
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a 12-membered cyclic siloxane possessing alkoxysilyl groups as a nanobuilding block and its use for preparation of gas permeable membranes†
Masashi Yoshikawa a,
Hiroya Shibaa,
Masakoto Kanezashib,
Hiroaki Wadaa,
Atsushi Shimojimaa,
Toshinori Tsurub and
Kazuyuki Kuroda*ac
a,
Hiroya Shibaa,
Masakoto Kanezashib,
Hiroaki Wadaa,
Atsushi Shimojimaa,
Toshinori Tsurub and
Kazuyuki Kuroda*ac
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Ohkubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: kuroda@waseda.jp
bDepartment of Chemical Engineering, Graduate School of Engineering, Hiroshima University, 1-4-1 Kagami-yama, Higashi-Hiroshima 739-8527, Japan
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo, 169-0051, Japan
First published on 17th October 2017
Abstract
A 12-membered cyclic siloxane possessing alkoxysilyl groups was synthesized as a nanobuilding block for siloxane-based materials by the alkoxysilylation of organometallasiloxane containing a 12-membered ring with Si–Me and Si–O− groups as the side groups. The cyclic structure was retained not only in the hydrolysis and condensation reactions (sol–gel process) of the alkoxysilyl groups but also in the xerogel and membrane preparation processes. The degree of condensation of the xerogel derived from the 12-membered ring siloxane was higher than that derived from alkoxysilane monomers, indicating that the alkoxysilylated cyclic oligosiloxane is useful for controlling siloxane networks. A membrane composed of the cyclic siloxane was prepared by coating the hydrolyzed solution onto a porous alumina tube for evaluating the gas permeation properties. The membrane showed a molecular sieving effect for H2/SF6.
Introduction
Siloxane-based porous materials have been used in practical applications, such as catalysis, separation, and adsorption,1,2 because of their thermal and chemical stability and high compatibility with other materials (such as polymers, metals, and metal oxides) for composite formation. The properties of such materials depend on the structure of the siloxane network; therefore, the structural control at a molecular level is important.The use of nanobuilding blocks with defined oligosiloxane structures is quite effective for controlling siloxane networks at the molecular level.3 This pathway provides unique materials that cannot be obtained from monomeric silicon compounds. Branched, cyclic, and cage-type oligosiloxanes have been synthesized and used as nanobuilding blocks for various siloxane-based nanomaterials.4,5 Among them, cyclic siloxanes are expected to act as nanobuilding blocks possessing inclusion properties similar to cyclic organic compounds such as cyclodextrins and crown ethers.6 Actually, inclusion compounds composed of cyclic penta-, hexa-, and hepta-siloxanes and metal ions have been reported,7–9 which indicates that the cavity within cyclic siloxanes is accessible to some guest species. The inner spaces provided by larger cyclic siloxanes are expected to show unique host–guest interactions with various molecular species. In contrast to many reports concerning the use of organic host compounds (carbon based), the effective utilization of the cavity of cyclic siloxanes remains largely unexplored.
There have been many reports concerning the synthesis of cyclic siloxanes with various ring sizes. In this study, we have chosen a 12-membered cyclic siloxane as a nanobuilding block. The inside diameter of the 12-membered siloxane ring is roughly estimated to be ca. 0.9 nm when the ring structure is fully extended and planar,‡ and the ring is larger than that of a benzene ring. The 12-membered cyclic siloxanes are easily obtained as complexes with metal cations by the hydrolysis and condensation of organotrialkoxysilanes in the presence of alkali metal hydroxide and transition metal cations.10,11 Despite the attractiveness of cyclic siloxanes, 12-membered cyclic siloxanes have rarely been used as nanobuilding blocks for the formation of siloxane-based materials to the best of our knowledge. Shchegolikhina et al.12,13 reported the preparation of layered compounds by the solid-phase condensation of 12-membered cyclic siloxanes possessing both hydroxy and phenyl groups. Zheng et al. prepared a porous polymer by the hydrosilylation polymerization of a 12-membered cyclic siloxane possessing both vinyl and hydrosilyl groups.14 Zheng et al. also reported a polymer by thiol–ene polymerization of a 12-membered cyclic siloxane possessing both vinyl and thiol groups.15 Unfortunately, these reports did not clarify the retention of the 12-membered cyclic siloxane structure in the products.
In this study, a 12-membered cyclic siloxane with alkoxysilyl groups as side groups was synthesized as a new nanobuilding block by alkoxysilylation of the complex between 12-membered cyclic siloxane and metal cations (Scheme 1). Hereafter, the obtained compound is referred to as 12MR-Me-TES (based on 12-membered ring molecule possesses Methyl groups and TriEthoxySilyloxy (TES) groups as side groups). The hydrolysis and polycondensation processes of this compound were studied in detail by NMR spectroscopies to confirm that the ring structure was retained after the reaction. Furthermore, the cyclic siloxane was hydrolyzed and polycondensed onto a porous alumina tube to prepare composite membranes, and the membrane gas permeation properties were investigated to discuss the usefulness of the cyclic siloxane as a nanobuilding block.
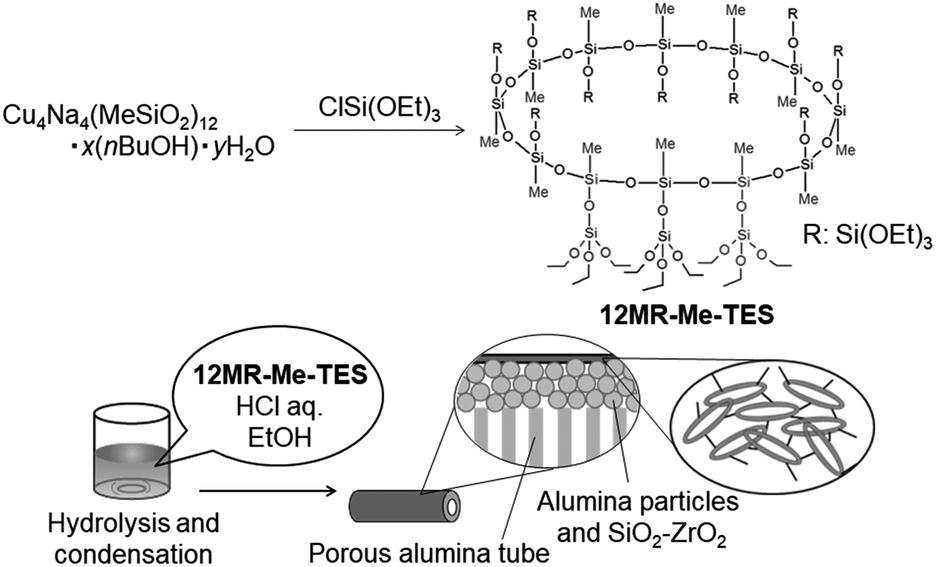 | ||
| Scheme 1 Synthesis of the cyclic siloxane with alkoxysilyl groups and the membrane preparation. | ||
Results and discussion
Characterization of 12MR-Me-TES
Fig. 1a shows the 29Si NMR spectrum of 12MR-Me-TES. The two T3 signals (Fig. 1a, A and B) observed at −67.43 ppm and −67.45 ppm can be assigned to the Si atoms constituting the 12-membered ring. No signal of the complex was observed in the T2 region (typically −56.8 ppm to −58.8 ppm (ref. 16–18)). The two Q1 signals (Fig. 1a, C and D) at −88.95 ppm and −89.04 ppm are due to the Si atoms of the TES groups. The two slightly different environments for both T3 and Q1 units can be attributed to the up-and-down arrangement of the side TES groups on the 12-membered ring siloxane, as shown in Fig. 1a. Please note that the arrangement of side groups against the 12-membered ring plane switches every three Si–O bonds. The intensity ratio of the 29Si NMR signals ((A + B)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C:D = 12:4:8) is consistent with the structure.§ These results strongly suggest that the alkoxysilylation of Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O proceeds with the retention of the 12-membered ring structure, including the up-and-down arrangement of side groups.
:C:D = 12:4:8) is consistent with the structure.§ These results strongly suggest that the alkoxysilylation of Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O proceeds with the retention of the 12-membered ring structure, including the up-and-down arrangement of side groups.
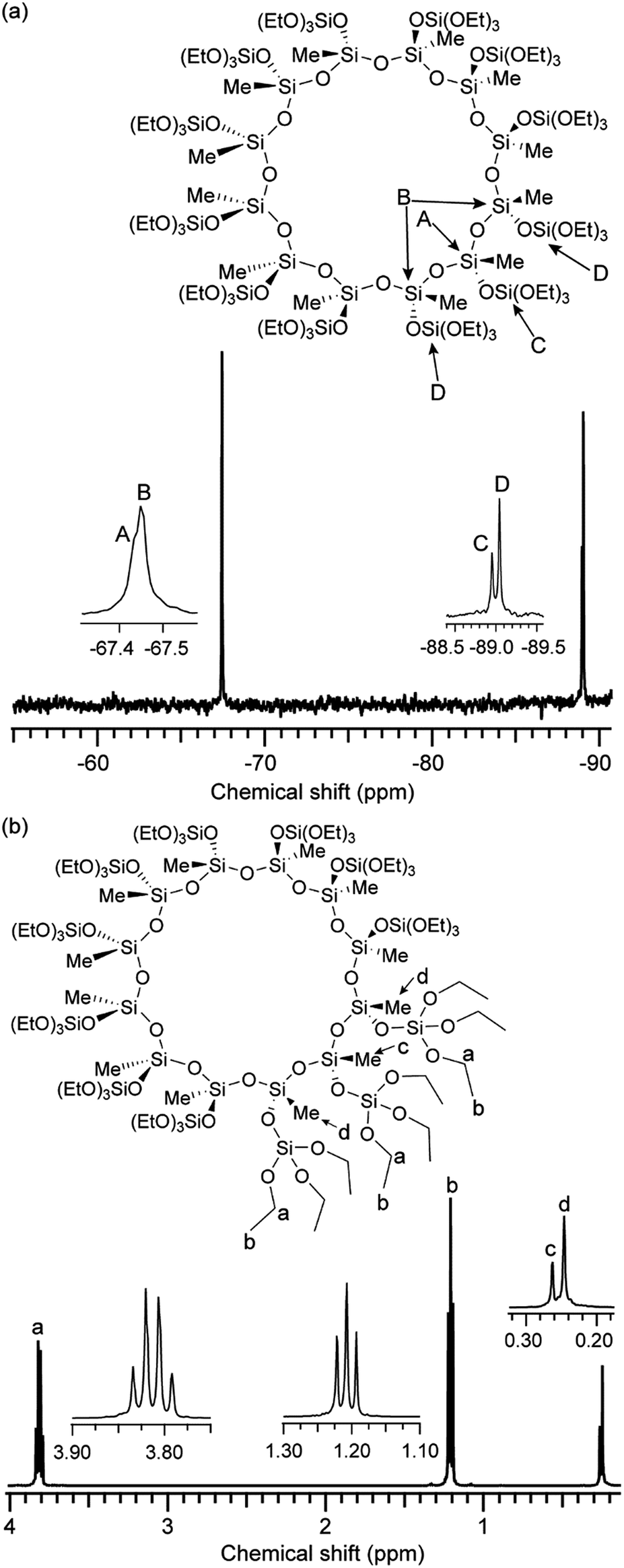 | ||
| Fig. 1 NMR spectra of 12MR-Me-TES. (a) 29Si and (b) 1H. | ||
The 1H NMR spectrum of 12MR-Me-TES (Fig. 1b) shows four signals assigned to ethoxy groups (3.83–3.79 ppm for –OCH2CH3, 1.22–1.19 ppm for –OCH2CH3) and methyl groups (0.26 ppm and 0.25 ppm). The intensity ratio of these signals is in accordance with the calculated ratio. The two different environments for the methyl carbons are consistent with the structure, as mentioned above. Regarding the ethoxy groups, the separation of the signals was too small to be observed clearly. These 1H NMR results, together with the 13C NMR results (Fig. S1 in the ESI†), also support the formation of 12MR-Me-TES.
The high-resolution electrospray ionization mass spectroscopy (ESI-MS) spectrum of 12MR-Me-TES shows a peak at m/z = 2879.8320, corresponding to the sodium adduct of 12MR-Me-TES (calcd. for 2879.8205), confirming that 12MR-Me-TES had been successfully synthesized. This is the first report concerning the synthesis of a 12-membered ring siloxane possessing alkoxysilyl groups that is available as a sol–gel precursor of siloxane-based materials. This synthetic procedure is applicable to the alkoxysilylation of metalorganosiloxanes possessing cyclic siloxane structures other than 12-membered rings,19–23 which could result in the variation of bond densities and angles of the formed siloxane networks.
Hydrolysis and polycondensation of 12MR-Me-TES
To investigate the behavior of 12MR-Me-TES in the sol–gel process, the hydrolysis and polycondensation of 12MR-Me-TES in solution was analyzed by liquid-state NMR spectroscopies. Fig. 2 shows the 13C NMR spectra of the hydrolyzed solution of 12MR-Me-TES. The signal intensity arising from the methylene carbon of the TES groups at 59.6 ppm decreased, and the intensity of the signal corresponding to the methylene carbon of ethanol (57.7 ppm) increased as the reaction time progressed. The signals arising from the TES groups almost disappeared after 3 h, which indicates the complete hydrolysis of the TES groups of 12MR-Me-TES. The signal arising from the –SiMe groups broadened with reaction time. The signal broadening is also confirmed by the 29Si NMR data (next paragraph).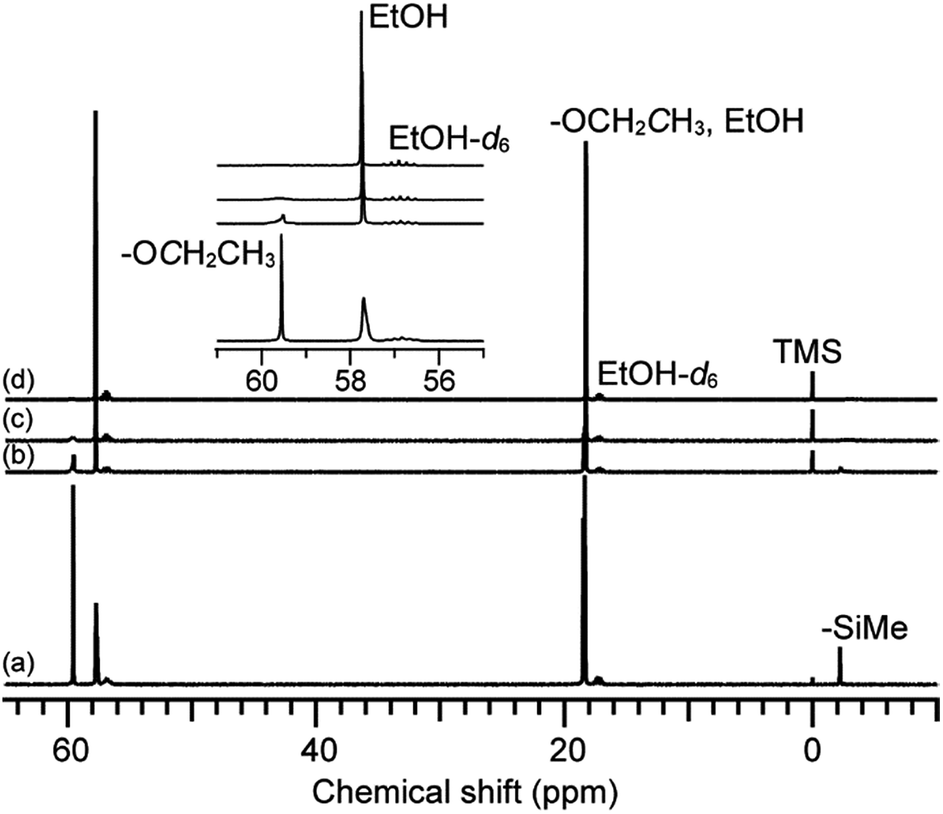 | ||
| Fig. 2 13C NMR spectra of the hydrolyzed solution of 12MR-Me-TES. (a) 0 h, (b) 1 h, (c) 3 h, and (d) 6 h. | ||
Fig. 3 shows the 29Si NMR spectra of the hydrolyzed solution of 12MR-Me-TES. Three new signals are observed in the Q region (−85 ppm to −93 ppm) after 1 h, in addition to the original signal arising from the TES groups of 12MR-Me-TES (−88.8 ppm). The new signals at −86.3 ppm, −90.7 ppm, and −92.8 ppm can be assigned to –SiOSi(OEt)2(OH), –SiOSi(OEt)(OH)(OSi), and –SiOSi(OH)2(OSi), respectively,17,24 which indicates that the partial hydrolysis and condensation of the TES groups of 12MR-Me-TES had occurred. Meanwhile, the T3 signals corresponding to the Si atoms of the 12-membered ring are observed. After 3 h and 6 h, all signals had weakened and broadened. This is caused by the following two factors: (i) the types of hydrolyzed and condensed molecules become more diverse with the progress of hydrolysis and condensation, and (ii) the molecular mobility of the condensed species is decreased by the increasing molecular weight as intermolecular condensation progresses or the rigidity of the cyclic siloxane increases through intramolecular condensation. During the reaction, no T2 signals arising from the cleavage of the Si–O–Si bonds appeared (the T2 signal of the Si atom possessing methyl group is generally observed at −56 ppm to −58 ppm), which suggests that the cyclic structure of 12MR-Me-TES is retained without rearrangement. Even if the rearrangement occurred too rapidly to observe by NMR spectroscopy, chemical shift of T3 silicon in reformed siloxanes bonds would not be shifted to downfield by 3 ppm. Regarding to the downfield shift of T3 signals, there are a couple of explanations. One probable reason is cyclization of adjacent TES groups to form four-membered rings. The other probable reason is the gradual progress of hydrolysis and condensation of TES groups, and this variation in the Q units must affect the electronic states of linked neighboring T3 silicons. Because –SiOH and –SiOSi groups show a more electron-withdrawing effect than TES group, the signals due to T3 silicon can be shifted to downfield. Therefore, the downfield shift observed in this study can be explained by the variations in the electron density during the sol–gel reaction. These results indicate that the cyclic structure of 12MR-Me-TES is retained after the sol–gel reactions, which is important for the preparation of separation/adsorption media using siloxane oligomers.
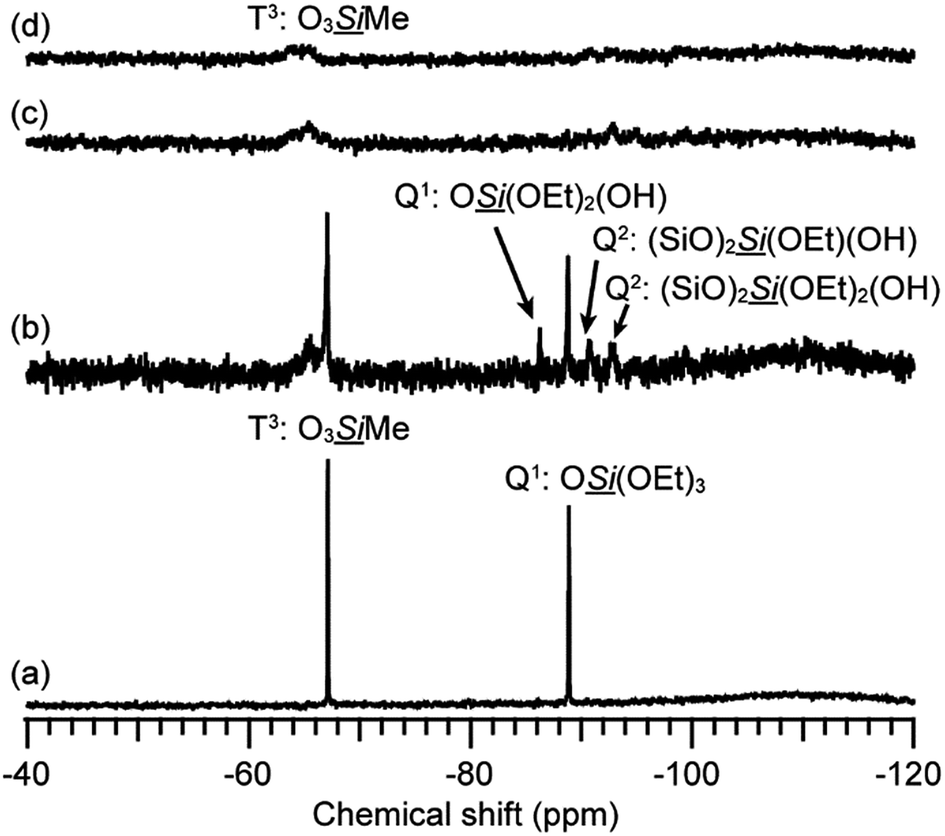 | ||
| Fig. 3 29Si NMR spectra of the hydrolyzed solution of 12MR-Me-TES. (a) 0 h, (b) 1 h, (c) 3 h, and (d) 6 h. | ||
Characterization of the 12MR-Me-TES-derived gels and tetraethoxysilane–methyltriethoxysilane-derived gels
The stability of the 12-membered ring units in the gels derived from 12MR-Me-TES was investigated. The hydrolyzed solution of 12MR-Me-TES was dried to obtain a xerogel (12MR-gel-as). Subsequently, the obtained gel was heated at 100 °C, 200 °C, or 300 °C under an argon atmosphere (12MR-gel-heat100, 12MR-gel-heat200, and 12MR-gel-heat300, respectively). These samples were analyzed by solid-state 29Si NMR spectroscopy. 12MR-gel-as was obtained by drying the solution containing hydrolyzed and partially polycondensed 12MR-Me-TES. Please note that the reaction conditions were different from those used for the investigation of hydrolysis process described in the previous section. The hydrolysis conditions in this section were the same conditions as those for the preparation of the 12MR-Me-TES-derived membrane precursor sol. The larger amount of solvent (ethanol) in these systems is favorable for the suppression of the hydrolytic cleavage of the 12-membered ring. The 29Si magic angle spinning (MAS) NMR spectrum of 12MR-gel-as shows T and Q signals (Fig. 4a). Three signals are observed in the Q region (Q2, Q3, and Q4), indicating the progress of hydrolysis and polycondensation of 12MR-Me-TES; however, the appearance of Q2 and Q3 signals indicates these reactions had not completed and –SiOEt and –SiOH groups remained. In the T region, only the T3 signal corresponding to the Si atoms in the 12-membered rings was observed, suggesting that the 12-membered ring structure of 12MR-Me-TES had been retained.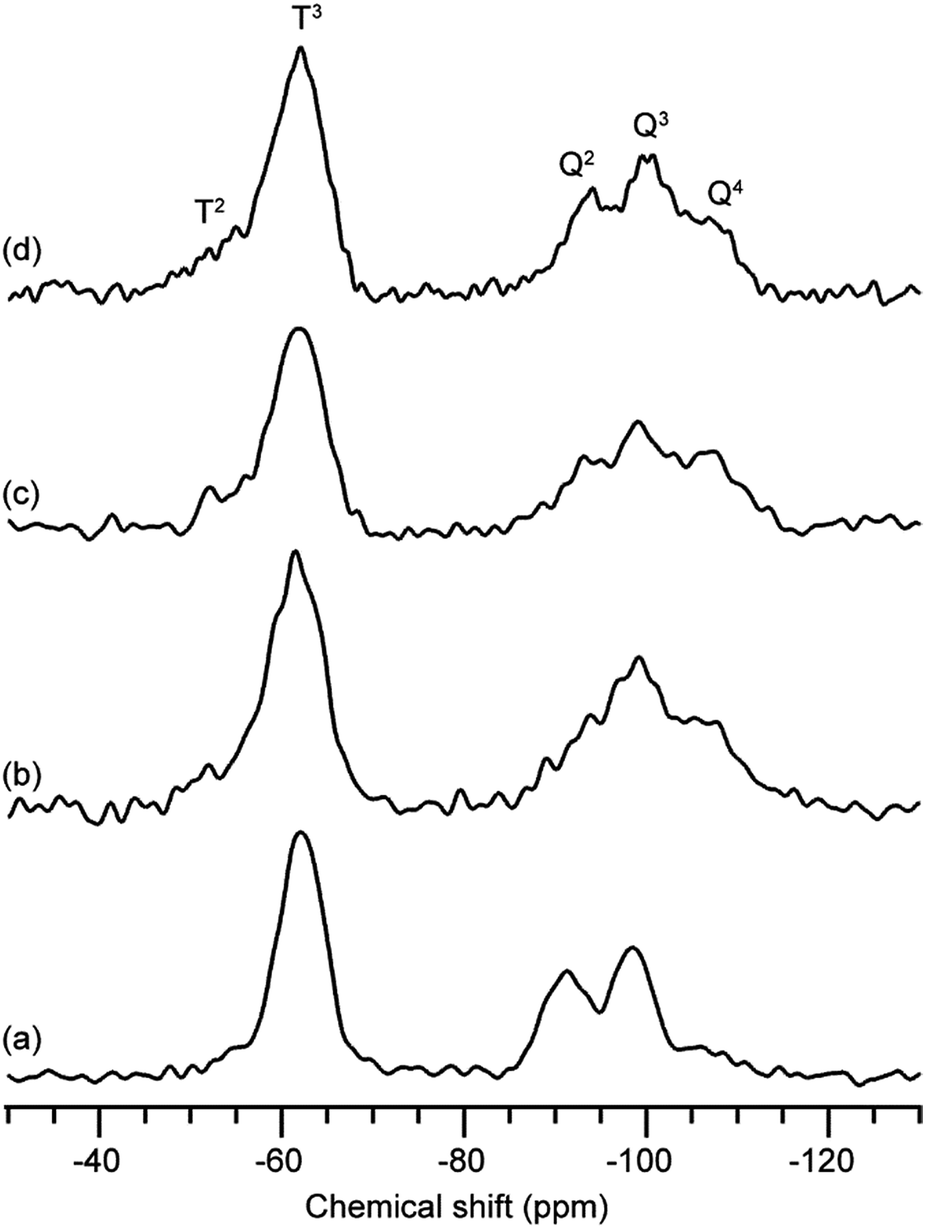 | ||
| Fig. 4 29Si MAS NMR spectra of 12MR-Me-TES-derived gel. (a) 12MR-gel-as, (b) 12MR-gel-heat100, (c) 12MR-gel-heat200, and (d) 12MR-gel-heat300. | ||
The signals assigned to ethoxy groups are observed in the 13C cross-polarization (CP)/MAS NMR spectrum (Fig. S2a in the ESI†), which indicates that the hydrolysis of 12MR-Me-TES had not completed. As described in the previous section, the hydrolysis reaction was completed under the conditions for the investigation of hydrolysis and polycondensation process of 12MR-Me-TES. Such a difference in the degree of hydrolysis can be explained by the difference in the concentrations of the HCl catalyst. The concentration of HCl in the reaction mixture for the preparation of the 12MR-Me-TES-derived gel (HCl/(EtOH + H2O) = 6.3 × 10−5) is much lower than that for the investigation of hydrolysis process of 12MR-Me-TES (HCl/(EtOH + H2O) = 3.0 × 10−3). Even the reaction time was increased from 1 d to 2 d, the ethoxy groups remained (13C CP/MAS NMR: Fig. S4 in the ESI†), and the partial cleavage of the 12-membered ring occurred (29Si MAS NMR: Fig. S5 in the ESI†). To avoid this cleavage, it is important to induce gelation after 1 d by evaporating the solvent.
After the heat treatment of 12MR-gel-as under an argon atmosphere, the polycondensation progress was confirmed by 29Si MAS NMR and Fourier-transform infrared (FT-IR) spectroscopies, which demonstrates the reduction in the number of SiOH groups (Fig. 4b–d and S3 in the ESI, respectively†), although some ethoxy groups remained (Fig. S2b–d in the ESI†). In the case of 12MR-gel-heat100, only the T3 signal was observed, in common with 12MR-gel-as (Fig. 4b), indicating that the 12-membered ring structure of 12MR-gel-as did not deteriorate at 100 °C. On the other hand, at higher temperatures, small T2 signals appeared in the 29Si MAS NMR spectra of both 12MR-gel-heat200 and 12MR-gel-heat300 (Fig. 4c and d, respectively). It is likely that the 12-membered rings were cleaved by increasing strain arising from the progress of polycondensation with heating.
For comparison, equimolar amounts of tetraethoxysilane (TEOS) and methyltriethoxysilane (MTES) were co-hydrolyzed and polycondensed to obtain a xerogel. The 29Si MAS NMR analysis confirmed that the signal intensity of the T2 silicon atom of TEOS–MTES-derived gel (Fig. S6 in the ESI†) is higher than that of 12MR-Me-TES-derived gel (Fig. 4). The lack of T2 silicon atoms in the 12MR-Me-TES-derived gel can be attributed to the usage of 12MR-Me-TES having only T3 silicon atoms as a precursor under controlled conditions. These results indicate that the cyclic oligosiloxanes, which are composed of silicon atoms with controlled condensation degree, are useful precursors to control the siloxane networks.
Gas permeation properties of 12MR-Me-TES-derived membranes and TEOS–MTES-derived membranes
The gas permeance of the 12MR-Me-TES-derived membranes was evaluated at 100 °C, and the influence of the heat treatment of the membranes was investigated. Fig. 5 shows the gas permeances of the 12MR-Me-TES-derived membranes heated at 100 °C, 200 °C, and 300 °C as a function of kinetic diameter. There are no significant changes in both the shape of these curves and the selectivity with heating temperature (e.g., H2/SF6: 550, 700, and 650 for the membranes heated at 100 °C, 200 °C, and 300 °C, respectively). The selectivity for H2/SF6 shows higher values than the expected value for Knudsen diffusion (H2/SF6 Knudsen selectivity: 6 (ref. 25)), indicating that the 12MR-Me-TES-derived membranes show a molecular sieving effect.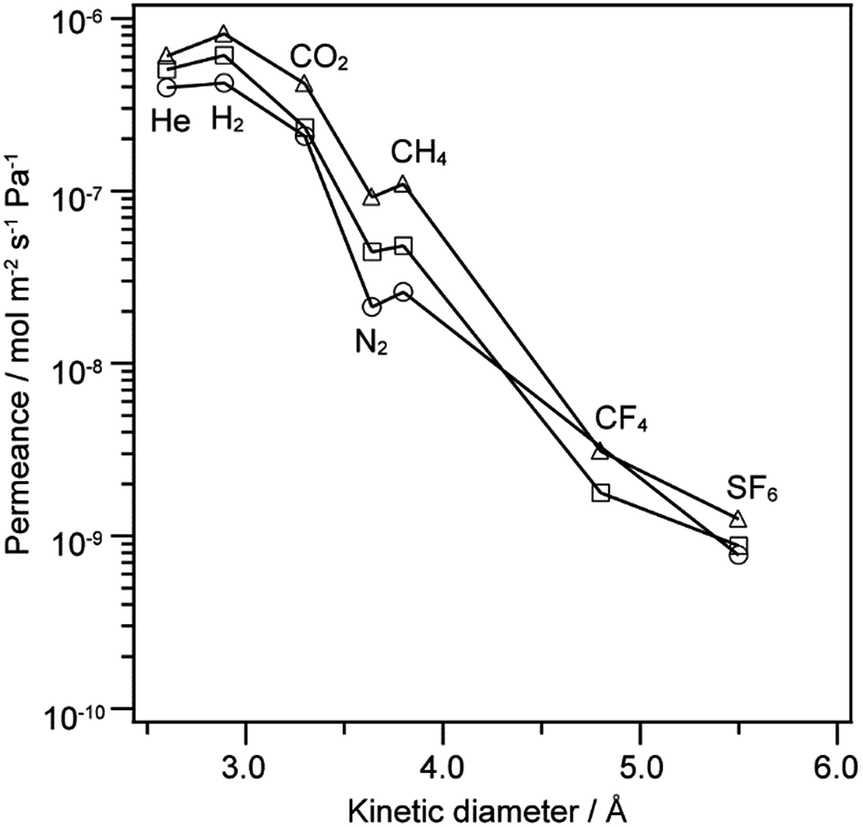 | ||
| Fig. 5 Gas permeance at 100 °C for 12MR-Me-TES-derived membranes as a function of molecular size. The membranes are heated at 100 °C (circle), 200 °C (square), and 300 °C (triangle). | ||
The slopes of the gas permeances against the kinetic diameter were almost the same, and the gas permeances were increased with increasing heat treatment temperature (Fig. 5). The former result indicates that the average pore diameter of the membrane does not depend on the heating temperature. Assuming that gas molecules pass through the inner space of 12-membered ring siloxanes, the slope of gas permeances against kinetic diameter would decrease with the cleavage of the 12-membered ring siloxanes. Actually, the 12-membered ring siloxanes are slightly cleaved by heat treatments at 200 °C and 300 °C, as shown in Fig. 4; however, the slopes were almost the same (Fig. 5). This could indicate that the relative positions of the Si atoms in the 12-membered ring siloxane of 12MR-Me-TES were not changed significantly by the cleavage of the cyclic siloxanes with heat treatment. The increase in the permeance with increasing heating temperature is probably due to the desorption of adsorbed water with heat treatment, which is often observed for sol–gel derived membranes.26
The physical adsorption of gas molecules affects the gas permeance measured at 100 °C (Fig. 5), but the effect can be ignored at 200 °C and higher. The gas permeation experiments at 100 °C show that the permeance of 12MR-Me-TES-derived membranes does not depend on their heating temperature; therefore, the gas permeance of the 12MR-Me-TES-derived membranes was evaluated at 200 °C.
TEOS–MTES-derived membrane was fabricated by hydrolysis and co-condensation of TEOS and MTES, and its gas permeance was also evaluated for comparison. Fig. 6a shows the gas permeances of the 12MR-Me-TES-derived membrane and TEOS–MTES-derived membrane as a function of kinetic diameter. Both membranes were heated at 300 °C after the coating of sols onto porous alumina tubes. Fig. 6b shows the dimensionless permeances based on the He permeance at 200 °C for these membranes. Compared with dimensionless permeance under the Knudsen mechanism, both membranes showed low dimensionless permeance between N2 and SF6, which indicates that both membranes showed a molecular sieving effect. The 12MR-Me-TES membrane showed approximately the same level of gas permeance and pore size distribution with that of TEOS–MTES membrane. There are two possible reasons for the similar permeances. (i) The flexibility of siloxane or (ii) the formation of smaller cyclic structure than the 12-membered ring among 12MR-Me-TES during the condensation of 12MR-Me-TES. (i) The shape of such a large cyclic siloxane easily changes; in fact, the precursor of the 12MR-Me-TES-derived membrane (Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O) contains a bending 12-membered ring siloxane. So, the inner space of the 12-membered cyclic siloxane may be narrowed and/or distorted by the bending of the ring structure during the sol–gel reaction. (ii) When cyclic siloxanes smaller than the 12-membered ring are formed between the intermolecular spaces of 12MR-Me-TES, the gas permeance is underestimated by the averaging of the 12-membered rings and the small intermolecular ring structures. The inclusion of some guest species into the ring before polymerization will be effective for the polymerization of extended 12-membered ring structures, which is now under investigation. In addition, intermolecular spacing among cyclic oligomers is quite important and should be further studied, although the present study suggests that there are no large spaces among the cyclic oligomers, as judged from the permeance data, which is promising for future research on the molecular design of nanobuilding block approach based on ring-type oligosiloxanes.
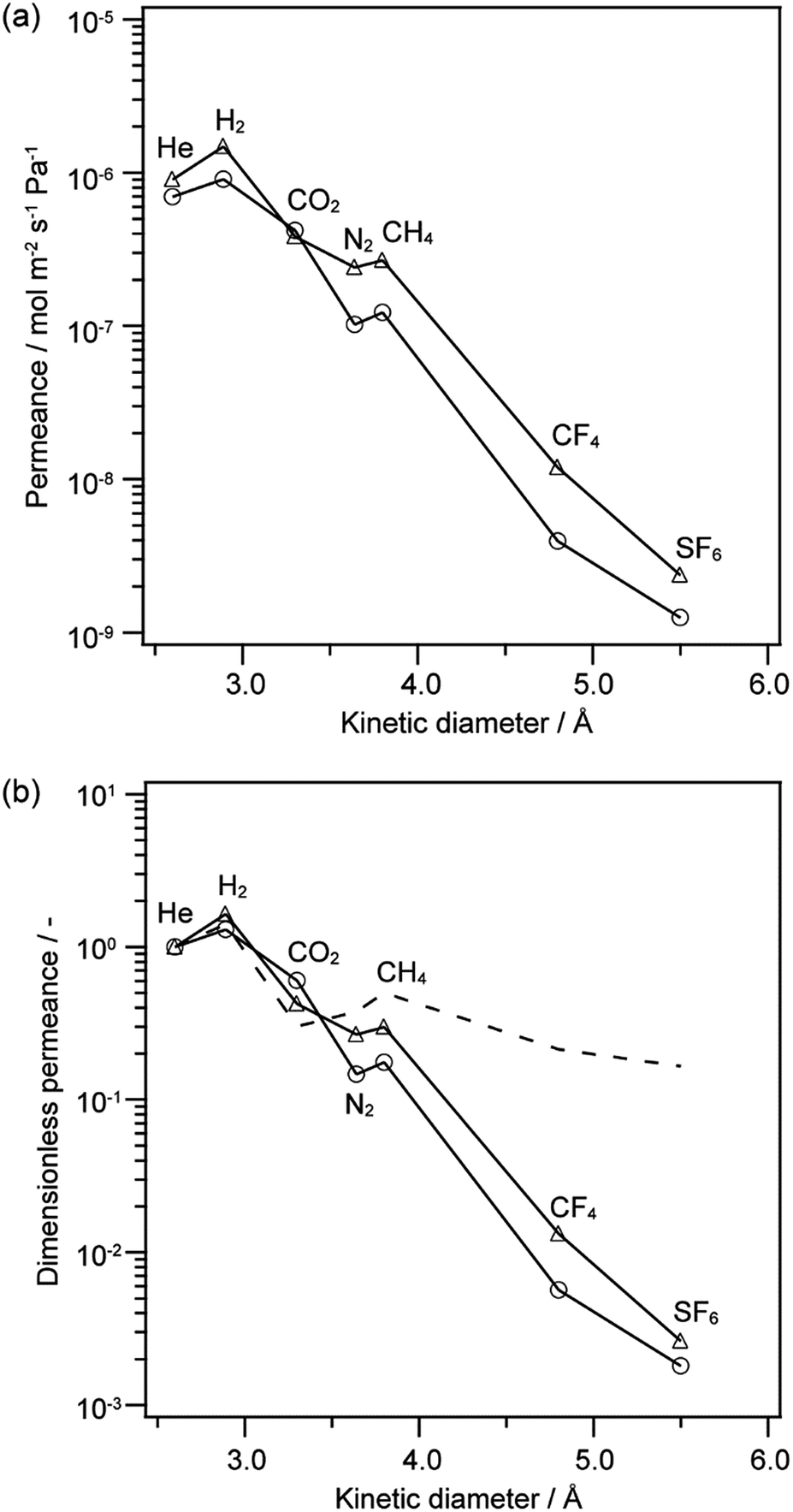 | ||
| Fig. 6 (a) Gas permeance and (b) dimensionless permeance based on He permeance at 200 °C as a function of molecular size. (circle) 12MR-Me-TES-derived membrane heated at 300 °C, (triangle) TEOS–MTES-derived membrane, and (broken line) calculated dimensionless permeance under Knudsen mechanism based on He (the calculation is based on ref. 25). | ||
Conclusions
An alkoxysilylated 12-membered ring siloxane was successfully synthesized as a nanobuilding block for siloxane-based materials. The cyclic structure of the molecule can be retained during the sol–gel reaction and membrane preparation processes, which indicates that the cyclic siloxane is a useful precursor for siloxane-based materials with controlled structures. A membrane composed of the cyclic siloxane was fabricated, and the membrane showed molecular sieving effect for H2/SF6. These results demonstrate that oligomeric siloxanes with relatively large ring structures are useful nanobuilding blocks for nanomaterials. Further studies on the control of extended ring structure with precisely controlled arrangements are underway for the creation of functional siloxane-based nanomaterials.Experimental
Materials
1-Butanol (>99.0%), chloroform (>99.0%), deuterated ethanol (EtOH-d6 >99.5%), ethanol (dehydrated >99.5%), hydrochloric acid (6 mol L−1 HCl aq.), sodium hydroxide (NaOH >97.0%), toluene (dehydrated >99.5%), and pyridine (dehydrated >99.5%) were purchased from Wako Pure Chemical Industries, Ltd. and used as received. Chlorotrimethylsilane (>98.0%), tetrachlorosilane (SiCl4 >98.0%), tetraethoxysilane (TEOS >96.0%), triethoxymethylsilane (MTES >98.0%), and zirconium tetrabutoxide (80% ZrBT in 1-butanol) were purchased from Tokyo Chemical Industry Co., Ltd. and used as received. Copper(II) chloride (CuCl2 >97%) was purchased from Sigma-Aldrich Co., LLC. and used as received. The α-alumina powders were purchased from Sumitomo Chemical Co., Ltd. The SiO2–ZrO2 sol was obtained by hydrolysis and condensation of tetraethoxysilane and zirconium tetrabutoxide. The detailed sol preparation procedure has been described previously.27,28 Porous α-alumina tubes (HU-A01, average pore size: 2.1 μm, outside diameter: 10 mm) were kindly supplied by the NIKKATO CORPORATION.Synthesis of Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O
Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O was synthesized according to the literature reported by Shchegolikhina et al.29 1-Butanol (80 mL), NaOH (2.01 g, 50.2 mmol), and water (2.72 mL, 150.9 mmol) were mixed in a three-neck flask equipped with a reflux condenser. After stirring the mixture for 30 min at room temperature, a solution of MTES (10 mL, 50.3 mmol) in 20 mL of 1-butanol was added to the mixture with vigorous stirring. Then, 7.24 mL of water was added to the mixture, and it was heated to reflux. A solution of CuCl2 (2.24 g, 16.7 mmol) in 60 mL of 1-butanol was added dropwise over ca. 20 min, and the mixture was refluxed for an additional 30 min. The hot solution was filtered to remove precipitates, and the filtrate was cooled to room temperature. The filtrate was evaporated in a rotary evaporator, and then the residue was dried in vacuo at 90 °C. A blue powder (Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O) was obtained. The X-ray diffraction (XRD) pattern of the obtained blue powder was not consistent with that of Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O reported by Shchegolikhina et al.29 (Fig. S8 in the ESI†), probably because of the difference in the number of solvated molecules (1-butanol and/or H2O). To confirm the formation of the 12-membered cyclic siloxane structure, the product was silylated with chlorotrimethylsilane (detailed method and characterization are shown in ESI†). The 1H, 13C, and 29Si NMR and MS data of the trimethylsilylated compound coincided with those of the reported compound;29 therefore, we concluded that the blue powder synthesized here possessed the 12-membered ring siloxane structure.Synthesis of chlorotriethoxysilane (ClSi(OEt)3)
ClSi(OEt)3 was synthesized by the alkoxylation of SiCl4 with ethanol under a nitrogen atmosphere. Tetrachlorosilane (16.5 mL, 143.7 mmol) was added to a Schlenk flask under a N2 atmosphere, and the flask was cooled to 0 °C. Dehydrated ethanol (29.3 mL, 501.8 mmol) was added dropwise with stirring at 0 °C. After the ethanol had been added, the mixture was stirred at 0 °C for 1 h and at room temperature for 4 h. A colorless clear liquid was obtained. In this procedure, an excess of ethanol (3.5 eq.) was used to avoid the formation of diethoxydichlorosilane (Cl2Si(OEt)2) because Cl2Si(OEt)2 leads to intermolecular crosslinking between Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O at the silylation step. Although tetraethoxysilane (TEOS) was also formed with ClSi(OEt)3 (approximately TEOS:ClSi(OEt)3 = 1:1), the mixture was used for the silylation without purification because TEOS is much less reactive than ClSi(OEt)3. The formation of ClSi(OEt)3 was confirmed by 1H, 13C, and 29Si NMR spectroscopies.
Synthesis of the 12-membered ring siloxane possessing alkoxysilyl groups (12MR-Me-TES)
Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O was alkoxysilylated with ClSi(OEt)3. Dehydrated toluene (40 mL) and dehydrated pyridine (17 mL, 210.6 mmol) were added to ClSi(OEt)3 in a Schlenk flask. Then, Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O (1.06 g) was added to the solution, and blue-green colored precipitates were formed. The mixture was stirred for 48 h at room temperature under N2 atmosphere. Then, an excess of dehydrated pyridine and dehydrated ethanol were added to the mixture for the ethoxylation of the remaining ClSi(OEt)3. The precipitates were filtered with a glass filter under N2 atmosphere. Then, the solvent, unreacted pyridine, ethanol, and the following two byproducts ((1) TEOS, generated by the ethoxylation of ClSi(OEt)3, and (2) hexaethoxydisiloxane, formed by the reaction between ClSi(OEt)3 and adsorbed water from Cu4Na4(MeSiO2)12·x(nBuOH)·yH2O) were removed in vacuo. A small amount of the precipitate was removed by syringe filtration. Finally, the alkoxysilylated 12-membered ring siloxane was isolated by gel permeation chromatography (GPC) using chloroform as an eluent (colorless clear viscous liquid, 1.8902 g, yield: 78%).12MR-Me-TES. δH (500.13 MHz; CDCl3; TMS) 0.25 (s, 24H, SiCH3), 0.26 (s, 12H, SiCH3), 1.21 (t, J = 7.0 Hz, 108H, OCH2CH3), 3.81 (q, J = 7.0 Hz, 71H, OCH2CH3); δC (125.76 MHz; CDCl3; TMS) −2.8 (SiCH3), 18.1 (OCH2CH3), 58.9 (OCH2CH3); δSi (99.36 MHz; CDCl3; TMS) −89.04 (Q1, 8Si, SiOSi(OEt)3), −88.95, (Q1, 4Si, SiOSi(OEt)3), −67.43 to −67.45 (T3, 12Si, O3SiMe (overlapping two signals)); HRMS (Electrospray ionization, 2 kV): calcd for C84H216O60Si24Na+ [M + Na]+: 2879.8205; found: 2879.8320 main paragraph text follows directly on here.
Hydrolysis and polycondensation of 12MR-Me-TES
12MR-Me-TES was dissolved in a mixture of dehydrated ethanol and deuterated ethanol (40 vol% of EtOH-d6). Then, water and 6 M hydrochloric acid were added to the solution. The molar ratio of 12MR-Me-TES:EtOH + EtOH-d6:H2O:HCl was 1:96:36:0.4. The mixture was analyzed by NMR spectroscopies after 1 h, 3 h, and 6 h of reaction.
Preparation of 12MR-Me-TES-derived gel
12MR-Me-TES was dissolved in dehydrated ethanol. Then, water and 6 M hydrochloric acid were added to the solution. The molar ratio of 12MR-Me-TES:EtOH:H2O:HCl was 1:6006:360:0.4 (EtO:H2O = 10), and the concentration of 12MR-Me-TES was 1 wt%. Please note that the molar ratio among those compounds is different from that for the investigation of hydrolysis and polycondensation process of 12MR-Me-TES. After the mixture had been stirred at 1200 rpm at room temperature for 24 h, the solution was cast on a Petri dish. Then, a colorless transparent xerogel was formed by drying at 100 °C for 10 min in air. A white powder was obtained by scraping the gel from the Petri dish for analysis (12MR-gel-as). The white powder was heated under an argon atmosphere at 100 °C, 200 °C, or 300 °C for 1 h (12MR-gel-heat100, 12MR-gel-heat200, and 12MR-gel-heat300, respectively) to investigate the possible structural changes of the siloxane network.
Preparation of TEOS–MTES-derived gel
The mixture of TEOS, MTES, ethanol, water, and 6 M hydrochloric acid was stirred at 1200 rpm at room temperature for 24 h. The molar ratio of TEOS:MTES:H2O:HCl was 1:1:70:0.4. The EtO:H2O molar ratio and mass concentration of alkoxides (TEOS + MTES) were the same as those for the aforementioned system of 12MR-Me-TES (EtO:H2O = 10 and 1 wt%, respectively). The hydrolyzed solution was cast on a Petri dish, and a colorless transparent gel was formed by drying at 100 °C for 10 min in air. A white powder was obtained by scraping from the Petri dish for analysis (TEOS–MTES-gel). The powder was heated under an argon atmosphere at 100 °C, 200 °C, or 300 °C for 1 h (TEOS–MTES-gel-heat100, TEOS–MTES-gel-heat200, and TEOS–MTES-gel-heat300, respectively).
Fabrication of membranes from 12MR-Me-TES or mixed TEOS–MTES
α-Alumina particles (a mixture of two types of particles with average diameters of 0.2 and 1.9 μm) were coated onto the outside of porous α-alumina tube by using a SiO2–ZrO2 sol as a binder. To obtain a smooth surface, the tube was calcined in air at 550–600 °C for 30 min. These procedures were repeated several times to prevent the formation of pinholes in the final membrane. Then, the diluted SiO2–ZrO2 sol (ca. 0.5 wt%) was coated onto the tube to form an intermediate layer (pore size: 2–3 nm).30 After calcination of the tube in air at 550–600 °C for 30 min, the 12MR-Me-TES-derived layer was fabricated by coating with the hydrolyzed solution of 12MR-Me-TES, which was prepared as the same way as 12MR-gel-as. Finally, the tube was dried and heated at 100 °C for 1 h under N2 atmosphere. A TEOS–MTES-derived membrane was fabricated in the same way as the 12MR-Me-TES-derived membrane by using the TEOS–MTES sol instead of the 12MR-Me-TES sol. The TEOS–MTES sol was prepared as shown above.Evaluation of single-gas permeation property
The experimental apparatus for a single-gas permeation measurement is shown in Fig. S9 in the ESI† as a schematic. A single gas (He, H2, CO2, N2, CH4, CF4, or SF6) was fed to the outside surface (upstream) of a cylindrical membrane at 200 kPa, and the downside was kept at atmospheric pressure. At first, the values for the single gas permeances were measured at 100 °C, and then the membrane was heat-treated at 200–300 °C under a N2 atmosphere. After confirming the attainment of a steady state at each temperature by measuring the time course of N2 permeance, the temperature was cooled to 100 °C and the gas permeance was measured. The permeation rate was measured by a bubble film meter. The deviation of the permeation data was less than 5%.Characterization
Solution 1H, 13C, and 29Si NMR spectra were recorded on AVANCE 500 (Bruker) or JNM-ECZ 500 (JEOL) spectrometers with resonance frequencies of 500.13 MHz, 125.76 MHz, and 99.36 MHz, respectively, at room temperature using 5 mm glass tubes. Tetramethylsilane (TMS) was used as an internal reference at 0 ppm. CDCl3 and ethanol-d6 were used to obtain lock signals. A small amount of Cr(acac)3 (acac: acetylacetonate) was added as a relaxation agent for 29Si nuclei. 13C NMR spectra were measured with a recycle delay of 2 s. 29Si NMR spectra were measured with a 45° pulse and a recycle delay of 10 s. Solid-state 29Si MAS NMR spectra were recorded on a JNM-ECX 400 (JEOL) spectrometer with a resonance frequency of 78.7 MHz at room temperature with a 45° pulse and a recycle delay of 250 s. The recycle delay was set at five times as long as longitudinal relaxation time (T1) to complete the relaxation of all nuclear spins. The samples were placed in 4 mm zirconia tubes and spun at 6 kHz. The chemical shifts were externally referenced to poly(dimethylsilane) at −33.8 ppm. Solid-state 13C CP/MAS NMR spectra were also recorded on a JNM-ECX 400 (JEOL) spectrometer with a resonance frequency of 99.5 MHz at room temperature with a recycle delay of 10 s and a contact time of 5 ms. The samples were put in 4 mm silicon nitride tubes and spun at 10 kHz. The chemical shifts were externally referenced to the methyl groups of hexamethylbenzene at 17.4 ppm. High-resolution electrospray ionization mass (HRMS) analysis was conducted by using an Exactive Plus (Thermo Fisher Scientific) instrument. Low-resolution electrospray ionization mass analysis was conducted by using a JMS-T100 CS AccuTOF (JEOL) instrument. Samples were dissolved in ethanol. Gel permeation chromatography (GPC) was carried out using a LC-9100 with a recycling preparative HPLC system, a refractive index (RI) detector (Japan Analytical Industry Co., Ltd.) and two types of crosslinked polystyrene packed columns (JAIGEL-1H and JAIGEL-2H; exclusion limits of 1000 and 2000, respectively, and theoretical plate of 13000). Chloroform was used as an eluent with a flow rate of 3.5 mL min−1. FT-IR spectra were recorded on a FT/IR-6100 (JASCO) spectrometer at ambient temperature. The FT-IR spectra were measured using KBr disk technique under vacuum conditions. Powder XRD patterns were recorded on a RINT-Ultima III (RIGAKU) diffractometer with Cu Kα radiation at 40 kV and 40 mA.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate Dr T. Shibue and Mr N. Sugimura (Materials Characterization Central Lab., Waseda University) for solid state NMR and HRMS measurements, respectively. The authors also thank Mr S. Saito (Waseda University) for his help on separation by GPC and Mr E. Yamamoto, Mr S. Mori, and Ms. S. Uchida (Waseda University) for their help in the preparation of the TEOS–MTES sol. M. Y. is thankful to the JX-Waseda Research Fund for Young Researchers and Japan Chemical Industry Association for a fellowship (Human Resources fostering Program in Chemistry). This work was supported in part by JSPS KAKENHI (Grant-in-Aid for Scientific Research(B), No. 15H03879).Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- T. Tsuru, J. Sol-Gel Sci. Technol., 2008, 46, 349–361 CrossRef CAS.
- C. Sanchez, G. J. d. A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061–3083 CrossRef CAS.
- A. Shimojima and K. Kuroda, Chem. Rec., 2006, 6, 53–63 CrossRef CAS PubMed.
- K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26, 211–220 CrossRef CAS.
- J. S. Ritch and T. Chivers, Angew. Chem., Int. Ed., 2007, 46, 4610–4613 CrossRef CAS PubMed.
- A. Decken, F. A. LeBlanc, J. Passmore and X. Wang, Eur. J. Inorg. Chem., 2006, 4033–4036 CrossRef CAS.
- A. Decken, J. Passmore and X. Wang, Angew. Chem., Int. Ed., 2006, 45, 2773–2777 CrossRef CAS PubMed.
- T. S. Cameron, A. Decken, I. Krossing, J. Passmore, J. M. Rautiainen, X. Wang and X. Zeng, Inorg. Chem., 2013, 52, 3113–3126 CrossRef CAS PubMed.
- M. Borsari, G. Gavioli, C. Zucchi, G. Pályi, R. Psaro, R. Ugo, O. I. Shchegolikhina and A. A. Zhdanov, Inorg. Chim. Acta, 1997, 258, 139–144 CrossRef CAS.
- Y. A. Molodtsova, Y. A. Pozdnyakova, I. V. Blagodatskikh, A. S. Peregudov and O. I. Shchegolikhina, Russ. Chem. Bull., 2003, 52, 2722–2731 CrossRef CAS.
- S. D. Korkin, M. I. Buzin, E. V. Matukhina, L. N. Zherlitsyna, N. Auner and O. I. Shchegolikhina, J. Organomet. Chem., 2003, 686, 313–320 CrossRef CAS.
- O. I. Shchegolikhina, E. V. Matukhina, A. A. Anisimov, Y. A. Molodtsova, M. I. Buzin, K. A. Lyssenko and A. M. Muzafarov, Macroheterocycles, 2016, 9, 11–16 CrossRef CAS.
- J. Han and S. Zheng, Macromolecules, 2008, 41, 4561–4564 CrossRef CAS.
- Y. Yi, N. Liu, L. Li and S. Zheng, RSC Adv., 2016, 6, 87802–87807 RSC.
- Y. Sugahara, S. Okada, S. Sato, K. Kuroda and C. Kato, J. Non-Cryst. Solids, 1994, 167, 21–28 CrossRef CAS.
- R. J. Hook, J. Non-Cryst. Solids, 1996, 195, 1–15 CrossRef CAS.
- D. A. Loy, B. M. Baugher, C. R. Baugher, D. A. Schneider and K. Rahimian, Chem. Mater., 2000, 12, 3624–3632 CrossRef CAS.
- Y. A. Pozdniakova, K. A. Lyssenko, A. A. Korlyukov, I. V. Blagodatskikh, N. Auner, D. Katsoulis and O. I. Shchegolikhina, Eur. J. Inorg. Chem., 2004, 1253–1261 CrossRef CAS.
- V. Pashchenko, B. Brendel, B. Wolf, M. Lang, K. Lyssenko, O. Shchegolikhina, Y. Molodtsova, L. Zherlitsyna, N. Auner, F. Schütz, M. Kollar, P. Kopietz and N. Harrison, Eur. J. Inorg. Chem., 2005, 4617–4625 CrossRef CAS.
- L. Zherlitsyna, N. Auner, M. Bolte, Y. Pozdniakova, O. Shchegolikhina, K. Lyssenko, V. Pashchenko, B. Wolf, M. Lang, F. Schütz, M. Kollar, F. Sauli and P. Kopietz, Eur. J. Inorg. Chem., 2007, 4827–4838 CrossRef CAS.
- I. V. Blagodatskikh, Y. A. Molodtsova, Y. A. Pozdnyakova, O. I. Shchegolikhina and A. R. Khokhlov, Colloid J., 2008, 70, 407–415 CrossRef CAS.
- A. N. Bilyachenko, A. N. Kulakova, M. M. Levitsky, A. A. Petrov, A. A. Korlyukov, L. S. Shul’pina, V. N. Khrustalev, P. V. Dorovatovskii, A. V. Vologzhanina, U. S. Tsareva, I. E. Golub, E. S. Gulyaeva, E. S. Shubina and G. B. Shul'pin, Inorg. Chem., 2017, 56, 4093–4103 CrossRef CAS PubMed.
- J. C. Pouxviel, J. P. Boilot, J. C. Beloeil and J. Y. Lallemand, J. Non-Cryst. Solids, 1987, 89, 345–360 CrossRef CAS.
- R. J. R. Uhlhorn and A. J. Burggraaf, in Inorganic Membranes Synthesis, Characteristics and Applications, Springer, Netherlands, Dordrecht, 1991, pp. 155–176 Search PubMed.
- J. Wang, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2012, 415–416, 810–815 CrossRef CAS.
- W. Puthai, M. Kanezashi, H. Nagasawa and T. Tsuru, Sep. Purif. Technol., 2016, 168, 238–247 CrossRef CAS.
- W. Puthai, M. Kanezashi, H. Nagasawa and T. Tsuru, J. Membr. Sci., 2017, 524, 700–711 CrossRef CAS.
- Y. A. Molodtsova, K. A. Lyssenko, I. V. Blagodatskikh, E. V. Matukhina, A. S. Peregudov, M. I. Buzin, V. G. Vasil’ev, D. E. Katsoulis and O. I. Shchegolikhina, J. Organomet. Chem., 2008, 693, 1797–1807 CrossRef CAS.
- M. Kanezashi, S. Miyauchi, H. Nagasawa, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2014, 466, 246–252 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. Characterizations of 12MR-Me-TES, 12MR-Me-TES-derivedgels, TEOS–MTES-derived gels, and precursor of 12MR-Me-TES. Experimental apparatus for gas permeation measurement. See DOI: 10.1039/c7ra09380b |
| ‡ The 12-membered cyclic siloxane of complex shows a saddle conformation before silylation because of the interaction among SiO− groups and metal cations. After silylation, the cyclic structure can take an extended and planar state because such strong interactions to regulate the configuration does not work. The inside diameter of 12-membered cyclic siloxane is calculated by using Chem3D. |
| § Two TES groups located in the end of the continuous three TES groups (Q unit), facing to the same direction against 12-membered ring plane, are set in the cis and trans positions against both of the adjacent TES groups. The center TES groups are in the cis position against both of the adjacent TES groups. Hence, the signals due to TES groups are observed at −88.95 ppm and −89.04 ppm, and signal intensity ratio of these signals is 1:2. In addition, magnification of the signal due to the Si atoms (T unit) of 12-membered ring exhibits the presence of the shoulder signal probably due to the conformation. |
| This journal is © The Royal Society of Chemistry 2017 |