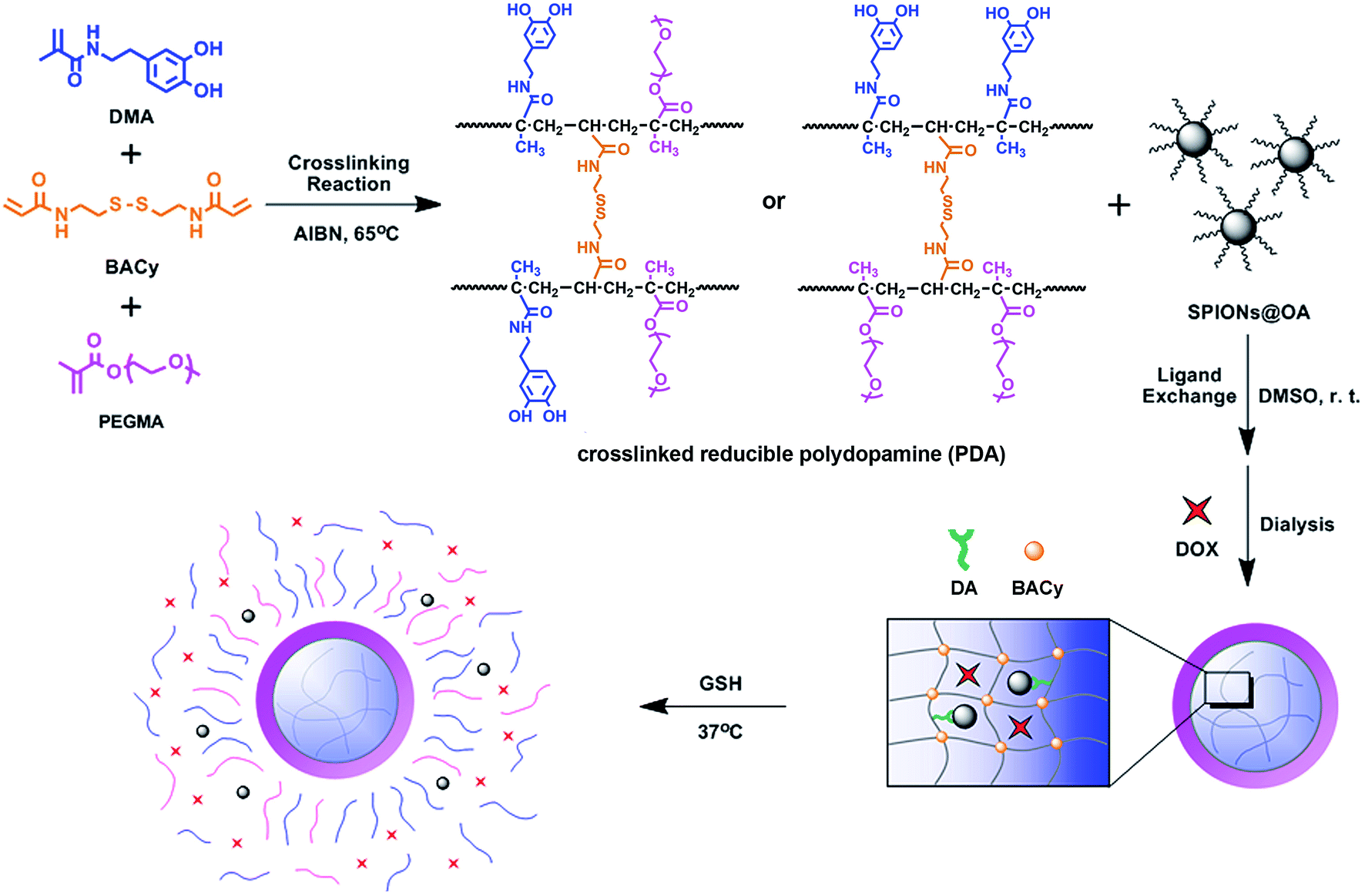 | ||
| Fig. 1 Illustration of the preparation and reduction-sensitive DOX release process of the magnetic nanoparticle. | ||
2. Experimental
2.1. Materials
PEG methyl ether methacrylate (PEGMA) (Mn = 300, 98%) was obtained from Sigma-Aldrich. Acryloyl chloride (98%) and cystamine dihydrochloride (96%) were obtained from J&K Chemical. Methacrylic anhydride (94%), dopamine hydrochloride (98%), GSH (98%), and DOX (98%) were purchased from Shanghai Aladdin Reagent. Dichloromethane, N,N-dimethylformamide, tetrahydrofuran, ethylene acetate, n-hexane, and ether were used after purified. The other chemicals were used as received.2.2. Preperation of SPIONs
FeCl3·6H2O (4.2 g, 15.5 mmol) and FeSO4 (2.3 g, 15.2 mmol) were dissolved in 30 mL of distilled water. 25 mL of ammonium hydroxide (25%) was added to this solution with vigorous agitation at 80 °C for 0.5 h under N2 atmosphere. 1 mL of 10% oleic acid was then added into the solution under a vigorous agitation for 1 h. With the evaporation of ammonia gas a distinct phase separation between the upper organic portion and the lower aqueous portion appeared. The aqueous phase was removed using a pipette, and oleic acid-coated magnetite nanocrystals were collected by magnetic separation, and then washed with 80 mL of ethanol to eliminate excess oleic acid. Finally, the products were separated by centrifugation. Ethanol was completely removed under reduced pressure at room temperature. The dried oleic acid-coated magnetite nanocrystals were obtained and dispersed in 10 mL of chloroform for future use.2.3. Synthesis of BACy
Cystamine dihydrochloride (2.3 g, 10.2 mmol) was dissolved in 18 mL deionized water, and added into a 100 mL three-necked flask equipped with a stirrer, a thermometer, and a dropping funnel. The mixture solution was cooled in ice-water bath for 30 min. NaOH (1.6 g, 40 mmol) at the same time was dissolved in 4 mL deionized water, and then cooled in ice-water bath for 30 min. Acryloyl chloride (1.8 mL, 21.5 mmol) was dissolved in dichloromethane and mixed with the as prepared ice-cooled NaOH solution, and then added dropwise slowly into the three-necked flask in 5 minutes. The three-necked flask was cooled in ice-water bath and stirred at 0 °C for 3 h, and then at room temperature for another 12 h. The organic phase was extracted with dichloromethane, and subsequently dried over anhydrous MgSO4. The solvent was removed under vacuum to obtain the raw product, and the BACy was finally purified by recrystallization from ethyl acetate/hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The 1H NMR of BACy in CDCl3 was shown in Fig. 2a. Yield: 1.9 g (72 wt%). 1HNMR (400 MHz): δ (ppm) = 2.89 (t, 4H, –CONHCH2CH2S–), 3.68 (q, 4H, –CONHCH2CH2S–), 5.66–6.31 (m, 6H, CHH
:1, v/v). The 1H NMR of BACy in CDCl3 was shown in Fig. 2a. Yield: 1.9 g (72 wt%). 1HNMR (400 MHz): δ (ppm) = 2.89 (t, 4H, –CONHCH2CH2S–), 3.68 (q, 4H, –CONHCH2CH2S–), 5.66–6.31 (m, 6H, CHH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO–), 6.61 (brs, 2H, –CONH–).
CHCO–), 6.61 (brs, 2H, –CONH–).
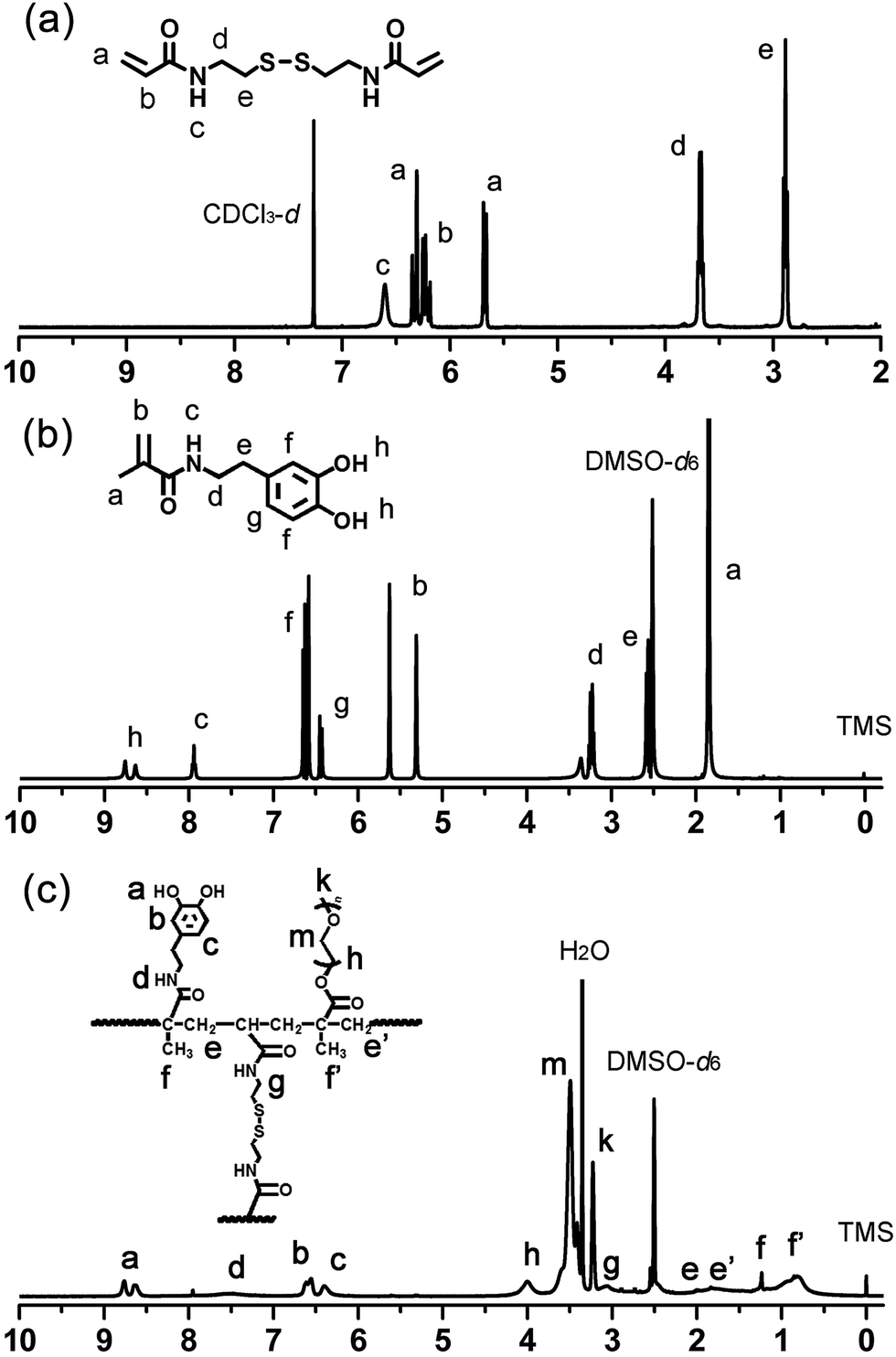 | ||
| Fig. 2 1H NMR spectra of BACy (a) in CDCl3, DMA (b), and PDA crosslinked copolymer (c) in DMSO-d6. | ||
2.4. Synthesis of DMA
DMA were synthesized following an already published procedure.43 Sodium borate (2.5 g, 6.6 mmol), sodium bicarbonate (1.0 g, 12.0 mmol), and 25 mL deionized water were added into three-necked flask, and then degassed with bubbling nitrogen for 20 min. 3,4-dihydroxyphenethylamine hydrochloride (1.0 g, 5.3 mmol) was added into the mixture solution. NaOH solution (1 M) was added drop-wise in order to keep the reaction mixture basic to around pH 8. Methacrylate anhydride (1.0 mL, 6.7 mmol) was added drop-wise into the above solution under gentle stirring at room temperature purged with nitrogen gas. After 6 h HCl solution was add to acidify the obtained aqueous solution to around pH 2. The organic phase was extracted three times from the acidified aqueous solution with 300 mL of ethyl acetate and dried over anhydrous MgSO4. The obtained solution was added to 200 mL of n-hexane with vigorous stirring to obtain a brownish solid, then purified by recrystallization from ethyl acetate/hexane mixture (2:1, v/v) to obtain a grey white powder. The product was finally dried in a vacuum overnight. The 1H NMR of DMA in DMSO-d6 was shown in Fig. 2b. Yield: 0.9 g (79 wt%).1H NMR (400 MHz): δ (ppm) = 1.84 (s, 3H, –COCCH3CH2), 2.55 (q, 2H, C6H3(OH)2CH2CH2CONH–), 3.21 (q, 2H, C6H3(OH)2CH2CH2CONH–), 5.29 and 5.61 (s, 2H, –COCCH3CHH), 6.42–6.64 (m, 3H, C6H3(OH)2–), 7.93 (t, 1H, –CONH–), 8.56–8.78 (s, 2H, C6H3(OH)2–).
2.5. Synthesis of PDA crosslinked copolymer
A typical procedure for synthesizing the crosslinked polymer is as following: molar ratio of comonomers was set at 1:1 (DMA:PEGMA). DMA (11.00 mg, 0.05 mmol), PEGMA (32.80 mg, 0.11 mmol), and BACy (7.80 mg, 0.03 mmol) were added to a 50 mL flask. 25 mL of DMF and AIBN (2% wt) was added, and the flask was purged with nitrogen gas, and proceeded at 65 °C for 24 h. The final product was purified by a dialysis method by dialysis membrane with molecular weight cut-off (MWCO) of 14000, and lyophilized into powder. The 1H NMR of DMA in DMSO-d6 was shown in Fig. 2c. Yield: 1.8 g. 1H NMR (400 MHz): δ (ppm) = 0.62–1.04 (CH3–, in polymer backbone of PEGMA unit), 1.07–1.27 (CH3–, in DMA unit), 1.42–2.06 (–CH2–, in polymer backbone), 3.04 (–CH2CH2SS–, in crosslinker), 3.32–3.56 (–CH2CH2O–, in PEGMA unit), 3.79–4.19 (–CH2CH2O–, next to the polymer backbone), 6.24–6.71 (C6H3(OH)2–), 7.25–7.72 (–CONH–, in DMA unit), 8.46–8.82 (C6H3(OH)2–).
2.6. Preparation of SPIONs@PDA and characterization
SPIONs@PDA were prepared via a ligand exchange reaction between the PDA crosslinked copolymer and SPIONs. 20 mg of as prepared SPIONs and 10 mg of PDA crosslinked copolymer were dissolved in 25 mL of DMF. Distilled water was dropwisely added into the mixture solution with sonication, and the resulting colloid was stirred vigorously for 24 h. After that, the raw product was separated by centrifugation, and the obtained SPIONs@PDA was washed with distilled water. The unbound copolymer was removed by dialysis method (MWCO 14000), and then the product solution was freeze-dried into a brown powder. The iron concentration of SPIONs@PDA was determined by ICP-OES.1H NMR spectra of BACy, DMA, and PDA were obtained using a Bruker 400 MHz spectrometer with CDCl3 or DMSO-d6 as solvent, and tetramethylsilane (TMS) as an internal standard. FT-IR spectra were gathered in solid state on a PE Spectrum One FT-IR spectrophotometer under ambient. Utilizing a resolution at 4 cm−1 resolution, the spectra were taken from 400 to 4000 cm−1. DLS (Malvern Zetasizer Nano-ZS90) was used to determine the nanoparticle size (Dh) of SPIONs@PDA in aqueous solution or culture media (DMEM, containing 10% FBS and 1% AA). Each sample of 1 mg mL−1 was conducted in a 1.0 mL quartz cuvette and measured at room temperature in three replicates, which yielded a size fraction distribution plots. Morphologies of magnetic nanoparticles were observed by transmission electron microscopy (Hitachi H-600, Japan). The samples were prepared by dropping the solution of magnetic nanoparticles onto carbon-coated copper grids and dried at room temperature overnight without staining before measurement. XRD patterns were collected on a Persee XD-6 diffractometer using Cu Kα radiation (λ = 0.154 nm) with an accelerating voltage of 36 kV and tube current 20 mA. Magnetic behavior was analyzed by a vibrating sample magnetometer (VSM) (MPMS-XL-7, Quantum Design Company, USA).
2.7. Drug loading and release profile
DOX was selected as a model anticancer drug to investigate the release behavior of the magnetic nanoparticles. The SPIONs@PDA was dissolved in DMF (6 mL). DOX (5 mg) was dissolved in DMSO (0.1 mL) and then TEA (0.7 mL) was added to keep the reaction mixture basic. The DOX solution was added drop-wise into SPIONs@PDA solution under stirring for 1 h. The mixture solution was transferred into a dialysis tube (MWCO 14000) and dialyzed against PBS buffer (pH 7.4, 10 mM, refreshed every 5 h) to remove the unloaded DOX for 72 h at room temperature. After dialysis, the DOX-loaded magnetic nanoparticle solution was lyophilized into a brown powder. The drug loading content (LC) and encapsulation efficiency (EE) was measured on a UV-Vis spectrophotometer at the absorbance of 480 nm according to a standard curve obtained from DOX/DMF solutions. The LC was defined as the weight ratio of loaded drug to the drug-entrapped nanoparticles. EE of DOX was obtained from the weight ratio of the drug incorporated in nanoparticles to that used in the preparation.In vitro release behavior was performed at 37 °C. DOX-loaded SPIONs@PDA at a concentration of 1 mg mL−1 in a dialysis tube (MWCO 14000) were incubated in different media: PBS (10 mM, pH 7.4) with 10 and 20 mM GSH, and PBS (10 mM, pH 7.4) without GSH under gentle stirring, respectively. At specified time intervals, the DOX content in the samples was measured on a UV absorbance at 480 nm. All DOX drug release data were averaged over three replicate measurements.
2.8. In vitro relaxivity measurement
In vitro relaxivity measurements were measured using a Varian (Palo Alto, California, USA) 4.0 cm inner diameter quadrature coil. The relaxation data and images were acquired at 7 T using a Varian Inova imaging and spectroscopy system. To measure T2 relaxation times a single slice, multi-echo spin echo sequence was set to TR = 2000 ms, TE = 11–110 ms (10 echoes, 11 ms increments), SL = 2 mm, FOV = 100 × 100 mm, MA = 128 × 128. Relaxation times were obtained by fitting the multi-echo data to a monoexponential decay curve using linearized least-squares optimization. Relaxivity values were calculated via linear least-squares fitting of 1/relaxation time (s−1) versus the iron concentration (mM Fe).2.9. In vitro cell assay
Cancer cell viability of the SPIONs@PDA was evaluated by CCK-8 assay. HeLa cells (human cervical carcinoma cells, cancer cells) were cultured in RPMI 1640 medium in a humidified atmosphere with 5% CO2 at 37 °C. For experiments, HeLa cells were seeded into 96-well plate at a density of 3 × 104 cells per well, and incubated 24 h to allow cell attachment. After 24 h, the medium was removed, and different concentrations of samples or free DOX were added into each well, and then incubated for 2 and 24 h. 10 μL of CCK8 solution were added into each well, and incubated for 3 h. After incubation, the absorbance was measured at 450 nm using a microplate reader to evaluate the cell viability. The cell viability was calculated as follows:| Cell viability (%) = (As/AControl) × 100% | (1) |
2.10. Confocal laser scanning microscopy
Human tubular epithelial cells (HK-2 cells) were kindly provided by Chengdu Medical College. HK-2 cells and HeLa cells were reseeded into an UV disinfected 6-well plate with cover glass at a density of 1 × 105 cells per well. After a 24 h incubation period different sample solution or free DOX were added into each well. At designated time intervals, the suspension was removed, and the samples were washed with PBS solution for three times, then mounted in fluorescent mounting medium to be observed by confocal laser scanning microscope (CLSM) with an imaging software.3. Results and discussion
3.1. Preparation and characterization of the SPIONs@PDA
To construct magnetic drug nano-carriers, homogeneous spherical SPIONs were prepared by the coprecipitation method. As the polymer coating to the SPIONs, the PDA crosslinked copolymer was synthesized via a free radical copolymerization, with AIBN as initiator, BACy as crosslinker, PEGMA and DMA as comonomers. The incorporation of disulfide-bonded crosslinker BACy was used to provide the reduction-sensitivity, and macromonomer PEGMA enhanced the colloidal stability in water, and DMA was utilized to anchor the magnetic nanoparticles. Finally, the SPIONs were incorporated into the PDA crosslinked copolymer by the strong bonds between SPIONs and DOPA moieties. The synthesis route of PDA crosslinked copolymer and formation of SPIONs@PDA nanoparticles were shown in Fig. 1. DOX was chosen as model drug to be encapsulated into magnetic nanoparticles by dialysis method to prepare the magnetic theranostic system. Subsequently, in the GSH reducing environment, it is expected that DOX can be effectively released from the DOX-loaded SPIONs@PDA nanoparticles due to the chemical cleavage of the disulfide linkages. In addition, SPIONs encapsulated in the nanoparticles could be used for the enhanced MRI detection.1H NMR spectra were used to confirm the chemical structures of the synthesized BAcy, DMA, and PDA crosslinked copolymer. The representative 1H NMR spectrum of BACy was depicted in Fig. 2a. The resonance signals of –CH2– (e and d) neighboring to the disulfide bond of the BACy molecule, vinyl group (a and b), and –NH– (c) was observed at δ = 2.89 (e), 3.68 (d), 5.66–6.31 (a and b), and 6.61 (c), respectively, as described in experimental section. 1H NMR spectrum of DMA (in DMSO-d6) was depicted in Fig. 2b. The resonance signals of methyl group (a), β-methylene group (e), α-methylene group (d), vinyl group (b), phenyl group (g and f), –NH– (c), and phenolic hydroxyl (h) appeared at δ = 1.84 (a), 2.55 (e), 3.21 (d), 5.29 and 5.61 (b), 6.42–6.64 (g and f), 7.93 (c), and 8.56–8.78 (h), respectively. The 1H NMR spectrum of PDA crosslinked copolymer (in DMSO-d6) was depicted in Fig. 2c. The resonance signals at 0.62–1.27 (f and f′), 3.04 (g), 3.32–3.56 (m), 6.24–6.71 (b and c), 7.25–7.72 (d), and 8.46–8.82 (a) were belong to the methylene group (f and f′) in polymer backbone, α-methylene group (g) of the BACy crosslinker, the glycol units in PEGMA (m), phenyl group (b and c), imino group (d), and phenolic hydroxyl (a) in DMA units. As seen in Fig. 2c, the appearance of the glycol unit in PEGMA, α-methylene signal in BACy, and phenolic hydroxyl in DMA unit indicated the successfully synthesis of the PDA crosslinked copolymer.
As shown in Fig. 3, FT-IR spectra were used to confirm the chemical structure of SPIONs, PDA crosslinked copolymer, and SPIONs@PDA. As shown in Fig. 3a and c, the absorption bands around 580 cm−1 can be ascribed to the Fe–O stretching vibration of Fe3O4, suggesting that the SPIONs were immobilized into the PDA crosslinked copolymer to form SPIONs@PDA. In Fig. 3b, the vibrational band of C–O group in PEGMA, C–S in BACy, and CO in DMA units occurred at 1112, 1270, and 1683 cm−1 indicating that the successful synthesis of PDA crosslinked copolymer. In Fig. 3c, Fe–O of Fe3O4, C–O group in PEGMA, C–S in BACy, and CO in DMA units were distinctively observed in SPIONs@PDA at 579, 1094, 1287, 1675 cm−1, respectively, indicating the successful preparation of SPIONs@PDA.
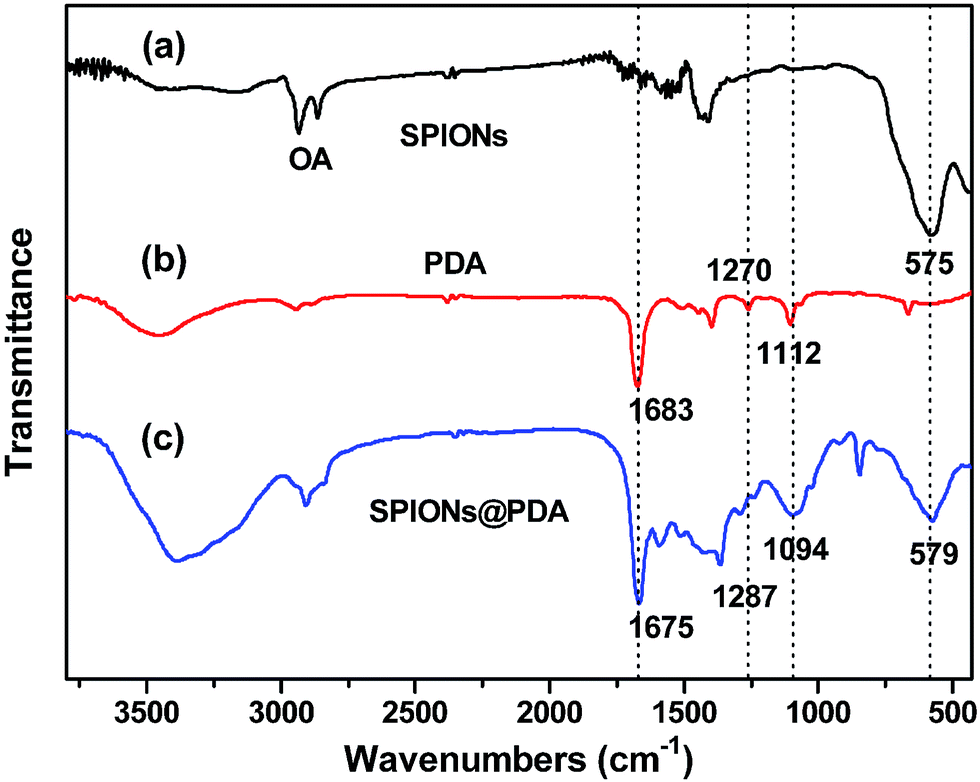 | ||
| Fig. 3 FT-IR spectra of (a) SPIONs, (b) PDA crosslinked copolymer, and (c) SPIONs@PDA. | ||
The as prepared SPIONs were composed of Fe3O4 magnetite nanoparticles coated with oleic acid with a size of about 10 nm (Fig. 4a), and dispersed well in chloroform due to their hydrophobic property. After addition of the synthesized PDA crosslinked copolymer, the copolymer was assembled onto the surface of SPIONs by a ligand exchange reaction between DOPA moieties and the oleic acid layer of SPIONs to form SPIONs@PDA, while the PEG chains provide colloidal stability in water by virtue of their high hydrophilicity. As shown in Fig. 4b, TEM photos showed that the SPIONs@PDA were well dispersed in water solution, and the spherical morphology was stable with an average diameter of 150 nm. The obtained SPIONs and water-soluble SPIONs@PDA were also characterized by DLS measurement in aqueous solution (shown in Fig. 4c), and the hydrodynamic size of SPIONs and SPIONs@PDA were about 22 and 170 nm, respectively, and no significant changes in size were observed for weeks, indicating that the SPIONs@PDA retained good stability. Fig. 4d showed that hydrophobic SPIONs were dispersed in chloroform while SPIONs@PDA were soluble in water.
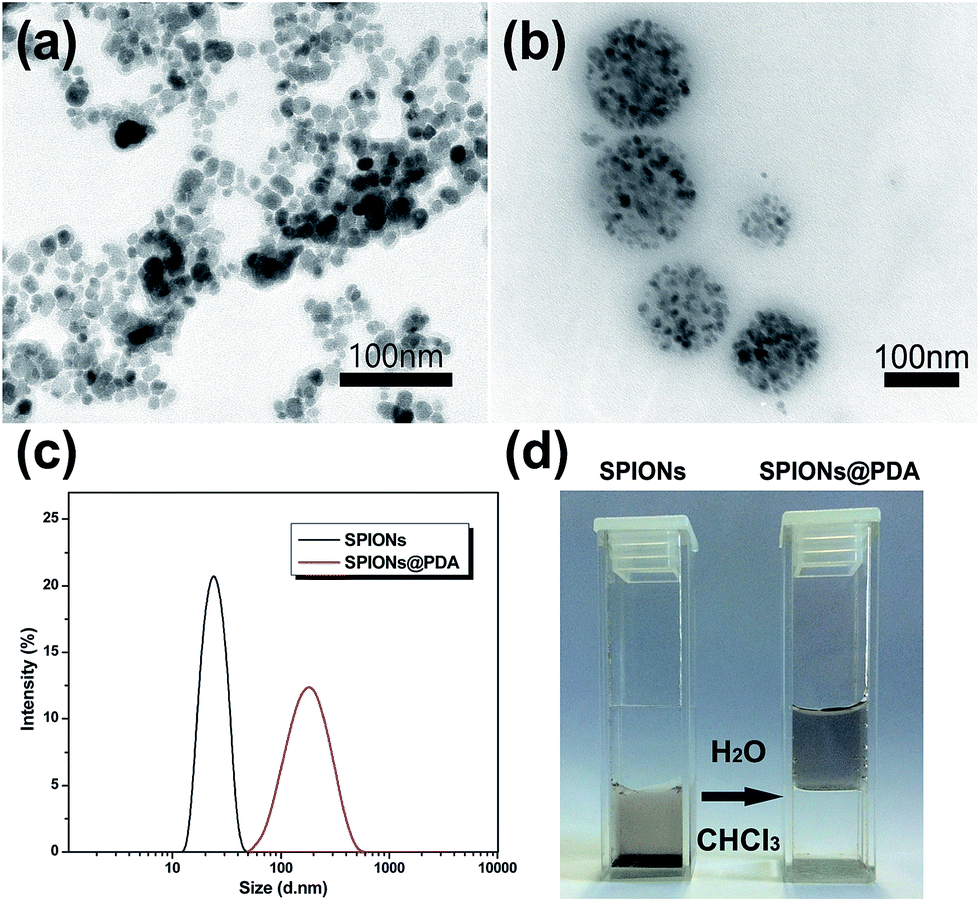 | ||
| Fig. 4 TEM photos of (a) SPIONs, and (b) SPIONs@PDA after the ligand exchange reaction; (c) DLS data showing the change of hydrodynamic size (in diameter) of SPIONs before, and after ligand exchange reaction with PDA crosslinked copolymer; (d) Photograph of hydrophobic SPIONs dispersed in chloroform (left) and water-soluble SPIONs@PDA (right). | ||
Additionally, the nanoparticle stability of SPIONs@PDA was measured by DLS in culture media (Fig. 5). DLS measurements indicated that the hydrodynamic size of SPIONs@PDA in DMEM was ∼150 nm initially, and no significant changes in size or aggregations were observed for up to a week, indicating the SPIONs@PDA remained good stability. The stability of SPIONs@PDA in culture media can be attributed to the presence of PEG on the nanoparticle surface, because the PEG molecules are very hydrophilic and can form coiled conformations in aqueous solutions, providing good© colloidal stability.
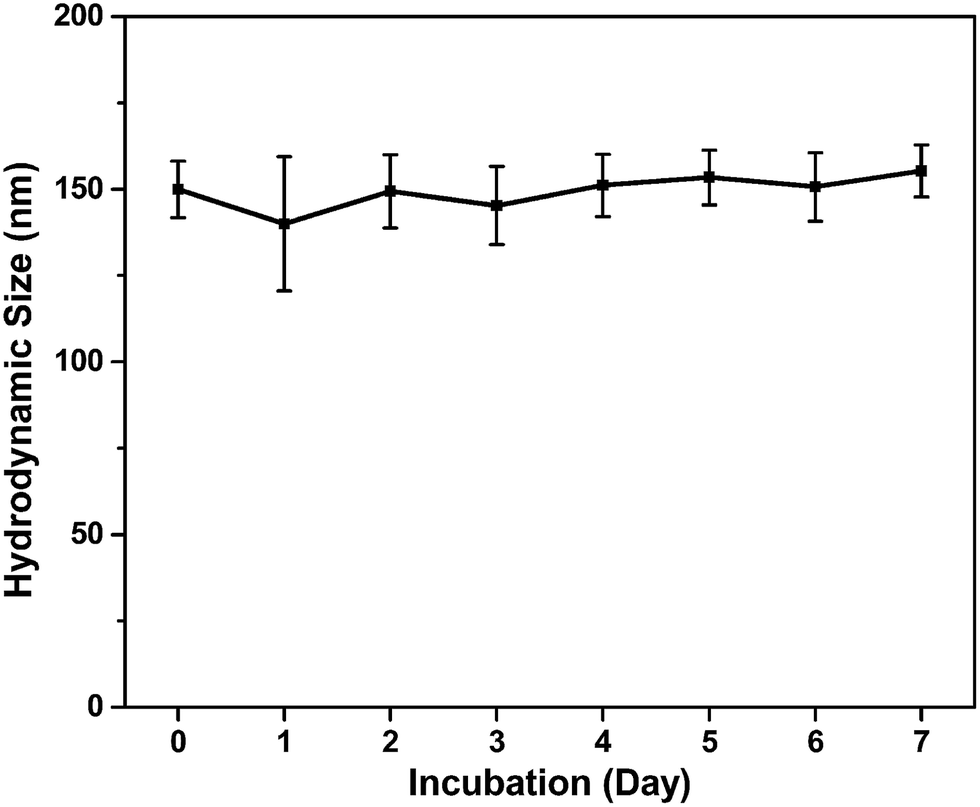 | ||
| Fig. 5 Hydrodynamic size of SPIONs@PDA in cell culture media (DMEM with 10% FBS) as a function of time. | ||
The diffractogram for SPIONs and SPIONs@PDA were shown in Fig. 6a, and all of the diffraction peaks were consistent with the database in JCPDS file. The presence of sharp and intense peaks confirmed the formation of highly crystalline Fe3O4. Besides the SPIONs diffraction peaks, the XRD pattern of SPIONs@PDA exhibits the clear reflections of Fe3O4 phase with low intensity compared with SPIONs. It was suggested that the lower crystallinity of the Fe3O4 particles in SPIONs@PDA samples due to the PDA modification. On the other hand, the magnetic properties of SPIONs and SPIONs@PDA were recorded with VSM. The magnetization of SPIONs and SPIONs@PDA was shown as a function of the variation of magnetic field which was shown in Fig. 6b. The saturation magnetization was around 57.0 and 9.4 emu g−1 for SPIONs and SPIONs@PDA, respectively. The decrease of saturation magnetization for SPIONs@PDA was due to the encapsulation of SPIONs into the polymer coating. However, the zero coercivity and remanence of the magnetization curve indicated that immobilization process didn't affect the supraparamagnetism of the SPIONs in SPIONs@PDA, and SPIONs@PDA remained the superparamagnetic nature.
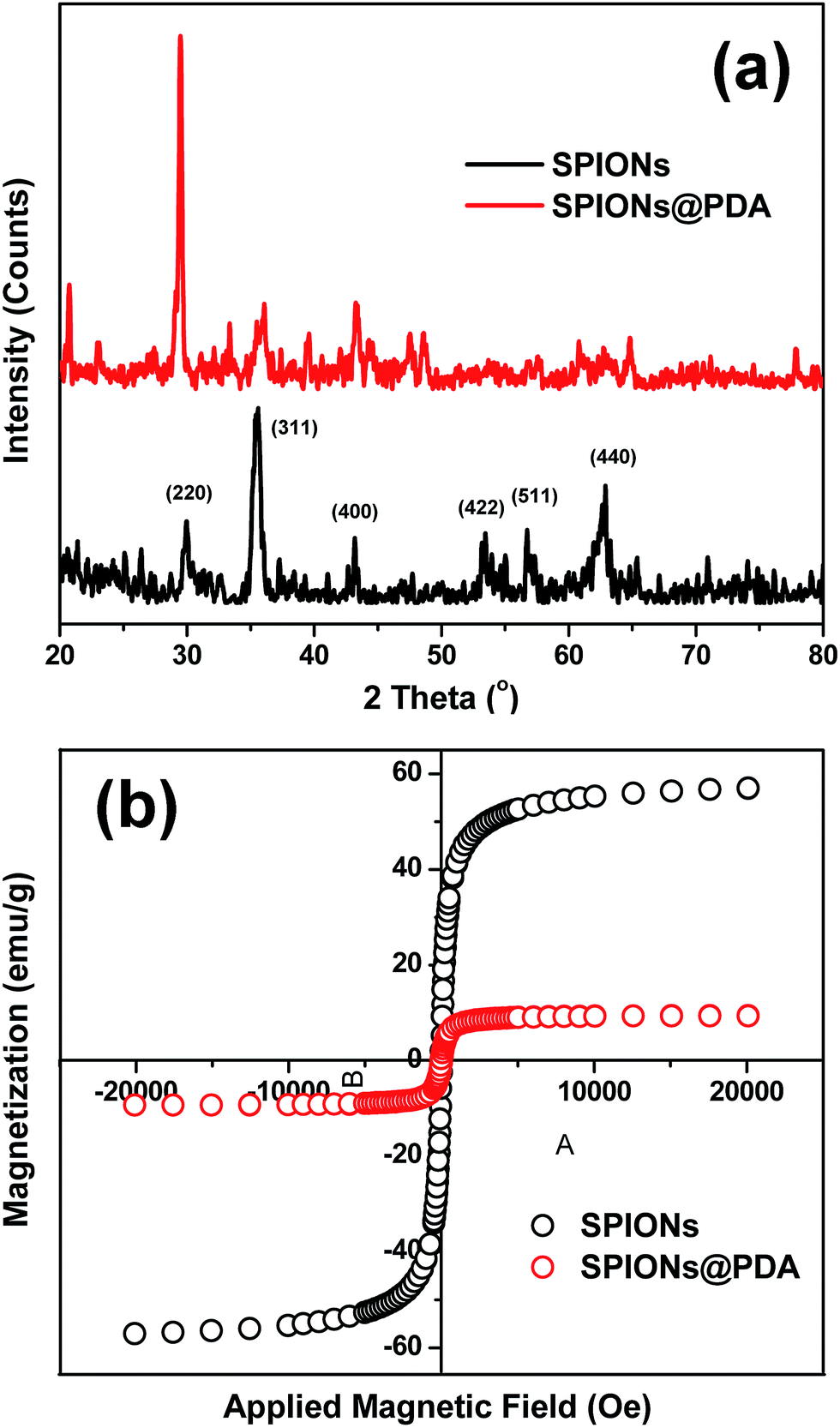 | ||
| Fig. 6 (a) XRD patterns of the SPIONs (black) and SPIONs@PDA (red). (b) Magnetization studies of SPIONs (black) and SPIONs@PDA (red). | ||
To confirm the reduction-sensitivity of the SPIONs@PDA, the degradation experiments were carried out by DLS. The size change of SPIONs@PDA in response to 10 and 20 mM GSH in PBS buffer (pH 7.4) at 37 °C was followed by DLS measurements. As shown in Fig. 7a and 6b, a rapid degradation was observed for SPIONs@PDA in the first 0.5 h, in which the particle size increased from about 170 nm to 800 nm, then reaching over 1100 nm after 2 h. It was indicated that an effectively reductive cleavage of the disulfide bonds resulted in a swelling structure in aqueous solution. On the contrary, the size decrease of the nanoparticles after 4 h can be explained by that SPIONs@PDA network was disassembled into small polymer chains and dissociated SPIONs due to the degradation of the SPIONs@PDA. However, no change in size of nanoparticle was discerned after 24 h in the absence of GSH.
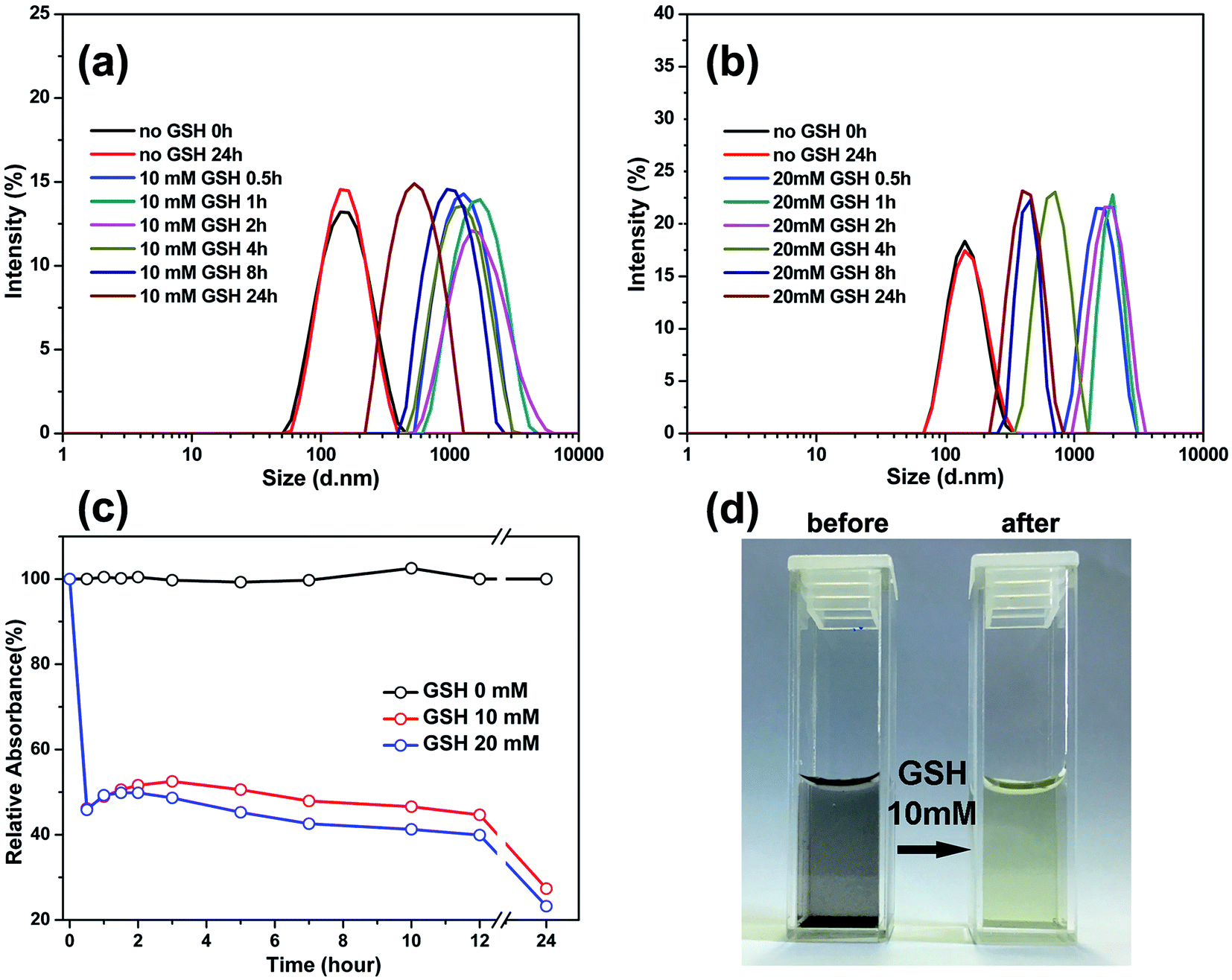 | ||
| Fig. 7 Investigations on the reduction-sensitive SPIONs@PDA degradation: The size change of SPIONs@PDA response to GSH of 10 (a) and 20 mM (b) by DLS measurement. (c) The relative turbidity in the presence or absence of 10 and 20 mM GSH. (d) The photograph of undegraded nanoparticles (left) and degraded nanoparticles (right). | ||
The turbidity measurement was also used to monitor the degradation behavior by calculating the ratio of the absorbance of the SPIONs@PDA to that of the initial nondegraded nanoparticles. Fig. 7c shows the degradation extent of the SPIONs@PDA by relative turbidity in phosphate buffer (pH 7.4) at 37 °C in the presence of 10 and 20 mM GSH. Upon the addition of GSH, the relative turbidity decreased quickly because of the degradation of the nanoparticles within half an hour. However, the nanoparticles kept stable in the solution without GSH, thus, the relative turbidity was almost 100%. As shown in Fig. 7d, after the addition of GSH, the turbid solution of SPIONs@PDA changed into a clear solution within half an hour, which was consistent with the turbidity measurement. The above results indicated that the SPIONs@PDA with disulfide bond were strongly sensitive against reducing environment. Therefore, it is expected that the degradation of nanoparticles can trigger the release of the encapsulated drug in a GSH reducing environment through disulfide-thiol reaction, thus, the SPIONs@PDA have great potential as intracellular and stimulus sensitive drug carriers for controlled drug delivery.
3.2. Relaxivity measurement
Because SPIONPs are generally used as a T2 contrast agent, in order to evaluate the effect of SPIONs@PDA the transverse relaxivities r2 are calculated through by the slopes of the linear least-squares fitting of 1/relaxation time (s−1) versus the iron concentration (mM Fe) two linear lines. The relaxation times of samples were measured at 7 T on an MRI scanner at room temperature. The DOX-loaded SPIONs@PDA showed effective T2 contrast under a T2-weighted imaging sequence with a calculated r2 value of 33.53 mM−1 S−1, as shown in Fig. 8.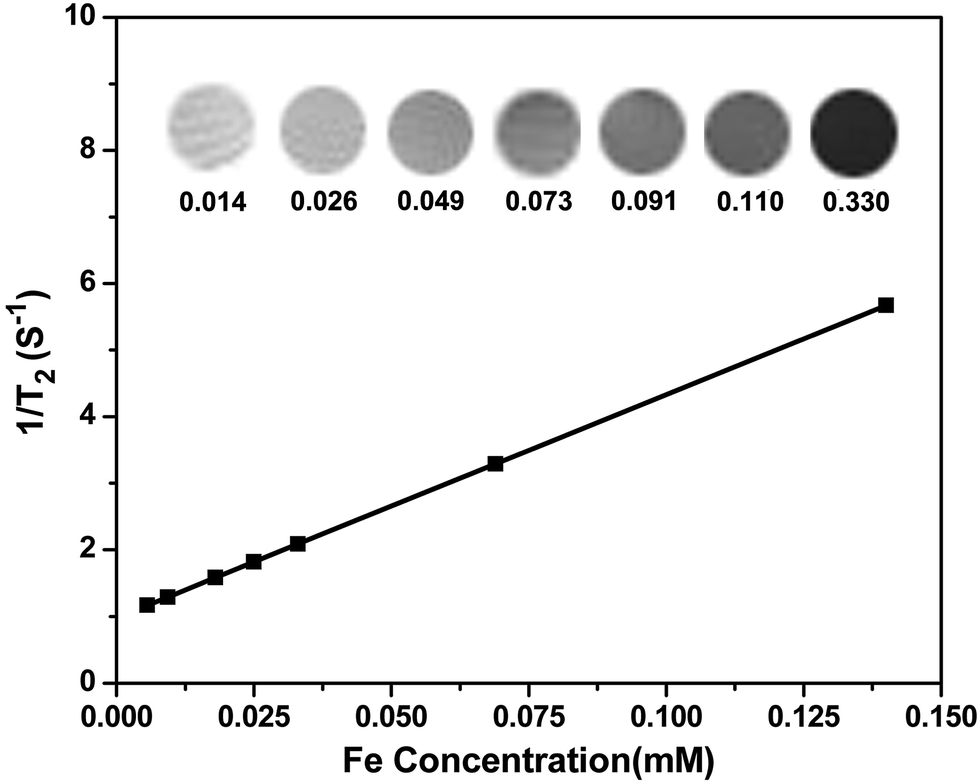 | ||
| Fig. 8 T2 relaxation rates (1/T2, s−1) as a function of iron concentration (mM) for the SPIONs@PDA. T2-weighted MRI images of the SPIONs@PDA were inserted. | ||
3.3. In vitro drug release
DOX is a wildly used DNA interacting drug in chemotherapy, thus, to test the loading and releasing capacity of the magnetic nanoparticles DOX was chosen as a hydrophobic model and entrapped into SPIONs@PDA. DOX can be encapsulated into the network of SPIONs@PDA by the hydrophobic interaction and hydrogen bond, because there are a sufficient amount of hydrophobic polymer chains in the PDA networks between inner SPIONs and hydrophilic PEG shell. The SPIONs@PDA were loaded with DOX in aqueous solution (pH 8.0) at room temperature, giving the DOX loading content of 24 wt% and encapsulation efficiency of 29 wt%. As shown in Fig. 9, the release behavior of the free DOX in PBS (pH 7.4) was investigated, and that of the entrapped DOX from the SPIONs@PDA were also performed with different concentrations of GHS (0, 10, and 20 mM) to induce the degradation of PDA crosslinked copolymer and enhance release of drugs at 37 °C. Compared with the burst release of free DOX in the first 6 h, a sustained drug release from SPIONs@PDA was observed. For DOX-loaded SPIONs@PDA, the release rate was only 22% with no GSH after 24 h, when the GSH concentration was set to 10 mM, a quick release of DOX from the nanoparticle was detected, where about 50% DOX released in 24 h. In addition, a higher cumulative release about 68% of DOX was released from SPIONs@PDA in presence of 20 mM GSH in PBS (pH = 7.4) at 37 °C. This effective drug release was likely because that following a degradation process DOX-loaded SPIONs@PDA were destabilized by the cleavage of disulfide bonds and the drug diffusion channels were formed, and then the drug released from nanoparticles, which could be potentially facilitated in the intracellular reducing environment.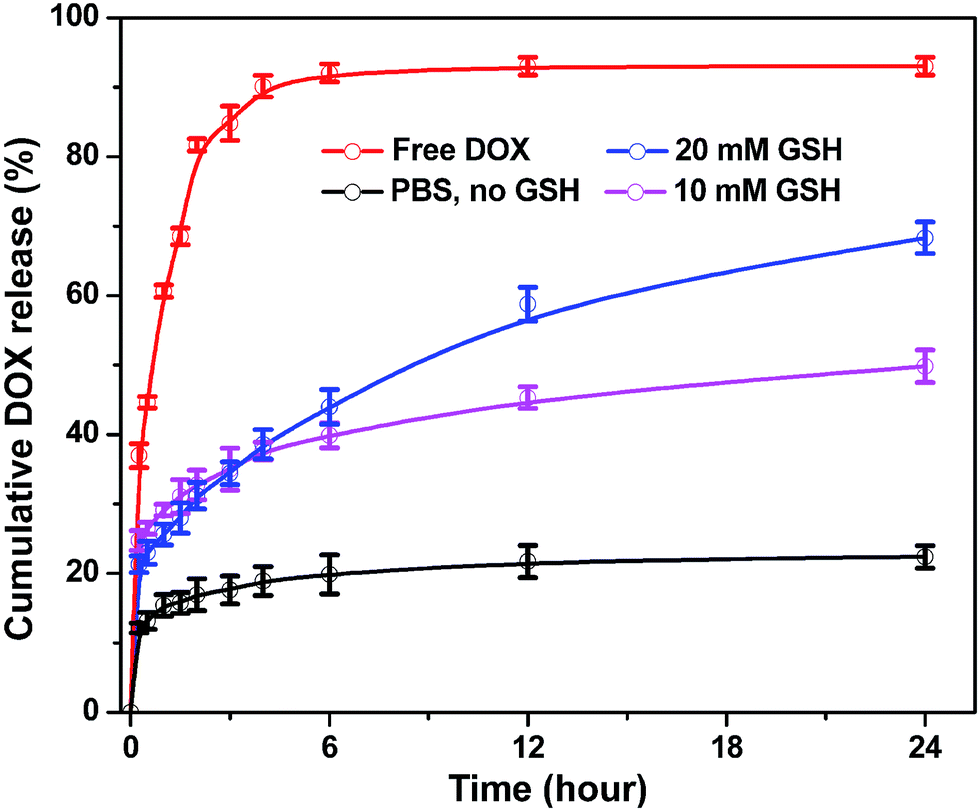 | ||
| Fig. 9 In vitro DOX release profiles of DOX-loaded SPIONs@PDA in different concentration of GSH. | ||
3.4. In vitro cell assays
It could be expected that DOX-loaded SPIONs@PDA would be degraded in cellular media due to the presence of GSH, thus, the released DOX would kill cancer cells. Based on this idea, the in vitro cell assays were performed to evaluate the cellular uptake and intra-cellular drug release of DOX-loaded SPIONs@PDA, and the cytotoxicity of free DOX, blank SPIONs@PDA, and DOX-loaded SPIONs@PDA with HeLa cells by CLSM and CCK8 assay.CLSM was used to characterize the cell uptake behavior. After 24 h of cell culture the HeLa cells were incubated with DOX-loaded SPIONs@PDA and free DOX. The fluorescence images of the SPIONs@PDA were taken at 2 and 24 h. In comparison to the free DOX that a weaker DOX fluorescence was observed in the HeLa cells after 2 h incubation with DOX-loaded SPIONs@PDA as shown in Fig. 10a2, indicating that only small amount of DOX-loaded SPIONs@PDA had been endocytized into the cells. The weaker red fluorescence than that of free DOX in Fig. 10c2 could be explained by the quicker diffuse rate into the cells of the free DOX, and the slow release kinetics of SPIONs@PDA. When incubation with DOX-loaded SPIONs@PDA lasted for 24 h, the gradually enhanced red fluorescence was observed in Fig. 10b2 indicating that more DOX-loaded SPIONs@PDA were endocytized into the cells and more DOX was released. These experimental results suggested that it took a period of time for SPIONs@PDA to release most loaded drugs into the cells. In summary, the prepared nano-carrier SPIONs@PDA has a potential application in chemotherapy for cancer treatment.
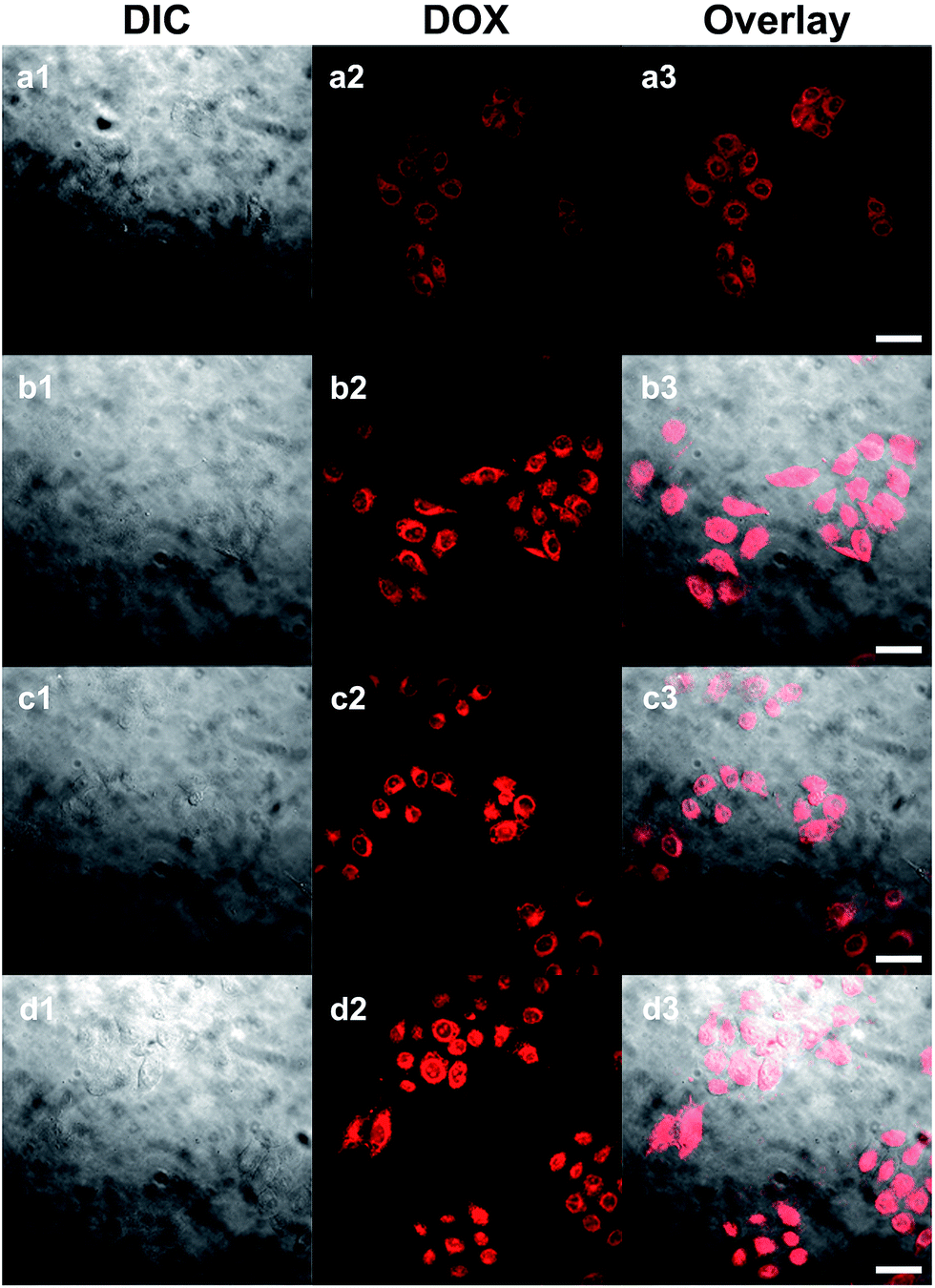 | ||
| Fig. 10 CLSM images of HeLa cells upon DOX-loaded SPIONs@PDA over incubation: (a) 2 h and (b) 24 h; cells cultured with free DOX: (c) 2 and (d) 24 h. I, the DIC images; II, DOX fluorescence images; III, overlay. The scale bar corresponds to 10 μm. | ||
Additionally, to study the tumor cell targeting property of the magnetic nanoparticles, the cellular uptake of DOX-loaded SPIONs@PDA was performed with CLSM using HK-2 cells (human tubular epithelial cells, normal cells). Shown in Fig. 11, a weaker DOX fluorescence was shown, and almost no DOX fluorescence was observed inside the HK-2 cells incubated with DOX-loaded SPIONs@PDA for 24 h, while the strong DOX fluorescence was shown inside the HeLa cells incubated with DOX-loaded SPIONs@PDA. The difference in DOX fluorescence is likely due to significantly faster intracellular release of DOX in HeLa cells, because disulfide linkages could be rapidly cleaved in the intracellular compartment caused by the presence of comparatively much higher GSH concentrations in HeLa cells than HK-2 cells. Based on the experimental results, the SPIONs@PDA nanoparticles can be used as potential nano-carriers for targeting drug delivery system.
 | ||
| Fig. 11 CLSM images of HK-2 cells upon DOX-loaded SPIONs@PDA over incubation of 24 h. | ||
The in vitro cell experiments were also conducted for evaluation of the cytotoxic activity for free DOX and encapsulated DOX in cancer cells. The cytotoxicity was assessed using CCK8 assay against HeLa cancer cells after 2 and 24 h incubation with DOX-loaded SPIONs@PDA, free DOX and blank SPIONs@PDA, respectively. As shown in Fig. 12a, DOX-loaded SPIONs@PDA showed similar cytotoxicity to free DOX at a DOX dose of about 14.4 μg mL−1 after 24 h of incubation. The experimental data showed that only 21% of the cells remained viable at a DOX dose of 14.4 μg mL−1 when treated with DOX-loaded SPIONs@PDA, while the blank SPIONs@PDA control showed almost no cytotoxic effect on the viability of HeLa cells. This observation indicated the effective internalization by the HeLa cells and cumulative drug release from DOX-loaded SPIONs@PDA due to the cleavage of disulfide bond within the reducing cytoplasmic environment. In Fig. 12b, the amount of DOX from DOX-loaded SPIONs@PDA required to achieve 50% of growth inhibition (IC50) for HeLa cells was 10.5 μg mL−1 at 2 h after treatment, and was much higher than that of free DOX (6.8 μg mL−1), which was likely due to the quicker diffuse rate into the cells of the free DOX and slow release kinetics of DOX-loaded SPIONs@PDA. Therefore, for DOX-loaded SPIONs@PDA, significant killing of cancer cell over what can be achieved by free DOX was not observed for the first 2 h. Conversely, after 24 h, the drug-nanoparticles conjugate achieved significantly better efficacy (6.9 μg mL−1, 34% reduction of IC50), which was similar to IC50 of free drug (5.7 μg mL−1).
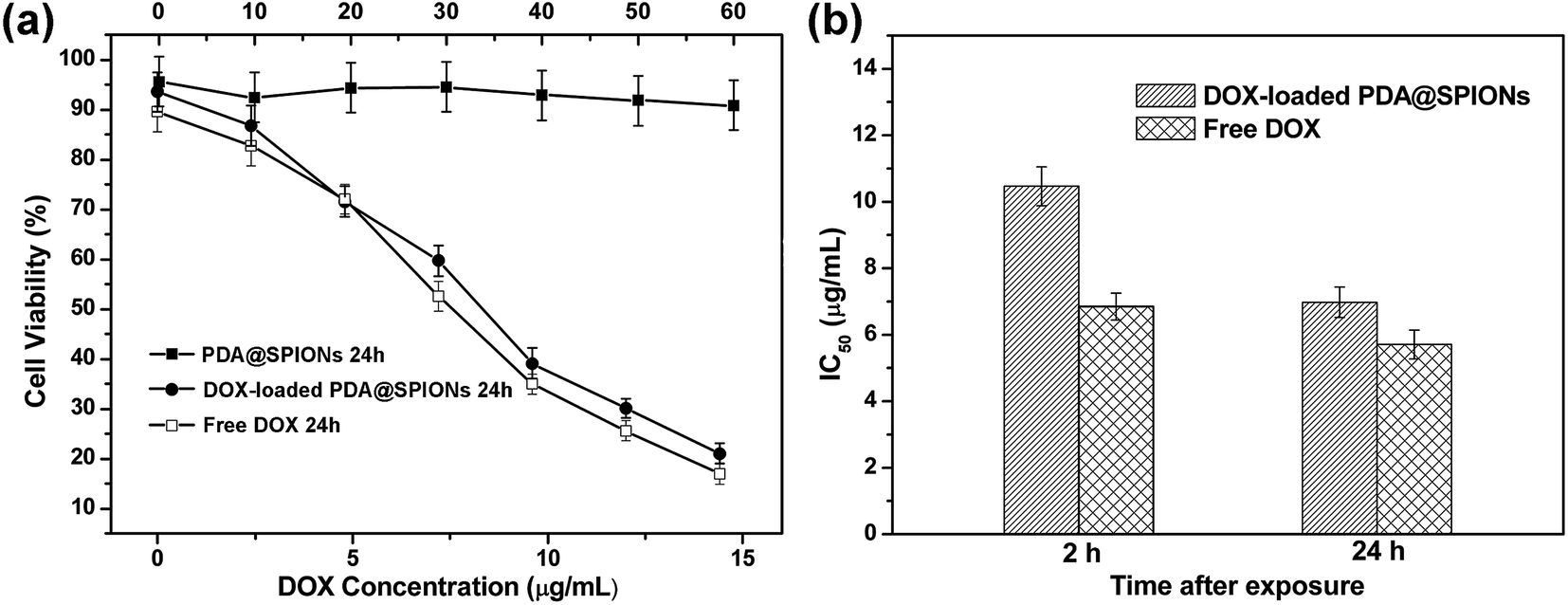 | ||
| Fig. 12 Cell survival assay of cells. (a) HeLa cells: (■) blank SPIONs@PDA, (●) DOX-loaded SPIONs@PDA, and (□) free DOX for 24 h. The concentration of blank SPIONs@PDA was shown on the top x-axis. (b) Graphic presentation for IC50 values (in μg mL−1) of free DOX and DOX-loaded SPIONs@PDA at 2 and 24 h after treatment of HeLa cells. | ||
Overall, we can conclude that the SPIONs@PDA exhibits many favorable traits, such as the biocompatibility and biodegradability, the capability of loading anticancer drugs, minimal drug release in a non-reductive extracellular condition, effective drug release in response to the reducing cytoplasmic environment, which makes the SPIONs@PDA an intriguing candidate for ideal drug carriers in the cancer therapy.
4. Conclusions
In summary, we demonstrated a facile method to prepare a reducible magnetic nanoparticle for both delivery of anti-cancer drug and MRI detection. The reduction-sensitive SPIONs@PDA exhibited a minimal drug release at neutral physiological environment compared to an effective drug release in response to the reducing environment. In addition, because of the encapsulation of the superparamagnetic iron oxide SPIONs@PDA exhibited good T2 contrast as potential MRI contrast agent. To further study the thermal behavior and the anti-fouling property of the nanoparticles and related molecules, DSC analysis and adsorption experiments of serum proteins are of great importance, and these experiments will be performed in future work.The cellular uptake and cytotoxicity of the DOX-loaded SPIONs@PDA against HeLa cells demonstrated that the nanoparticles could be internalized by HeLa cell and kill the cells effectively. We are convinced that this reducible magnetic nanoparticle with supraparamagnetism, good biocompatibility, excellent stability, and reduction-sensitivity holds great promise for constructing safe and effective targeting system of drug delivery and MRI detection in cancer theranostics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 21404086), and the Fundamental Research Funds for the Central Universities (2017NZYQN05), and the research project of Sichuan Education Office (16ZA0284).References
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48 CrossRef CAS PubMed.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. Von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- Q. Liu, L. Song, S. Chen, J. Gao, P. Zhao and J. Du, Biomaterials, 2017, 114, 23–33 CrossRef CAS PubMed.
- A. O. Elzoghby, A. L. Hemasa and M. S. Freag, J. Controlled Release, 2016, 243, 303–322 CrossRef CAS PubMed.
- J. Chen, M. Shi, P. Liu, A. Ko, W. Zhong, W. Liao and M. M. Xing, Biomaterials, 2014, 35, 1240–1248 CrossRef CAS PubMed.
- C. Fang, F. M. Kievit, O. Veiseh, Z. R. Stephen, T. Wang, D. Lee, R. G. Ellenbogen and M. Zhang, J. Controlled Release, 2012, 162, 233–241 CrossRef CAS PubMed.
- Y. Huang, K. Mao, B. Zhang and Y. Zhao, Mater. Sci. Eng., C, 2017, 70, 763–771 CrossRef CAS PubMed.
- I. Karimzadeh, M. Aghazadeh, M. R. Ganjali, P. Norouzi, S. Shirvani-Arani, T. Doroudi, P. H. Kolivand, S. A. Marashi and D. Gharailou, Mater. Lett., 2016, 179, 5–8 CrossRef CAS.
- L. Zhang, Y. Li and C. Y. Jimmy, J. Mater. Chem. B, 2014, 2, 452–470 RSC.
- L.-m. Huang, L.-d. Li, L. Shang, Q.-h. Zhou and J. Lin, React. Funct. Polym., 2016, 107, 28–34 CrossRef CAS.
- S. Cao, Y. Liu, H. Shang, S. Li, J. Jiang, X. Zhu, P. Zhang, X. Wang and J. Li, J. Controlled Release, 2017, 256, 182–192 CrossRef CAS PubMed.
- Q. Zhou, J. Lin, J. Wang, F. Li, F. Tang and X. Zhao, Prog. Nat. Sci.: Mater. Int., 2009, 19, 1529–1536 CrossRef CAS.
- L. Li, L. Shang, K. Chen, Q. Wang, J. Luo, Q. Zhou and J. Lin, J. Nanosci. Nanotechnol., 2017, 17, 4532–4541 CrossRef.
- X. Chen, W. Wu, Z. Guo, J. Xin and J. Li, Biomaterials, 2011, 32, 1759–1766 CrossRef CAS PubMed.
- Y. Wang, J. Nie, B. Chang, Y. Sun and W. Yang, Biomacromolecules, 2013, 14, 3034–3046 CrossRef CAS PubMed.
- K. Zhu, Z. Deng, G. Liu, J. Hu and S. Liu, Macromolecules, 2017, 50, 1113–1125 CrossRef CAS.
- K. Ulbrich, K. i. Holá, V. Šubr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- M. Ghadiri, E. Vasheghani-Farahani, F. Atyabi, F. Kobarfard, F. Mohamadyar-Toupkanlou and H. Hosseinkhani, J. Biomed. Mater. Res., Part A, 2017, 105, 2851–2864 CrossRef CAS PubMed.
- M. C. Kim, M. M. Lin, Y. Sohn, J. J. Kim, B. S. Kang and D. K. Kim, J. Biomed. Mater. Res., Part B, 2017, 105, 145–154 CrossRef CAS PubMed.
- Q. Hu, C. J. Rijcken, E. van Gaal, P. Brundel, H. Kostkova, T. Etrych, B. Weber, M. Barz, F. Kiessling and J. Prakash, J. Controlled Release, 2016, 244, 314–325 CrossRef CAS PubMed.
- C. Yao, J. Liu, X. Wu, Z. Tai, Y. Gao, Q. Zhu, J. Li, L. Zhang, C. Hu and F. Gu, J. Controlled Release, 2016, 232, 203–214 CrossRef CAS PubMed.
- Q. Hu, C. J. Rijcken, R. Bansal, W. E. Hennink, G. Storm and J. Prakash, Biomaterials, 2015, 53, 370–378 CrossRef CAS PubMed.
- Q. Fu, J. Xu, K. Ladewig, T. Henderson and G. Qiao, J. Controlled Release, 2015, 6, 35–43 CAS.
- Y. Shi, C. F. van Nostrum and W. E. Hennink, ACS Biomater. Sci. Eng., 2015, 1, 393–404 CrossRef CAS.
- W. Stark, P. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- Q.-H. Zhou, J. Lin, L.-D. Li and L. Shang, Colloid Polym. Sci., 2015, 293, 2291–2300 CAS.
- K. Yang, L. Feng and Z. Liu, Adv. Drug Delivery Rev., 2016, 105, 228–241 CrossRef CAS PubMed.
- M. A. Ghaz-Jahanian, F. Abbaspour-Aghdam, N. Anarjan, A. Berenjian and H. Jafarizadeh-Malmiri, Mol. Biotechnol., 2015, 57, 201–218 CrossRef CAS PubMed.
- C.-C. Chuang and C.-W. Chang, ACS Appl. Mater. Interfaces, 2015, 7, 7724–7731 CAS.
- L. L. Israel, E. Lellouche, S. Ostrovsky, V. Yarmiayev, M. Bechor, S. Michaeli and J.-P. Lellouche, ACS Appl. Mater. Interfaces, 2015, 7, 15240–15255 CAS.
- J. Wang, B. Zhang, L. Wang, M. Wang and F. Gao, Mater. Sci. Eng., C, 2015, 48, 416–423 CrossRef CAS PubMed.
- X. Ma, A. Gong, B. Chen, J. Zheng, T. Chen, Z. Shen and A. Wu, Colloids Surf., B, 2015, 126, 44–49 CrossRef CAS PubMed.
- G. Kandasamy and D. Maity, Int. J. Pharm., 2015, 496, 191–218 CrossRef CAS PubMed.
- G. Marcelo, A. Muñoz-Bonilla and M. Fernández-García, J. Phys. Chem. C, 2012, 116, 24717–24725 CAS.
- A. L. Volsi, D. J. de Aberasturi, M. Henriksen-Lacey, G. Giammona, M. Licciardi and L. M. Liz-Marzán, J. Mater. Chem. B, 2016, 4, 1150–1155 RSC.
- A. Gulzar, S. Gai, P. Yang, C. Li, M. B. Ansari and J. Lin, J. Mater. Chem. B, 2015, 3, 8599–8622 RSC.
- P. C. Liang, Y. C. Chen, C. F. Chiang, L. R. Mo, S. Y. Wei and W. Y. Hsieh, Int. J. Nanomed., 2016, 11, 2021–2037 CAS.
- C. Scialabba, R. Puleio, D. Peddis, G. Varvaro, P. Calandra and G. Cassata, Nano Res., 2017, 10, 3212–3227 CrossRef CAS.
- M. Arshad, R. A. Pradhan and A. Ullah, Mat. Sci. Eng. C-Mater., 2017, 76, 217–223 CrossRef CAS PubMed.
- M. Arshad, S. Saied and A. Ullah, Rsc Adv., 2014, 4, 26439–26446 RSC.
- S. Laurent, A. A. Saei, S. Behzadi, A. Panahifar and M. Mahmoudi, Expert Opin. Drug Del., 2014, 11, 1449–1470 CrossRef CAS PubMed.
- S. Zhang, M. Arshad and A. Ullah, Nat. Nanotechnol., 2015, 26, 415703 CrossRef PubMed.
- Y.-S. Choi, H. Kang, D.-G. Kim, S.-H. Cha and J.-C. Lee, ACS Appl. Mater. Inter., 2014, 6, 21297–21307 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |