
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivity and stability analysis of covalent conjugated lysozyme-single walled carbon nanotubes: potential biomedical and industrial applications
Z. Borzooeian*a,
M. E. Taslima,
G. Borzooeianb,
O. Ghasemi†
c and
M. Aminlarid
aDepartment of Mechanical and Industrial Engineering, College of Engineering, Northeastern University, Boston, MA, USA. E-mail: z.borzooeian@northeastern.edu; borzooeianm@gmail.com
bDepartment of Biology, Payame Noor University of Isfahan, Iran
cTakeda Pharmaceuticals USA Inc., Cambridge, MA, USA
dDepartment of Biochemistry, School of Veterinary Medicine, Shiraz University, Shiraz, Iran
First published on 17th October 2017
Abstract
Conjugated protein–carbon nanotubes possess unique physicochemical properties that make them attractive to a wide range of applications. We explored the effects of covalent conjugation of lysozymes with single-walled carbon nanotubes (SWCNTs) on its activity and stability. Lysozyme was conjugated onto carboxyl-functionalized SWCNTs through carbodiimide method. Post conjugation changes of the enzyme were investigated using fluorescence measurements to generate plots of protein unfolding at 0 and 6 M guanidine hydrochloride (Gn-HCl) concentrations. Free lysozyme showed a remarkable increase in the fluorescence intensity at 287 nm in addition to a red shift from 343 to 352 nm. The emission spectrum of conjugated lysozyme showed a significant increase in the fluorescence intensity at 287 nm and a significant decrease in intensity at 348 nm. These results confirmed that both tryptophan and phenylalanine residues have an important role in the fluorescence bulk of both free and conjugated lysozymes. Kinetic parameters of free and conjugated lysozyme activities (KM and Vmax), optimum pH, thermal and pH stability, and stability in the presence of denaturant agents such as urea and salts were investigated. Changes were observed in KM and Vmax from 4.8 to 5.6 mM and 193 to 197 nmol min−1, respectively. Conjugated lysozyme showed a remarkable increase in pH stability in a range of 3.0 to 10.0 at 70 °C. The results showed first-order inactivation kinetics for both forms of lysozyme with a rate constant of about 0.139 min−1 for 10 minutes of incubation at 70 °C in the presence of KCl and KSCN.
Introduction
Nanobiotechnology is revolutionizing many scientific fields by providing a platform for co-assembly of biology and material sciences and by exploration of the interface between functional hybrid nanomaterials involving biomolecule–nanotube composites.1–3 Little research has been directed to the study of covalent interaction of nanotubes with macromolecules such as biocompatible synthetic polymers,4,5 DNA,6–8 and proteins.9–13During recent decades, the main focus has been on macromolecular conformational modifications through adsorption on nanotubes.14–16 In regenerative medicine, specifically in tissue regeneration, hybrid nanomaterials such as conjugated protein/biomimetic peptide-nanomaterials17,18 have been investigated to provide scaffold-based delivery systems for signalling molecules. Molecules such as low-molecular-weight drugs, oligonucleotides, and proteins stimulate cell migration, growth and differentiation.19 The resulted scaffolds of conjugated proteins with nanomaterials can be used in cell encapsulation in order to control and track where the enzymes and signalling proteins go and exactly what they do when they get there. Therefore, a wide range of research have focused on immobilizing proteins/biopeptides such as collagen,20–22 gelatin,23 laminin,24–26 chitosan,27–29 and Arg–Gly–Asp (RGD)-containing peptides30,31 onto the surfaces of polymeric scaffolds to enhance the biocompatibility of the surface and improve their cytocompatibility.32 In addition, drug delivery, diagnostic imaging and bio-sensing are promising new technologies that are resulted from creating protein–nanomaterials conjugates. For design and fabrication of biocompatible polymers such as high-performance coronary artery stents, nanomaterial–protein conjugates have been evaluated and optimized.33
In biotechnology, protein and especially enzyme immobilization provides technical and economic advantages since the molecules can be used multiple times for the same reaction. Immobilized enzymes, in comparison with their soluble counterparts, demonstrate enhanced stability under extreme conditions of temperature, pH, salts, and denaturing solvents. Therefore, they have longer half-lives since they degrade less and recover better and they are suitable for continuous processes.34 With physical or covalent immobilization,35 enzyme catalytic stability is achieved without contaminating the final product with enzymes thus saving cost and reducing process steps for purification.36 Over the last decade, enzyme immobilization on nanomaterials has become more important in order to develop a better understanding of enzyme–nanomaterial interactions, and to design functional protein–nanomaterial engraftments.5 Immobilization of biomolecules on nanomaterials, which is achieved either by glutaraldehyde or carbodiimide is the most common methods for covalent conjugation.6,7 Several researchers have investigated immobilization of lipase on carbon nanotubes through adsorption in non-aqueous medium,8–11 lysozyme adsorption on mica,37 polyethylene surfaces,38 and silica surface,39 horseradish peroxidase adsorption on silica surfaces,40 polysaccharide-decorated particles,41 and onto titanate nanowires.42
Single-walled carbon nanotubes (SWCNTs) are promising for robust supports for enzyme immobilization and stabilization since they possess unique structural, mechanical and thermal properties, chemical reactivity and ease of surface modification.12 They are remarkable candidates because of their high aspect ratio which leads to high enzyme loading. They have potential biotechnological applications in biosensors and nano-scaffolds development.13–15 A number of papers have appeared with regard to the particular aspect of stabilization and protein–nanotubes interactions with the effect of non-adsorbed protein.16 However, there is a need to further improve our knowledge on the effect of nanotubes on activity and stability of covalent immobilized proteins.
In our previous study, we evaluated the conjugation of chicken egg white lysozyme (EWL) with single-walled carbon nanotubes (SWCNTs) structurally.44 Lysozymes are a family of enzymes with antimicrobial activity. They are present in saliva, tears, egg white, and many animal fluids. They are characterized by their ability to damage the cell wall of bacteria. In the present contribution, enzyme-induced post immobilization changes of activity and stability were analysed using various spectroscopy techniques. Conjugated EWL-SWCNTs were chemically and biochemically compared with free enzyme using scanning electron microscopy (SEM) and fluorescence spectroscopy. The fluorescence changes of free lysozyme and conjugated EWL-SWCNTs were characterized by tryptophan, tyrosine, and phenylalanine fluorescence measurements. The catalytic efficiency of the immobilized lysozyme, in terms of thermal and pH stability, and stability in the presence of denaturing agents such as guanidine hydrochloride, urea and salts were also studied.
Materials and methods
Materials
Lyophilized chicken egg white lysozyme (EC 3.2.1.17), EWL, were purchased from Inovatech Inc., (Abbotsford, BC, Canada). Micrococcus lysodeikticus cells (as salt-free, dry powders), MES [2-(N-morpholino)ethanesulfonic acid] buffer, N-ethyl-N0-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC) and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Highly purified activated SWCNTs were synthesized by the plasma-enhanced chemical vapour deposition (PECVD) process.44 The SWCNTs' outer diameters were 1–3 nm.Enzyme attachment onto SWCNTs
Conjugated lysozyme–SWCNTs solution was prepared using a modified carbodiimide method.13,43 Activated SWCNTs (1.0 mg) were dispersed in 1.0 ml of 50 mM of MES [2-(N-morpholino)ethanesulfonic acid] buffer solution at pH 6.2 and then added to equal volume of 400 mM N-hydroxy succinimide (NHS) in MES [2-(N-morpholino)ethane sulfonic acid] buffer. After sonication of the mixture (MSE Ultrasonic Disintegrators, 150W, England) for 30 minutes, 20 mM N-ethyl-N′-(3-(dimethyl amino)propyl)carbodiimide hydrochloride (EDC) was added to promote the coupling of NHS to the carboxylic functional groups of SWCNTs. The resulting mixture was centrifuged at 7000 rpm for 15 minutes after stirring at 200 rpm for 30 minutes. Then MES buffer was added to the remaining precipitate followed by removing supernatant. The enzyme solution (10 mg ml−1 in 10 mM phosphate buffer, pH 8) was mixed in the nanotubes solution and sonicated for ca. 1 minute to re-disperse the SWCNTs. The solution was then shaken overnight on an orbital shaker at 200 rpm at room temperature to conjugate enzyme to the SWCNTs. The enzyme–SWCNTs suspension was then centrifuged. To remove all non-specifically adsorbed enzymes, washing was achieved three times with triply distilled water and once with 1% (v/v) Tween-20. Control solution of nanotube–enzyme conjugates was prepared using the identical procedure in the absence of EDC and NHS. Conjugation was tracked by scanning of supernatant of EWL-SWCNTs solution using a spectrophotometer (ULTEROSPEC PLUS, Pharmacia LKB Biotechnology, Sweden) for wavelength range 200–500 nm. The chemical stability of conjugation was characterized using Fourier transform infrared spectroscopy (FTIR), (Shimadzu FTIR 8300 spectrophotometer). In our previous work, we have shown the related results.44 The size and diameter of the conjugated EWL-SWCNTs was prepared using a scanning electron microscope (SEM, Oxford, S360).Fluorescence measurements of free and conjugated lysozyme
The excitation wavelength of the fluorescence was set at a wide range around 225–282 nm for free lysozyme and conjugated form of enzyme at room temperature with resolution 0.5 nm, and scan rate 500 nm min−1 with an incident angle of 90°. The emitted fluorescence was measured from 225 to 500 nm for all forms of enzyme at different concentrations of Gn-HCl using a fluorescence spectrometer (PerkinElmer (Waltham, MA) LS 50 Luminescence Spectrometer). The protein concentration in all measurements was 36 μM using Bradford protein assay.Determination of free and conjugated enzyme activity
The activities of free and conjugated enzymes were measured using Imoto and Yagishita method.43 Nine milligrams of dried M. lysodeikticus cell walls was dissolved in 30 ml potassium phosphate buffer (0.1 M, pH 7.0) solution. The cell wall suspension (2.9 ml) was poured into a cuvette and incubated at 25 °C for 5 min. Free lysozyme and conjugated form of the enzyme, at a concentration of 1 mg protein, were dissolved in one ml phosphate buffer (pH 7) and added to the cuvette. Absorbance at 450 nm was then recorded. One unit of activity of lysozyme is defined as the decrease in the absorbance at 450 nm of 0.001 min−1 at pH 7.0 and 25 °C using M. lysodeikticus cells as a substrate.Kinetic characterization of conjugated and free enzyme
Kinetic parameters of free and conjugated lysozymes were determined using 0, 0.04, 0.06, 0.08, 0.15, 0.25 and 0.6 mg ml−1 of M. lysodeikticus cell wall in 0.1 M potassium phosphate buffer (pH 7.0) at room temperature, respectively. The Michaelis constants, KM and Vmax for the free and conjugated lysozyme preparations were determined using Lineweaver–Burk plot. Then the turnover number (Kcat) was determined.Optimum pH and thermal stability of free and conjugated lysozyme
The optimum pH for lysozyme activity was studied over a pH range of 3 to 8.5 to determine the activity of free as well as conjugated enzyme. Citrate buffer (pH 3–6), phosphate buffer (pH 7–8), and bis–Tris propane buffer (pH 6.3–9.5) were used to determine enzyme activity. The stability of the enzyme was determined by preincubating the enzyme for 30 min at 37 °C with various buffers. After incubation, the residual enzyme activity (%) was measured using phosphate buffer (0.1 M, pH 7.0). The thermal stability of the enzyme was studied by preincubating the enzyme at 70 °C for 0–20 minutes range at optimum pH.Effect of KCl and KSCN on thermostability of free and conjugated lysozymes
Heat-induced aggregation and inactivation of free and conjugated lysozyme in the presence of KCl and KSCN (as a strong chaotropic salt) was evaluated by heating stock solution containing 1.0 mg ml−1 of lysozymes in 0.1 M phosphate, pH 7.0 and 1.0 M salts at 70 °C for various time intervals.Effect of urea on stability of free lysozyme and EWL-SWCNTs
400 μl from lysozyme and EWL-SWCNTs solutions (1 mg ml−1) were incubated in the presence of 2, 4, 6 or 8 M urea in 0.1 M phosphate buffer, pH 7.0. Enzymes were assayed after 0, 30, 60, 90 and 120 minutes. Solutions containing urea were prepared fresh each time.Results and discussion
The main objective of this study was to investigate the influence of covalent conjugation of lysozyme onto SWCNTs on the enzyme structure, function, and stability in order to evaluate the efficiency of functionalized nanotubes as a support for enzyme immobilization.Enzyme loading onto nanotubes (covalently or noncovalently) was confirmed by the disappearance of the absorption peak at 280 nm in the supernatant of conjugated enzyme solution after precipitation of conjugated EWL-SWCNTs in the cuvette. In addition, the efficient conjugation have been evaluated using analytical techniques such as TEM, agarose gel electrophoresis, XRD and FTIR spectroscopy which we reported in our previous work.44 We determined that lysozyme would covalently bind to the surface of the SWCNTs through amino groups of lysozyme side chains. Fig. 1 shows the SEM micrographs and the size of conjugated EWL-SWCNTs.
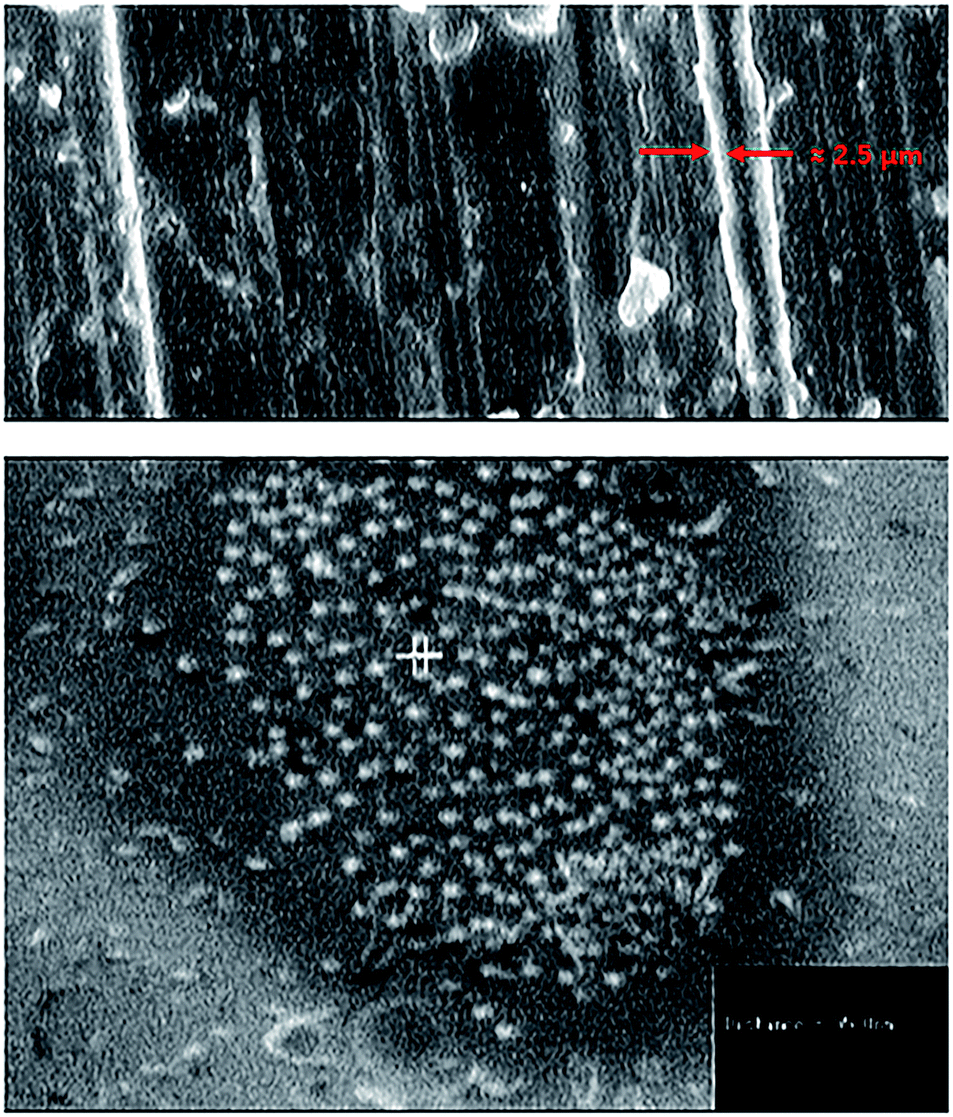 | ||
| Fig. 1 Scanning electron micrographs of conjugated lysozyme-SWCNTs (a) diameter of a bundle of lysozyme-SWCNTs ≈ 2.5 μm, (b) average diameter of each conjugated lysozyme-SWCNTs = 95 nm. | ||
Increase in the thickness of the sidewalls of the functionalized SWCNTs, after covalently conjugated enzyme, confirmed the presence of enzyme. Similar observations of adsorption of lysozyme molecules onto the hydrophilic silica surface have been reported.45
Fluorescence measurements
To complete the characterization of enzyme conjugation, we compared fluorescence measurements using tryptophan, tyrosine and phenylalanine as important intrinsic fluorescent probes, which can be used to estimate the nature of microenvironment of these amino acids and probe for protein folding.46 The main purpose was to study the effect of guanidine hydrochloride (Gn-HCl) on the florescent intensity of lysozyme in its free lysozyme and conjugated form. The fluorescence intensity and maximum emission wavelength (λmax) are affected by the polarity and charge densities surrounding tryptophan, tyrosine and phenylalanine residues.46 Imoto et al. showed two buried tryptophan residues at positions 62 and 108 which interpret fluorescence changes in the protein. Because of these aromatic residues, fluorescence changes have more direct correlation with major tertiary structural changes in the protein.47 For lysozyme, however, presence of multiple tryptophan, tyrosine and phenylalanine complicates the interpretation of fluorescence changes. Therefore, there is not a direct correlation between fluorescence changes and bulk tertiary changes of the protein. This egg white protein has six tryptophan residues, several of which are known to be near the active site, and two tyrosines and two phenylalanines.48Surfactants, denaturants, or other amphiphilic molecules can change the microenvironment of the aromatic residues. Microenvironment alteration of tryptophan and phenylalanine residues in free and conjugated lysozymes has been monitored by studying the changes (λmax) in 0 and 6 M Gn-HCl concentrations (Fig. 2a–c). Emission efficiency was reached in the wavelength range of 225–282 nm excitation for both free and conjugated enzymes. By selecting a range of excitation, the possibility of missing the optimum emission peaks for both forms of proteins is reduced. For free lysozyme, to our surprise, we found that the fluorescence intensity at 286 nm, which is the maximum emission wavelength (λmax) of phenylalanine,49 significantly increased at 6 M of Gn-HCl, Fig. 2a. Indeed, there are no existing investigations to figure out the exact reason for the intensity change at 286 nm. This subject should be the focus of future research to determine fluorescence of lysozyme using emission spectra of tryptophan, tyrosine, and phenylalanine residues and from energy migration.
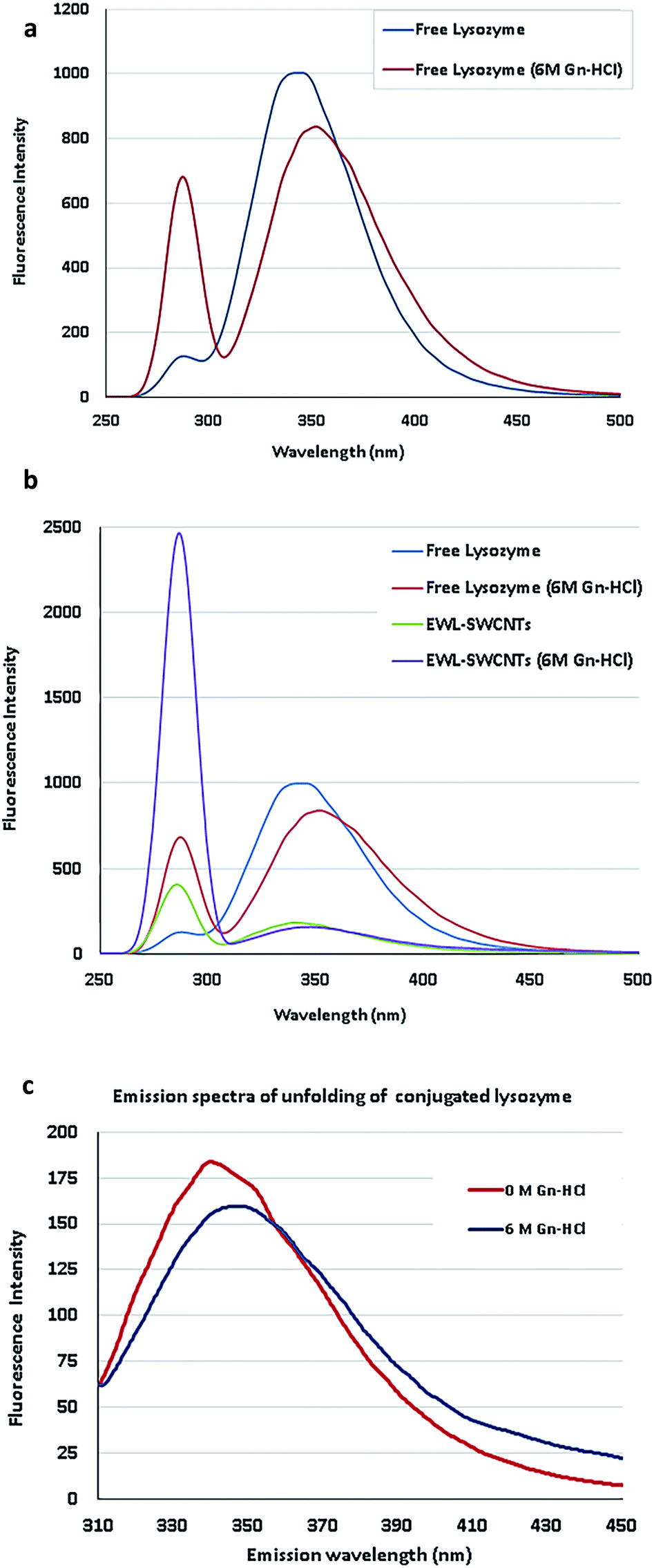 | ||
| Fig. 2 (a) Emission spectra of lysozyme unfolding. (b) Emission spectra of free and conjugated lysozymes in 0 and 6 M concentrations of Gn-HCl. (c) Spectral shift of conjugated EWL-SWCNTs unfolding. | ||
In Fig. 2b, the emission spectrum of EWL-SWCNTs showed significantly increased fluorescence intensity at 286 nm and a decreased intensity at 340 nm in comparison with the free lysozyme.
The first observation is related to the peak at around 350 nm associated with tryptophan, in which the conjugated lysozyme has much lower emission distribution than the free protein. We speculate that SWCNTs change the structure of the protein, specifically at tryptophan sites, such that the emission intensity decreases by over 5 folds. Moreover, the emission maximum wavelength (λmax) is red shifted for both form of enzymes as the protein encounters high Gn-HCI concentration. This effect can be due to more exposure of the tryptophan residues to an aqueous environment in the presence of high concentration of denaturant that leads to the higher polarity of the tryptophans' microenvironment.
Second, the aforementioned structural change does not affect the emission spectrum of phenylalanine. As shown in the figure and described in the previous section, by adding Gn-HCI, the intensity of the peak at 286 nm for both free and conjugated form of lysozyme increases by around 5 and 6 folds, respectively. The results indicate that conjugation with SWCNTs completely converted microenvironments of phenylalanine residues (an increased intensity in 283 nm) and tryptophan residues (a decreased intensity) in conjugated form, which leads to more exposure of these amino acids to a more polar environment. This fact indicates that the protein conformation is getting altered in both forms. To figure out the reasons for fluorescence intensity changes of EWL-SWCNTs, more extensive research are needed to determine the locations of lysozyme fluorescence bulks.
Since the emission peaks of SWCNTs are higher than 900 nm,50 we believe that this remarkable increase of intensity at 286 nm is attributed to the fluorescence change of phenylalanine 20 and 53. Phenylalanine is one of the three fluorescent amino acids in proteins. Its use, however, is limited in protein structural studies because of its low fluorescence quantum yield (about 0.02).48 Birch et al.49 showed the decays of excite measurable fluorescence for this weakly fluorescent amino acid using a 265 nm AlGaN light-emitting diode (LED) from 343 to 352 nm at 6 M of Gn-HCl. Fig. 2c shows the spectra of EWL-SWCNT at 6 M of Gn-HCl.
The emission spectra of EWL-SWCNTs resulting from 282 nm of excitation wavelength used to measure transient fluorescence spectra at 0 and 6 M concentrations of Gn-HCl. The results shows that decrease in the intensity at 340 nm due to increased concentration of Gn-HCl indicates quantum yield change of those buried phenylalanine residues in the hydrophobic cores in the structure of conjugated enzyme. Therefore, there is a change in enzyme folding under these conditions. In our work, the reading of red (340 nm) increased and shifted from 340 for free lysozyme to 347 nm at 6 M concentration of Gn-HCI. This reduced shift of wavelength is probably due to protein unfolding or/and absorption of the emitted red fluorescence by the SWCNTs. One possible reason is that the tryptophan residues 62 and 108 in conjugated lysozyme were less accessible to the solvent.
Fig. 3 represents maximum fluorescence intensity monitored in excitation at 286 nm (peak 1) and 340 nm (peak 2) for free and conjugated enzymes in 0 and 6 M Gn-HCl concentrations. The figure shows that by adding Gn-HCI, there is not a significant change in the maximum fluorescence intensity of any forms of lysozyme at peak 2 due to the hydrophobic environment experienced by tryptophan. On the other hand, the maximum intensity increased dramatically at peak 1 by adding Gn-HCI to the compound. Adding Gn-HCI increases the intensity of peak 1 for both free and conjugated form of lysozyme by around 5 and 6 folds, respectively. It is also observed that peak 1 in the conjugated form of lysozyme at 6 M Gn-HCI is almost 4 folds higher than that of free lysozyme.
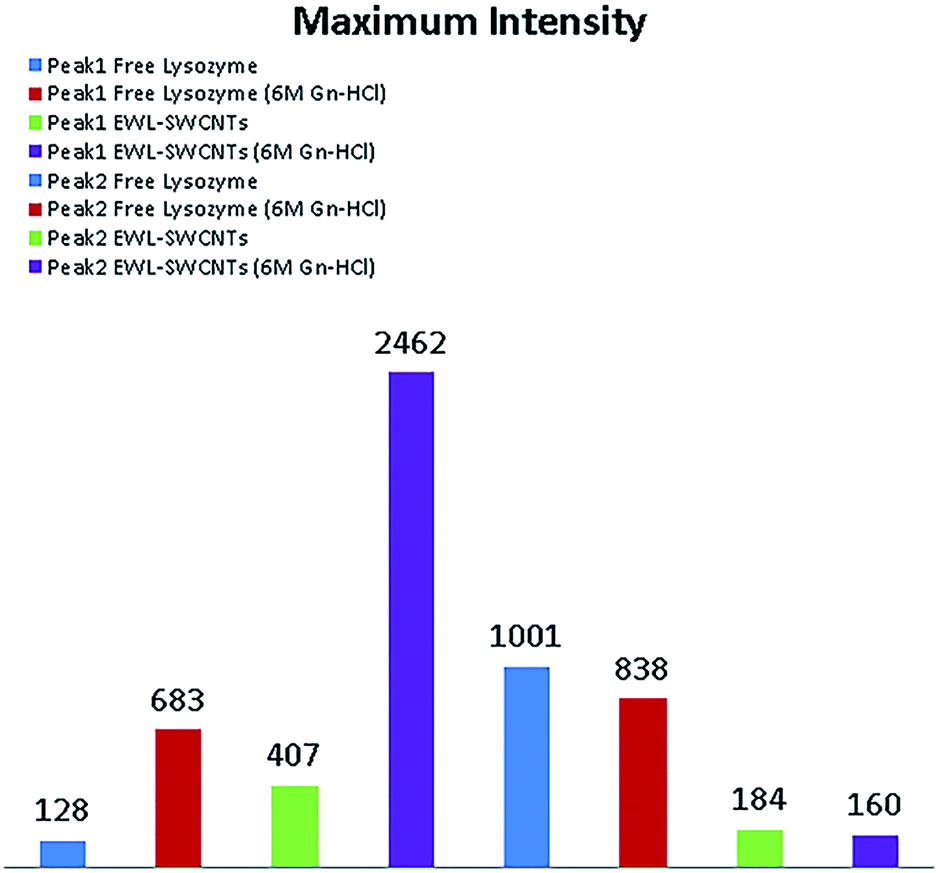 | ||
| Fig. 3 Maximum intensity of free and conjugated form of lysozyme at peak 1 and 2. | ||
Determination of enzyme activity
Lysozyme activity assay was performed with the M. lysodeikticus cell walls as substrate. For EWL-SWCNTs, the activities were lower than that of free enzyme, probably due to the partially unfolded 3D structure. Covalent interaction between lysozyme and SWCNTs could significantly affect the active site situation, structure, and function of lysozyme.51 The ionizable groups in active site consist of the side chain of glutamic acid and aspartic acid at positions 35 and 52, respectively,52 and aspartic acid groups at position 66 and 101.53 Steric hindrance and change in the position of carboxylic groups of active site might be responsible for the decrease in lytic activity of conjugated lysozyme. Basically, it is well documented that the surface properties of molecules change due to the attachment of groups to proteins leading to changes in protein conformation, unfolding, molecular reorientation and aggregation.54,55Kinetic characterization of immobilized and free enzyme
Kinetic parameters of free and conjugated lysozyme were determined by measuring initial reaction rates for each form by varying the amount of substrates (M. lysodeikticus cell walls). Conjugated and free lysozymes exhibited Michaelis–Menten kinetic behavior (Fig. 4a and b). The maximum velocity (Vmax) and KM were calculated from the y-intercept and x-intercept from Lineweaver–Burke graph. KM value of the EWL-SWCNTs was 5.6 mM, and for free enzyme was 4.8 mM. Indeed, conjugated enzyme showed a higher KM value (P < 0.05) with respect to the free enzyme, indicating lower affinity of enzyme for its substrate. Maximum velocity of the free and conjugated enzymes were 193 and 197 nmol min−1, respectively. These changes in the kinetic parameters indicate that covalent binding of lysozyme onto SWCNTs resulted in change of affinity for the substrate. This may be due to moderately decreased substrate access to the active site of the conjugated enzyme. Conformational changes in the protein, which can result in changes in enzyme activity and affinity for the substrate occur because of covalent binding of lysozyme during immobilization.57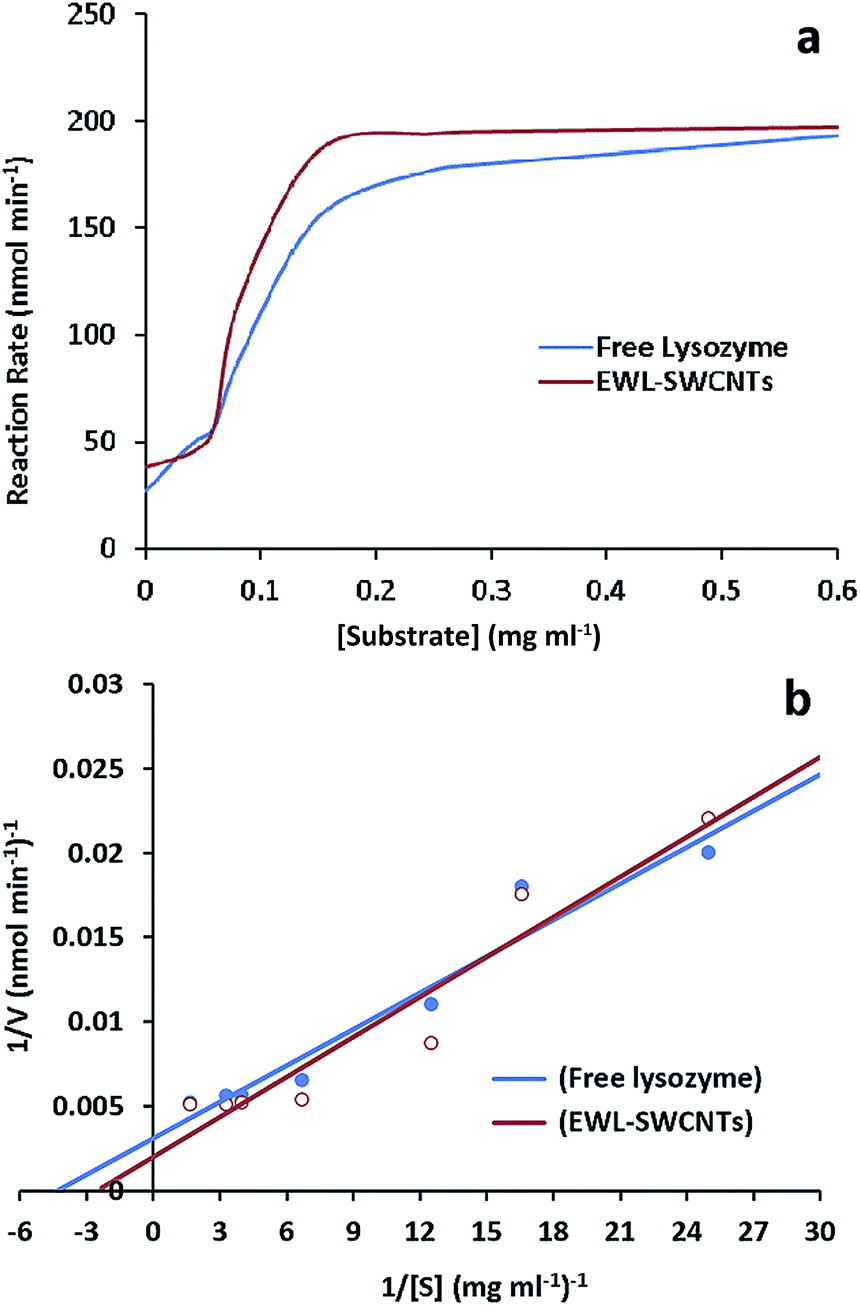 | ||
| Fig. 4 (a) Effect of substrate concentration on the specific activity of free lysozyme and EWL-SWCNTs. (b) Double reciprocal plot of velocity and substrate concentration for determination of KM and Vmax values of lysozyme in both free and conjugated enzymes. | ||
Thermal stability of free and conjugated lysozyme
Relative activities of free and conjugated lysozymes before and after thermal treatment at 70 °C for 20 minutes are shown in Fig. 5. The results showed that the thermal stability of the lysozyme was improved through conjugation onto SWCNTs. Residual activity of the free enzyme was approximately 20% after 20 minutes of incubation at 70 °C whereas the conjugated lysozyme retained 60% of its initial activity after 20 minutes of incubation. Predominantly, immobilization enhances the enzyme thermal stability due to a decrease in the protein structure mobility and an increase in rigidity protects enzyme from stability, specifically thermal stability of enzymes.56 The increased thermal stability for the conjugated enzyme can be due to the effects of the environment.57,58 Limited structural mobility due to covalent bonds and anchorage to the nanotubes increases thermal stability of immobilized lysozyme. In this case, a study showed that the thermal stability and catalytic activity of enzymes were significantly improved by enzyme–carbon nanotube interactions.7 We previously reported44 that the effect of amid bonds in conjugation with coupling agent (carbodiimide) to SWCNTs played an important role in improving the catalytic property of immobilized enzyme. The results obtained in this study suggest that covalent conjugation of lysozyme to SWCNTs can sterically prevent enzyme from heat-induced unfolding.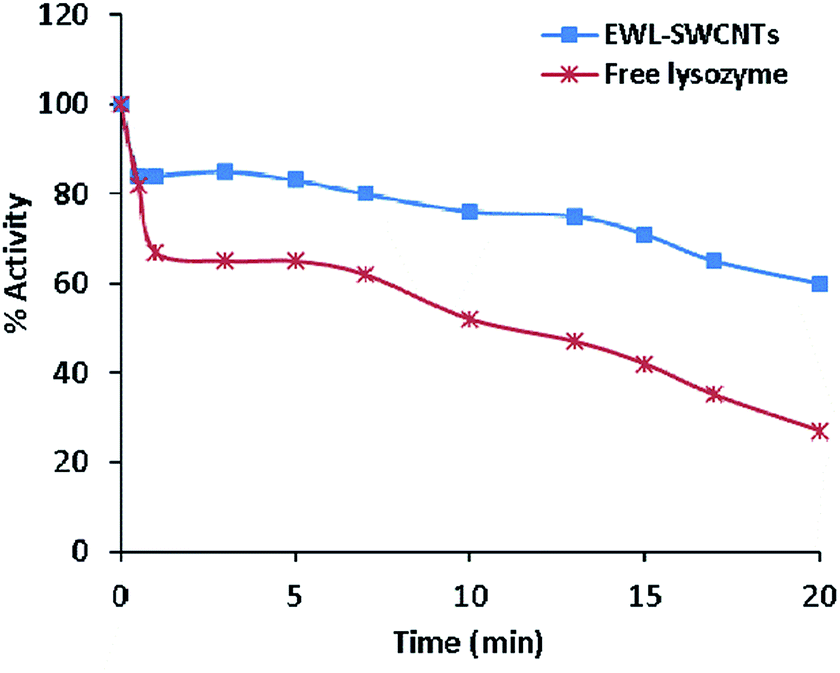 | ||
| Fig. 5 Thermostability of free lysozyme and conjugated EWL-SWCNTs. The enzymes solutions were incubated at 70 °C. Residual activities were measured using Imoto and Yagishita method. The initial activity is denoted as 100%. | ||
Optimum pH of free and conjugated lysozyme
The pH activity profile of free and conjugated lysozymes are shown in Fig. 6. The optimum pH of these forms of enzymes was studied at a pH range of 5.5–8.5. The results showed that the optimum pH values of free and conjugated lysozymes were 7.0 and 6.5, respectively. The pH shift towards the acidic value upon conjugation is speculated to be due to amide bonds between the amino groups of enzyme and the carboxylic groups of SWCNTs. Amphoteric properties of the enzyme depends on the ionizable groups in its active site. The charges on these groups depend on their accessibilities to the environment in different pH ranges. Variations in the pH lead to the changes in the ionic form of the active site and, consequently, changes in the activity of the enzyme. It seems that conjugation with SWCNTs affects the total net charge of the enzymes, distribution of charge on their exterior surfaces, and reactivity of the active groups. These effects are especially important in the vicinity of the active sites. The changes in lysozyme activity due to conjugation in different pH ranges can be explained by the change of charges of the four ionizable groups in the active site which are involved during catalysis. The ionizable groups consist of the side chain of glutamic acid at positions 35 and 66, and aspartic acid at positions 52 and 101, respectively.53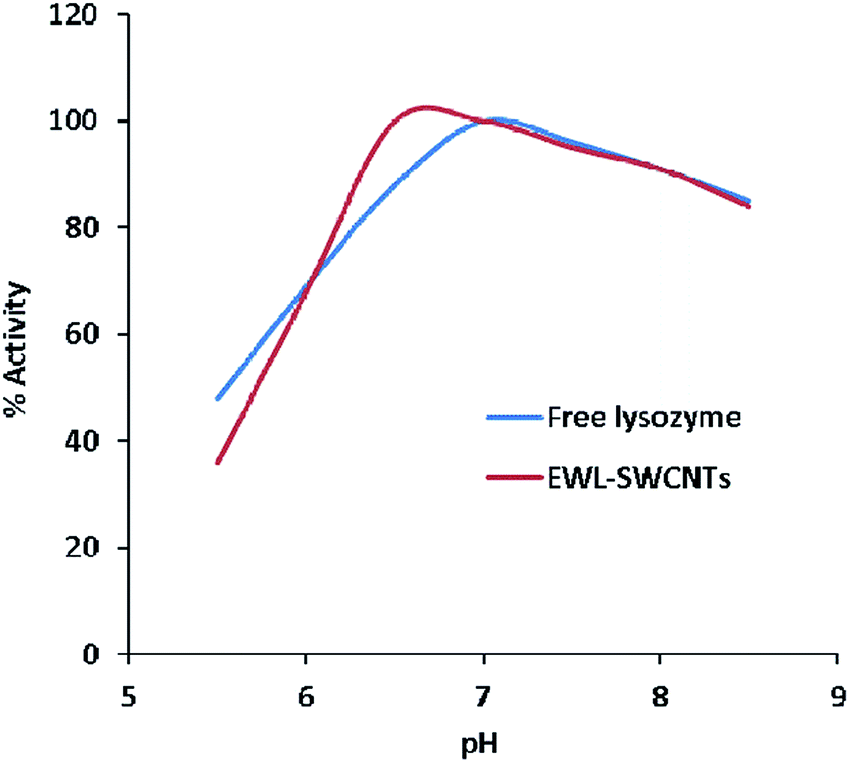 | ||
| Fig. 6 The optimal pH for free and conjugated lysozymes. | ||
Evaluation of pH stability of the free and conjugated enzymes
The pH stability of free and conjugated lysozymes were compared at different pH values (2.0, 3.0, 5.0, 7.0, and 10.0) after incubation for 1, 2, and 3 hours. Enzyme activity was measured at the pH 7 (Fig. 7a–c). The results indicated that conjugated enzyme had the highest stability at pH 2.0, 7.0, and 10.0 after 1 hour of incubation. At pH 2.0, however, the highest stability retained after 2 and 3 hours of incubation. The free lysozyme, however, had the highest stability around pH 5 to 7 after 1, 2, and 3 incubation hours. Significant increase in stabilization of the conjugated enzyme in the wide pH range of 2 to 10, after 3 hours shows the role of the SWCNTs in stabilizing the enzyme. The increased stability confers some level of protection to the enzyme during the reaction either in acidic or alkaline environment. Different factors affect pH stability, including temperature, ionic strength, enzyme concentration, chemical nature of the buffer, concentration of various preservatives (e.g. glycerol and sulfhydryl compounds), concentration of substrates and cofactors. In addition, pH stability of proteins is altered by multiple mechanisms including salt-bridge forming/breaking, hydrogen bond interactions, and like-charge repulsion effects.59 Salt bridges are pH-dependent and non-covalent bonds between oppositely charged residues that are adequately close to each other to create electrostatic bonds. In tertiary structure of proteins, salt bridges are constituted from negative charges from aspartic acid, glutamic acid, tyrosine, cysteine and the C-terminal carboxylate group, and of positive charges from lysine, arginine and the N-terminal amino groups.60 We believe that increased stability of EWL-SWCNTs can occur due to the formation of more interior salt bridges and also change of charge distribution on the exterior surfaces of the protein. Cannon et al.61 showed a statistically significant clustering of charged residues as ion-pairs and salt-bridge networks. Thermophilic proteins have more salt bridges than their mesophilic counterparts.62,63 In their study, pKa shifts showed that in buried salt bridges, stability decreased by 3–5 kcal mol−1 by removing a charge from one residue.64,65 It indicates that immobilization impact on stability was caused by charge change in the structure of proteins. Our results demonstrate that conjugation with SWCNTs have impact on residue charges, electrostatic bonds, the number of salt bridges and ion-pairs, geometrical orientation of the charged side chain in the salt bridges, and, consequently, on the folding and 3D structure of lysozyme.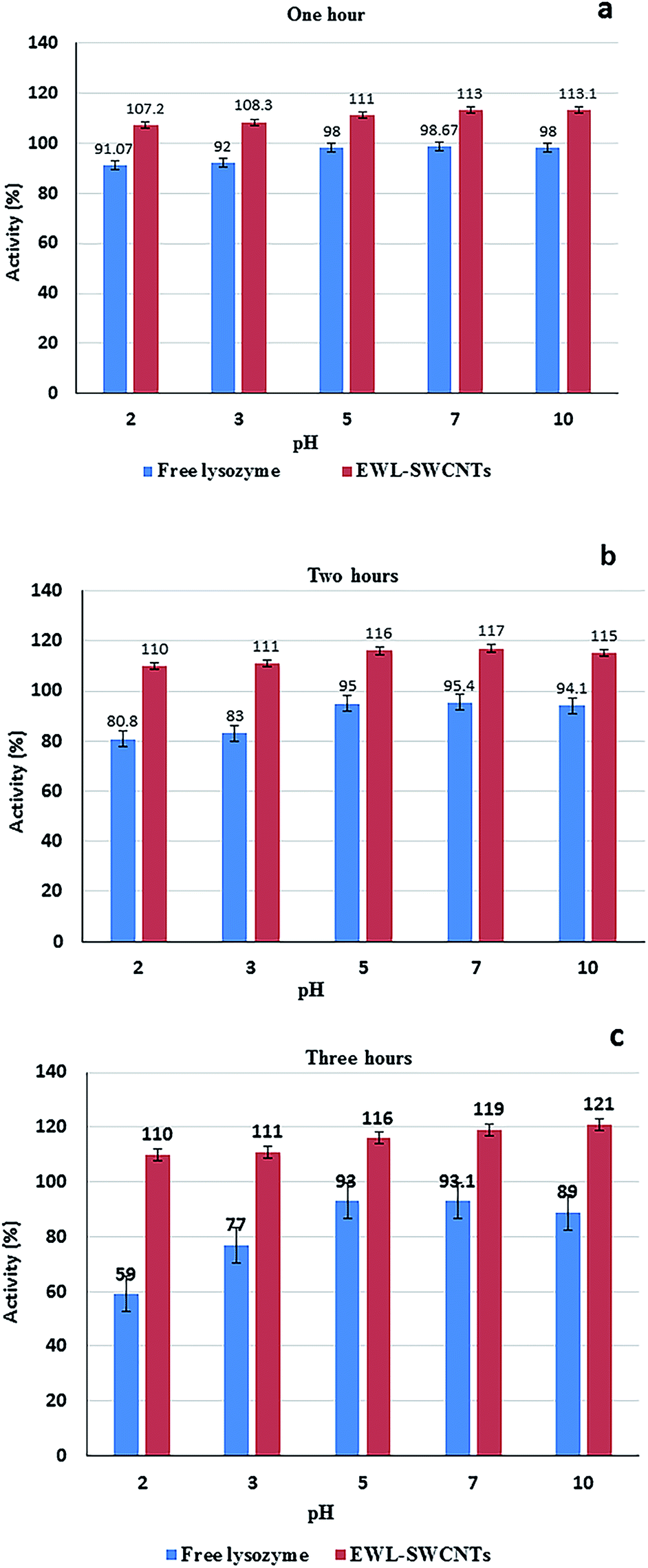 | ||
| Fig. 7 Evaluation of pH stability of free and conjugated lysozymes. The solutions in mix buffer (Tris-base, MES, and acetate) were incubated at pH 2, 3, 5, 7, and 10 in 70 °C for (a) one, (b) two, and (c) three hours. Enzyme activity was measured at pH 7. | ||
Effect of KCl and KSCN on the thermostability of free and conjugated lysozymes
Fig. 8a and b show representative data for the thermostability of lysozyme (1.0 mg ml−1 phosphate buffer, 0.1 M and pH 7) in the presence of KCl and KSCN (1.0 M) at 70 °C.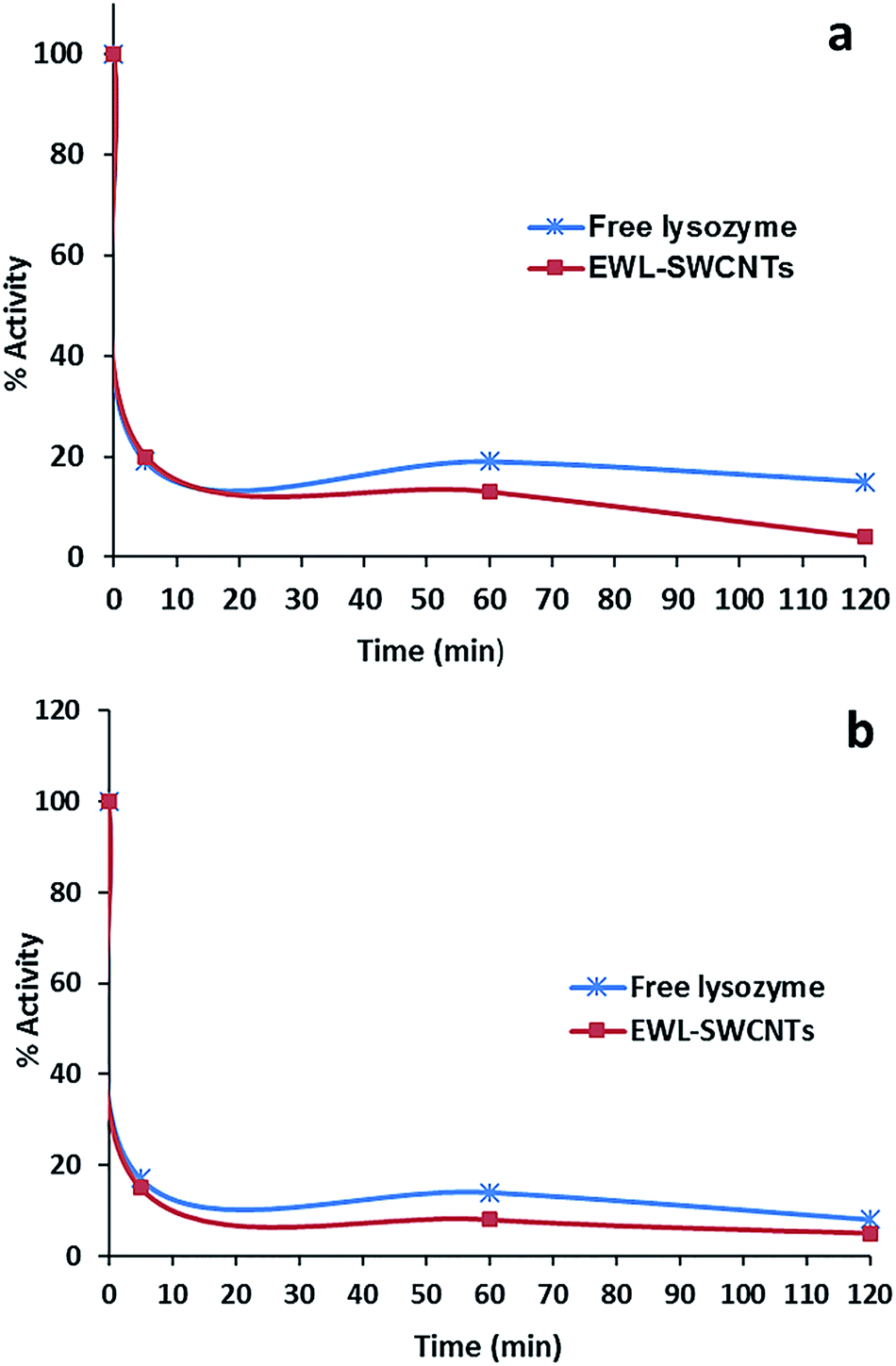 | ||
| Fig. 8 Effect of (a) KCl (1 M) and (b) KSCN (1 M) on thermostability of free lysozyme and EWL-SWCNTs at 70 °C. | ||
After heat treatment for 10 minutes, both free lysozyme and conjugated EWL-SWCNTs retained 20% and 10% activities in the presence of KCl and KSCN, respectively. Both free and conjugated forms of enzyme were inactivated with first-order kinetics at a rate constants of about 0.139 min−1 for 10 minutes. There are a number of research studies focusing on the role of salts during the heat induced proteins unfolding.56,60–64 However, the role of salts on heat-induced unfolding of conjugated enzyme with SWCNTs has not been studied to the same extent. To study the role of anions on thermal stability of proteins in free and SWCNT-conjugated forms, unfolding reaction should be investigated. These reactions consist of breaking noncovalent bonds between polypeptide chains (which include a combination of hydrogen bonds, van der Waals forces, and salt bridges), hydrating the newly exposed hydrophobic core of the protein and finally denaturing the secondary structure sections – alpha helices and beta pleated sheets, held together by hydrogen bonds.65 At pH 7, free lysozyme has a positive net charge of around +8.66 Positively charged amino acid side chains of lysine, arginine and histidine may electrostatically be covered by the anions (Cl− and SCN−) if they are accessible and not sterically hindered. The effect of these interactions on the structural stability of the protein depends on local conditions of the protein surface. Bye et al.67 suggest three mechanisms – electrostatic interaction at low concentrations, weak interaction with polar residues, and competition for water of hydration, by which anions regulate lysozyme stability. At high concentration of salts, anions compete for water with the enzyme molecules. Ionic dispersion potential is an important source of ion specificity which acts between each ion and protein.62 Boström et al. showed that this attractive potential is much stronger for SCN− than it is for Cl−. Our results also confirmed more ionic dispersion potential for KSCN (Fig. 8b).
Effect of urea on stability of free and conjugated lysozymes
The activity measurements of free and conjugated lysozyme in aqueous solutions in the presence of urea are reported in Fig. 9a and b. The comparison of these figures indicates that free lysozyme retained 30% activity, whereas the conjugated enzyme retained 10% activity in the presence of 2, 4, 6 and 8 molar urea. Both free and conjugated forms of the enzyme were inactivated with first-order kinetics at a rate constants of about 0.069 min−1 for 20 min. A second important observation from Fig. 9 is that, in comparison with free lysozyme, the EWL-SWCNTs showed a higher loss of activity (about 90%) for all urea concentrations. Hédoux et al.68 showed a lysozyme tertiary structure transformation into a softer structure without unfolding of the secondary structure because of binding urea molecules to hydrophilic residues. The significant decrease in conjugated lysozyme activity in all urea concentrations is due the changes in both positions and charges of hydrophilic residues which results in a significant conformational structure change of conjugated lysozyme. A number of researchers have proposed two mechanisms, “direct” and “indirect” models, to explain how urea induces the unfolding of the protein and changes the protein–water/protein–protein interaction balance.68–71 Urea molecules are absorbed by the charged hydrophilic residues on the surface, which causes the protein swelling. In addition, urea intrudes the protein followed by water entering the hydrophobic core. Therefore, it causes exposure of hydrophobic residues to the polar environment72 thus unfolding of the protein. Tirado-Rives et al.73 showed that a model in which urea directly binds to the amide units via hydrogen bonds and consequently denatures proteins by decreasing the hydrophobic effect. Our results suggest that conjugation with SWCNTs leads to more accessibility of charged hydrophilic residues on the surface through conjugation and consequently more interaction with urea molecules. This idea is confirmed by molecular dynamics simulations72 as well as Raman spectroscopy by Bennion et al.74 who detected the strong enhancement of isotopic exchanges in the presence of a high urea content (>4 M) in the amide I region. These results can be considered as evidences of direct interaction of urea with polar residues and the lysozyme backbone. Consequently, there will be high density of urea molecules on the surface of the protein, which results in more intense unfolding. The conformational changes in the conjugated lysozyme structure needs more studies using Raman spectroscopy.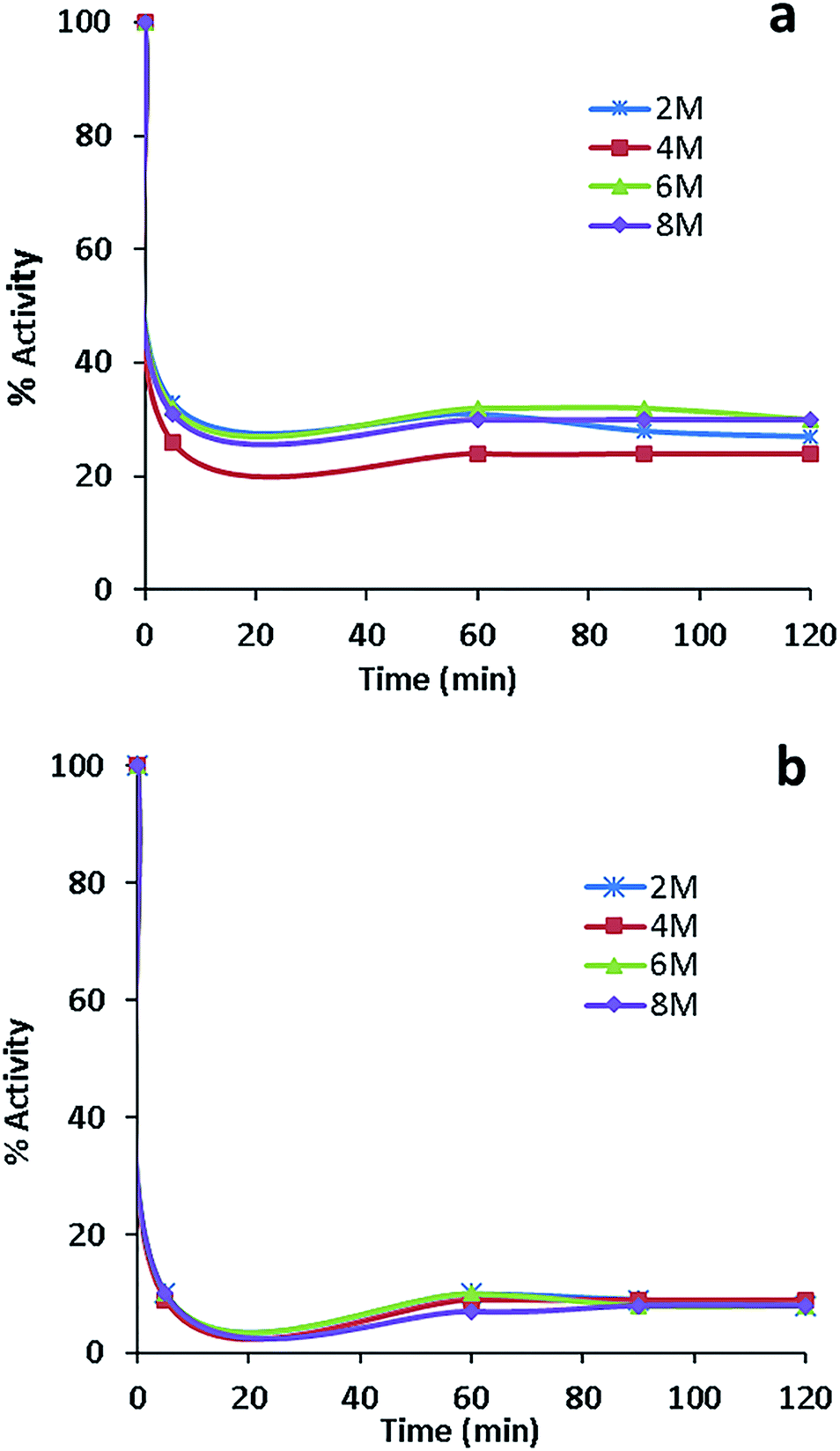 | ||
| Fig. 9 Effect of urea on the activity of (a) free lysozyme and (b) conjugated lysozyme. | ||
Conclusions
Conjugated protein–carbon nanotubes are extensively used as biophysical and biochemical tools in the area of biomedicine and biotechnology. Covalent conjugation of proteins on carbon nanotube surfaces, while retaining their structure, function and stability is a significant process in bionanofabrication technology. High chemical stability of conjugated lysozyme was confirmed by FTIR, SEM, and agarose gel electrophoresis.44 The results of this study confirmed the excellent potential of the SWCNTs as a support for enzyme immobilization. The fluorescence spectra confirmed, rather surprisingly, the role of both phenylalanine and tryptophan residues in the fluorescence bulk of the conjugated lysozyme. Compared with the free lysozyme, the emission spectrum of conjugated EWL-SWCNTs showed a significant increase in the fluorescence intensity at 286 nm (emission wavelength (λmax) of phenylalanine75) and a significant decrease in intensity at 340 nm (emission wavelength (λmax) of tryptophan47). All kinetic parameters of the conjugated form of lysozyme changed, probably due to the structural modifications in the tertiary structure, particularly in the active site. Successful conjugation of lysozyme on SWCNT's improves its thermostability and pH stability thus making this nanobiocatalyst system an excellent candidate for biotechnology and biomedical applications. More interactions of urea with hydrophilic residues in conjugated lysozyme resulted in disrupting more intramolecular hydrogen bonds and protein denaturation. Our results showed more ionic dispersion potential for a chaotropic salt, KSCN, in comparison with KCl. At 70 °C and for a given rate constant, inactivation of both forms of lysozyme were under a first order kinetic. It should be noted that there is a great need for research in the area of SWCNT support for enzyme conjugation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Special thanks to Mrs Maryam Tavana for buffer preparation.References
- I. Willner, Science, 2002, 298, 2407–2408 CrossRef CAS PubMed.
- D. F. Emerich and C. G. Thanos, Expert Opin. Biol. Ther., 2003, 3, 655–663 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Chem. Commun., 2011, 47, 10182–10188 RSC.
- J. C. Cruz, P. H. Pfromm, J. M. Tomich and M. E. Rezac, Colloids Surf., B, 2010, 79, 97–104 CrossRef CAS PubMed.
- M. Puri, C. J. Barrow and M. L. Verma, Trends Biotechnol., 2013, 31, 215–216 CrossRef CAS PubMed.
- I. V. Pavlidis, T. Vorhaben, T. Tsoufis, P. Rudolf, U. T. Bornscheuer, D. Gournis and H. Stamatis, Bioresour. Technol., 2012, 115, 164–171 CrossRef CAS PubMed.
- I. Pavlidis, T. Tsoufis, A. Enotiadis, D. Gournis and H. Stamatis, Adv. Eng. Mater., 2010, 12, B179–B183 CrossRef.
- S. Shah, K. Solanki and M. N. Gupta, Chem. Cent. J., 2007, 1, 1 CrossRef PubMed.
- S. Boncel, A. Zniszczoł, K. Szymańska, J. Mrowiec-Białoń, A. Jarzębski and K. Z. Walczak, Enzyme Microb. Technol., 2013, 53, 263–270 CrossRef CAS PubMed.
- T. Raghavendra, A. Basak, L. M. Manocha, A. R. Shah and D. Madamwar, Bioresour. Technol., 2013, 140, 103–110 CrossRef CAS PubMed.
- A. van Blaaderen, Nature, 2006, 439, 545–546 CrossRef CAS PubMed.
- P. Asuri, S. S. Bale, R. C. Pangule, D. A. Shah, R. S. Kane and J. S. Dordick, Langmuir, 2007, 23, 12318–12321 CrossRef CAS PubMed.
- Q. Shi, D. Yang, Y. Su, J. Li, Z. Jiang, Y. Jiang and W. Yuan, J. Nanopart. Res., 2007, 9, 1205–1210 CrossRef CAS.
- S. H. Lee, T. T. N. Doan, K. Won, S. H. Ha and Y.-M. Koo, J. Mol. Catal. B: Enzym., 2010, 62, 169–172 CrossRef CAS.
- F. Bomboi, A. Bonincontro, C. La Mesa and F. Tardani, J. Colloid Interface Sci., 2011, 355, 342–347 CrossRef CAS PubMed.
- L. Zhang, S. Ramsaywack, H. Fenniri and T. J. Webster, Tissue Eng., Part A, 2008, 14, 1353–1364 CrossRef CAS PubMed.
- L. Sun, D. Li, U. D. Hemraz, H. Fenniri and T. J. Webster, J. Biomed. Mater. Res., Part A, 2014, 102, 3446–3451 CrossRef PubMed.
- K. Lee, E. A. Silva and D. J. Mooney, J. R. Soc., Interface, 2011, 8, 153–170 CrossRef CAS PubMed.
- Z. Cheng and S.-H. Teoh, Biomaterials, 2004, 25, 1991–2001 CrossRef CAS PubMed.
- Z. Ma, C. Gao, J. Ji and J. Shen, Eur. Polym. J., 2002, 38, 2279–2284 CrossRef CAS.
- Y. Zhu, C. Gao, Y. Liu and J. Shen, J. Biomed. Mater. Res., Part A, 2004, 69, 436–443 CrossRef PubMed.
- Y. Zhu, C. Gao and J. Shen, Biomaterials, 2002, 23, 4889–4895 CrossRef CAS PubMed.
- A. Matsuda, H. Kobayashi, S. Itoh, K. Kataoka and J. Tanaka, Biomaterials, 2005, 26, 2273–2279 CrossRef CAS PubMed.
- L. Y. Santiago, R. W. Nowak, J. P. Rubin and K. G. Marra, Biomaterials, 2006, 27, 2962–2969 CrossRef CAS PubMed.
- H. Koh, T. Yong, C. Chan and S. Ramakrishna, Biomaterials, 2008, 29, 3574–3582 CrossRef CAS PubMed.
- X. Wang, D. Li, W. Wang, Q. Feng, F. Cui, Y. Xu and X. Song, Int. J. Biol. Macromol., 2003, 33, 95–100 CrossRef CAS PubMed.
- Z. Ding, J. Chen, S. Gao, J. Chang, J. Zhang and E. Kang, Biomaterials, 2004, 25, 1059–1067 CrossRef CAS PubMed.
- J.-D. Liao, S.-P. Lin and Y.-T. Wu, Biomacromolecules, 2005, 6, 392–399 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- H. Sun, A. Wirsén and A.-C. Albertsson, Biomacromolecules, 2004, 5, 2275–2280 CrossRef CAS PubMed.
- W. Mattanavee, O. Suwantong, S. Puthong, T. Bunaprasert, V. P. Hoven and P. Supaphol, ACS Appl. Mater. Interfaces, 2009, 1, 1076–1085 CAS.
- S. Smart, A. Cassady, G. Lu and D. Martin, Carbon, 2006, 44, 1034–1047 CrossRef CAS.
- A. Illanes, Enzyme biocatalysis: principles and applications, Springer Science & Business Media, 2008 Search PubMed.
- L. Chronopoulou, G. Kamel, C. Sparago, F. Bordi, S. Lupi, M. Diociaiuti and C. Palocci, Soft Matter, 2011, 7, 2653–2662 RSC.
- R. C. Barabino, D. N. Gray and M. H. Keyes, Clin. Chem., 1978, 24, 1393–1398 CAS.
- D. T. Kim, H. W. Blanch and C. J. Radke, Langmuir, 2002, 18, 5841–5850 CrossRef CAS.
- T. Wei, M. A. Carignano and I. Szleifer, Langmuir, 2011, 27, 12074–12081 CrossRef CAS PubMed.
- K. Kubiak-Ossowska, M. Cwieka, A. Kaczynska, B. Jachimska and P. A. Mulheran, Phys. Chem. Chem. Phys., 2015, 17, 24070–24077 RSC.
- E. S. Kirkor and A. Scheeline, J. Phys. Chem. B, 2001, 105, 6278–6280 CrossRef CAS.
- R. A. Silva, A. M. Carmona-Ribeiro and D. F. S. Petri, Int. J. Biol. Macromol., 2007, 41, 404–409 CrossRef CAS PubMed.
- J. V. Nicolini, N. S. de Resende and H. C. Ferraz, J. Chem. Technol. Biotechnol., 2015, 90, 739–746 CrossRef CAS.
- T. Imoto and K. Yagishita, Agric. Biol. Chem., 1971, 35, 1154–1156 CrossRef CAS.
- Z. Borzooeian, A. Safavi, M. Hossain Sheikhi, M. Aminlari and M. Mahdi Doroodmand, J. Exp. Nanosci., 2010, 5, 536–547 CrossRef CAS.
- H.-M. Ding, L. Shao, R.-J. Liu, Q.-G. Xiao and J.-F. Chen, J. Colloid Interface Sci., 2005, 290, 102–106 CrossRef CAS PubMed.
- J. R. Lakowicz and B. R. Masters, J. Biomed. Opt., 2008, 13, 029901 CrossRef.
- T. Imoto, L. S. Forster, J. Rupley and F. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 1151–1155 CrossRef CAS.
- R. Cundall, Time-resolved fluorescence spectroscopy in biochemistry and biology, Springer Science & Business Media, 2013 Search PubMed.
- C. D. McGuinness, A. M. Macmillan, K. Sagoo, D. McLoskey and D. J. Birch, Appl. Phys. Lett., 2006, 89, 063901 CrossRef.
- H. Huang, M. Zou, X. Xu, F. Liu, N. Li and X. Wang, TrAC, Trends Anal. Chem., 2011, 30, 1109–1119 CrossRef CAS.
- K.-C. Kao, T.-S. Lin and C.-Y. Mou, J. Phys. Chem. C, 2014, 118, 6734–6743 CAS.
- A. J. Kirby, Nat. Struct. Mol. Biol., 2001, 8, 737 CAS.
- C. Blake, L. N. Johnson, G. Mair, A. North, D. Phillips and V. Sarma, Proc. R. Soc. London, Ser. B, 1967, 167, 378–388 CrossRef CAS.
- S. Damodaran, J. Food Sci., 2005, 70, R54–R66 CrossRef CAS.
- V. Fainerman, E. Lucassen-Reynders and R. Miller, Colloids Surf., A, 1998, 143, 141–165 CrossRef CAS.
- C. H. Ramos and R. L. Baldwin, Protein Sci., 2002, 11, 1771–1778 CrossRef CAS PubMed.
- E. Taqieddin and M. Amiji, Biomaterials, 2004, 25, 1937–1945 CrossRef CAS PubMed.
- H. H. Weetall, Science, 1969, 166, 615–617 CAS.
- R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 62, 197–212 CrossRef CAS.
- X. Tadeo, M. Pons and O. Millet, Biochemistry, 2007, 46, 917–923 CrossRef CAS PubMed.
- W. R. Cannon, N. D. Talley, B. A. Danzig, X. Liu, J. S. Martinez, A. P. Shreve and G. MacDonald, Biophys. Chem., 2012, 163, 56–63 CrossRef PubMed.
- A. J. Richard, C.-C. Liu, A. L. Klinger, M. J. Todd, T. M. Mezzasalma and V. J. LiCata, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1546–1552 CrossRef CAS PubMed.
- L. A. Sikkink and M. Ramirez-Alvarado, Biophys. Chem., 2008, 135, 25–31 CrossRef CAS PubMed.
- R. Komsa-Penkova, R. Koynova, G. Kostov and B. G. Tenchov, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1297, 171–181 CrossRef.
- M. Boström, D. Williams and B. W. Ninham, Biophys. J., 2003, 85, 686–694 CrossRef.
- M. Bončina, J. Reščič and V. Vlachy, Biophys. J., 2008, 95, 1285–1294 CrossRef PubMed.
- J. W. Bye and R. J. Falconer, Protein Sci., 2013, 22, 1563–1570 CrossRef CAS PubMed.
- A. Hédoux, S. Krenzlin, L. Paccou, Y. Guinet, M.-P. Flament and J. Siepmann, Phys. Chem. Chem. Phys., 2010, 12, 13189–13196 RSC.
- Y. Nozaki and C. Tanford, J. Biol. Chem., 1963, 238, 4074–4081 CAS.
- D. B. Watlaufer, S. K. Malik, L. Stoller and R. L. Coffin, J. Am. Chem. Soc., 1964, 86, 508–514 CrossRef.
- D. R. Robinson and W. P. Jencks, J. Am. Chem. Soc., 1965, 87, 2462–2470 CrossRef CAS PubMed.
- E. Finer, F. Franks and M. Tait, J. Am. Chem. Soc., 1972, 94, 4424–4429 CrossRef CAS.
- J. Tirado-Rives, M. Orozco and W. L. Jorgensen, Biochemistry, 1997, 36, 7313–7329 CrossRef CAS PubMed.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5142–5147 CrossRef CAS PubMed.
- C. Albrecht, Anal. Bioanal. Chem., 2008, 390, 1223–1224 CrossRef CAS.
Footnote |
| † Current affiliation is Takeda Pharmaceuticals, but this work was completed before my employment at Takeda Pharmaceuticals Company Ltd. |
| This journal is © The Royal Society of Chemistry 2017 |