
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfiguration and stability of naturally occurring all-cis-tetrahydrofuran lignans from Piper solmsianum†
Clécio S. Ramosa,
Harrald V. Linnertb,
Marcilio M. de Moraesb,
João H. do Amaralb,
Lydia F. Yamaguchib and
Massuo J. Kato *b
*b
aDepartment of Chemistry, Rural Federal University of Pernambuco, Rua Dom Manoel de Medeiros, s/n, 52.171-030 Recife, PE, Brazil
bInstitute of Chemistry, University of São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000 São Paulo, SP, Brazil. E-mail: majokato@iq.ups.br
First published on 4th October 2017
Abstract
The study of variability of the tetrahydrofuran lignan (−)-grandisin in leaves of Piper solmsianum (Piperaceae) revealed two unknown compounds, that were isolated and determined to be the all-cis tetrahydrofuran lignans 1a [rel-(7R,8S,7′S,8′R)-3,4,5,3′,4′,5′-hexamethoxy-7,7′-epoxylignan] and 1b [rel-(7R,8S,7′S,8R′)-3′,4′-methylenedioxy-3,4,5,5′-tetramethoxy-7,7′-epoxylignan]. Their structures were determined by spectroscopic analysis including 1D and 2D-NMR while their configurations were determined by ECD associated to the density functional theory (DFT) at the B3LYP/6-31G(d,p) level. The hydrogen bonds between methoxy groups in trimethoxyphenyl rings stabilizes the all-trans tetrahydrofuran lignan grandisin by 6.5 kcal mol−1 as compared to the corresponding all-cis isomer of grandisin. The occurrence of all-cis tetrahydrofuran lignans as natural products is a very rare event.
Introduction
Lignans are dimers of phenylpropanoid units widely found in the plant kingdom and with a variety of biological properties such as cytotoxic, antioxidant, antimicrobial, anti-inflammatory, antiviral and immunosuppressive activities.1,2 The dimerization process in their formation requires an oxidative coupling mechanism forming a β–β bonding, which can be followed by further reactions yielding eight different sub-classes including tetrahydrofuran, furofuran, dibenzylbutane, dibenzylbutyrolactone, aryltetralin, arylnaphthalene, dibenzocyclooctadiene and dibenzylbutyrolactol lignans.3–5 Tetrahydrofuran (THFs) lignans can be further sub-divided in 7,7′-monoepoxy-, 7,9′-monoepoxy- and 9,9′-monoepoxy-lignans. The THFs lignans have been described so far from several plant species and their structural diversity results from the type of substituent in the aromatic ring that can be hydroxy, methoxy, methylenedioxy and a combination of them. The relative configuration among aryls and methyl groups can give rise to further stereochemistry diversity. In case of having the same aromatic rings, ten isomers are possible consisting of four diastereomeric pairs besides two meso forms.6 The (−)-grandisin is an example of the all-trans tetrahydrofuran lignan, which was formerly isolated from the Lauraceae plants, Litsea grandis7 and then from Licaria aurea8 and Cryptocarya crassinevia.9 Grandisin has also been isolated from monocotiledoneous Araceae,10 from other Angiosperms families such as Piperaceae,11 Myristicaceae,12 Magnoliaceae,13 Annonaceae14 but also from Rosids including Meliaceae,15 Violaceae species,16 and other plant species. The THFs lignans have displayed several biological activities such as anti-PAF,17 antiprotozoal,18–21 larvicidal,22,23 cytotoxic,24,25 estrogenic,25 anti-inflammatory,26 and NF-κB inhibitory activity.27 Thus, some studies have also been addressed to generate analogues.28–31 To date, the natural occurrence of all-cis tetrahydrofuran lignan has only been described from Anogeissus acuminata (Combretaceae)32 and from Illicium floridanum (Illiciaceae).33 Besides, a synthetic version of all-cis had been obtained by catalytic hydrogenation of a furan derivative.34 Additionally, a series of synthetic tetrahydrofuran lignans with various stereochemistry were prepared, including the all-cis tetrahydrofuroguaiacin B, in order to evaluate their plant growth inhibitory activity.31 Herein we report on the isolation and structural determination of two novel all-cis tetrahydrofuran lignans 1a and 1b from the leaves of two specimens of Piper solmsianum. Besides, two known lignans 2b and 3a were also isolated (Fig. 1). The structures of the last two tetrahydrofuran lignans had been only reported without the assignment of their configurations or optical rotations.10,15,35 The determination of configuration of THFs lignans (1a, 1b, 2b and 3a) were carried out using electronic circular dichroism (ECD) having (−)-grandisin as reference data since its configuration was previously reported based on crystallographic studies.14 Additionally, since natural all-cis tetrahydrofuran lignans, such as 1a and 1b, are of rare occurrence a study of structural stability were undertaken using density functional theory (DFT) at the B3LYP/6-31G(d,p) level. The relative energies of lignans 1a and 3a were compared to that of all-trans lignan (−)-grandisin (2a). Solvent effects were analysed by the self-consistence reaction field (SCRF) method, and the energy difference in water and cyclohexane environments were determined with the polarizable continuum model PCM(B3LYP/6-31G(d,p)) method.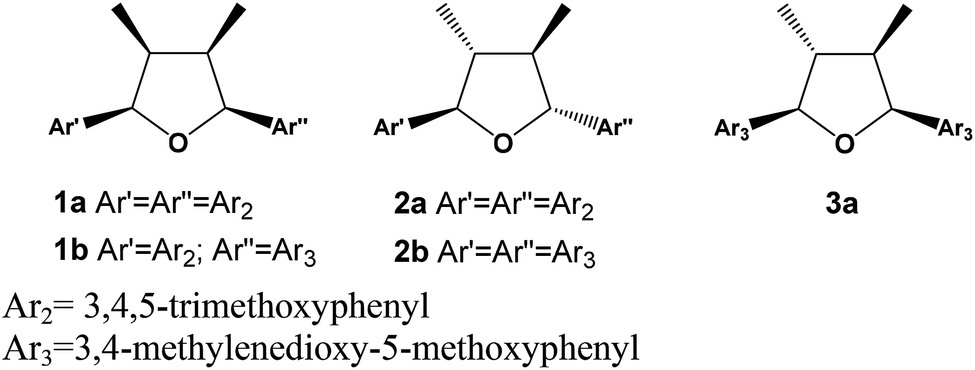 | ||
| Fig. 1 Tetrahydrofuran lignans from P. solmsianum. | ||
Results and discussions
Analysis of crude extracts from P. solmsianum
The tetrahydrofuran lignan (−)-grandisin has displayed several bioactivities and thus, the initial approach was to determine its variability in specimens of P. solmsianum occurring in several sites in São Paulo State. The initial survey in samples from leaves of different sites revealed common profiles based on the occurrence of phenylpropanoids apiole–dillapiole and grandisin (2a). Then, the analysis of chemical variability of a population of P. solmsianum occurring in the Campus of University of São Paulo was undertaken and several samples collected presented an unusual chromatographic profile with one major peak with shorter retention time than that of grandisin. These extracts were submitted to 1H NMR analysis, which suggested the occurrence of a major tetrahydrofuran lignan (1a) with similar oxygenation pattern in the aromatic rings with six methoxy groups. Crude extracts from different P. solmsianum individuals collected in Alberto Löfgren State Park analysed by HPLC indicated a second tetrahydrofuran lignan, having methoxy and 3,4-methylenedioxy groups (1b). Thus, both compounds were isolated and had their structures determined by spectroscopic analysis.Structural determination of lignans 1a, 1b, 2b and 3a
The lignan 1a was isolated as a pale white powder with purity of 99.6% by HPLC. Its HR-ESIMS spectrum displayed a quasi-molecular ion [M + H]+ peak at m/z 433.2232 corresponding to the molecular formula C24H33O7. The IR of 1a showed no absorptions near 3400 cm−1, but an intense band at 1128 cm−1 suggesting C–O–C functions. The simplicity of the 1H and 13C NMR spectra (Fig. S1 and S2; ESI†) indicated that 1a is a symmetrical compound (Table 1). The dimethyl-tetrahydrofuran moiety could be readily recognized in the 1H NMR spectrum by the doublets at δH 0.64 (6H, J = 6.7 Hz, H-9/H-9′) and 5.11 (2H, J = 6.7 Hz, H-7/H-7′), and a multiplet at δH 2.70 (2H, H-8/H-8′). The singlet at δH 6.74 (H-2/H-2′, H-6/H-6′) was assigned to the two equivalent ortho-methoxy aromatic hydrogens, while the intense singlets at δH 3.88 (4 × OCH3; 3,3′,5,5′) and 3.86 (2 × OCH3; 4,4′) corresponded to the six methoxy groups. The structure of 1a bearing two 3,4,5-trimethoxyphenyl groups was firmly supported by further 13C NMR analysis (Table 1).3,35,36 All NMR assignments were confirmed by HSQC and HMBC correlations in which cross peaks were observed between the aromatic and aliphatic nuclei (Fig. S3 and S4†).| Position | 1a | 1b | ||||||
|---|---|---|---|---|---|---|---|---|
| δ 1H | COSY | δ 13C | HMBC | δ 1H | COSY | δ 13C | HMBC | |
| a 500 MHz for 1H and 125 MHz for 13C; compounds dissolved in CDCl3; δ in ppm relative to TMS, J in Hz. | ||||||||
| 1 | — | — | 137.0 | C6, C2 | — | — | 135.3 | C6, C2 |
| 2 | 6.67 (s, 1H) | H7 | 103.6 | C6, C7, C8, C4, C3 | 6.65 (s, 1H) | H7 | 103.5 | C6, C7 |
| 3 | — | — | 153.1 | C2, OCH3-3 | — | — | 153.0 | C2, OCH3-3 |
| 4 | — | — | 136.0 | C-6, OCH3-4 | — | — | 137.5 | C6, OCH3-4 |
| 5 | — | — | 153.1 | C-6, OCH3-5 | — | — | 153.0 | C6, OCH3-5 |
| 6 | 6.67 (s, 1H) | H7 | 103.6 | C2, C7, C8, C4, C5 | 6.65 (s, 1H) | H7 | 103.5 | C2, C7 |
| 7 | 5.11 (d, J 6.7 Hz, 1H) | H8, H9, H2, H6 | 82.9 | C9, C8′, C2, C3, C1 | 5.09 (d, J 7.0 Hz, 1H) | H8, H9 H2, H6 | 82.8 | C9, C2, C3 |
| 8 | 2.70 (m, 1H) | H7, H9 | 41.6 | C7, C7′, C8′, C9, C9′, C1 | 2.67 (m, 1H) | H7, H9 | 41.5 | C7, C7′, C8′, C9, C9′ |
| 9 | 0.64 (d, J 6.7 Hz, 3H) | H8, H7 | 11.6 | C9′, C8, C8′, C7, C1 | 0.64 (d, J 6.9 Hz, 3H) | H8, H7 | 11.6 | C8, C8′, C7 |
| 1′ | — | — | 137.0 | C6′, C2′ | — | — | 136.1 | C6′, C2′ |
| 2′ | 6.67 (s, 1H) | H7′ | 103.6 | C6′, C7′, C8′, C4′, C3′ | 6.63 (s, 1H) | H7′ | 105.8 | C6′, C7′ |
| 3′ | — | — | 153.1 | O2CH2, C2′ | — | — | 143.4 | O2CH2, C2′ |
| 4′ | — | — | 136.0 | O2CH2, C6′ | — | — | 139.8 | O2CH2, C6′ |
| 5′ | — | — | 153.1 | C6′, OCH3-5′ | — | — | 148.8 | C6′, OCH3-5′ |
| 6′ | 6.67 (s, 1H) | H7′ | 103.6 | C2′, C7′, C8′, C4′, C5′ | 6.61 (s, 1H) | H7′ | 100.7 | C2′, C7′ |
| 7′ | 5.11 (d, J 6.7 Hz, 1H) | H8′, H9′, H2′, H6′ | 82.9 | C9′, C8, C2′, C3′, C1′ | 5.09 (d, J 7.0 Hz, 1H) | H8′, H9′, H2′, H6′ | 82.9 | C9′, C2′, C3′ |
| 8′ | 2.70 (m, 1H) | H7′, H9′ | 41.6 | C7, C7′, C8, C9, C9′, C1′ | 2.67 (m, 1H) | H7′, H9′ | 41.6 | C7, C7′, C8, C9, C9′ |
| 9′ | 0.64 (d, J 6.7 Hz, 3H) | H8′, H7′ | 11.6 | C9, C8, C8′, C7′, C1′ | 0.62 (d, J 6.9 Hz, 3H) | H8′, H7′ | 11.7 | C8, C8′, C7′ |
| OCH3/3 | 3.88 (s, 3H) | — | 56.1 | C3 | 3.88 (s, 3H) | 56.1 | — | |
| OCH3/3′ | 3.88 (s, 3H) | — | 56.1 | C3′ | — | — | — | |
| OCH3/4 | 3.86 (s, 3H) | — | 60.9 | C4 | 3.86 (s, 3H) | 60.9 | — | |
| OCH3/4′ | 3.86 (s, 3H) | — | 60.9 | C4′ | — | — | — | |
| OCH3/5 | 3.88 (s, 3H) | — | 56.1 | C5 | 3.88 (s, 3H) | 56.1 | — | |
| OCH3/5′ | 3.88 (s, 3H) | — | 56.1 | C5′ | 3.91 (s, 3H) | 56.6 | — | |
| O2CH2 | 5.98 (s, 2H) | 101.3 | — | |||||
Considering the symmetry observed in the 1H and 13C NMR spectra and that 1a is optically inactive, the compound was determined as a meso isomer with configuration similar to the all-cis (1c) or trans–cis–trans (5) (Table S1†). The determination of the relationship between the methyl and the aryl groups was based on the chemical shifts of methyl (H-9/H-9′) and oxymethine (H-7/H-7′) protons, and of the oxybenzylic carbons (C-7/C-7′) (Tables 1 and S1†).3,8,32 The protons at H-9/H-9′ and H-7/H-7′ of 1a showed signals at δH 0.64 and 5.11, respectively and the coupling constants of J 6.7 Hz for the H-7/H-8 (H-7′/H-8′) hydrogens is compatible with a dihedral angle of approximately 30°, indicating all-cis relationship between the methyl and aryl substituents.32 The up-field shifted methyl carbons at δC 11.6 in the 13C NMR is contrasting to the all-trans isomer grandisin (2a), which appeared at δC 13.9. This shielding effect has been assigned to the anisotropic effect of the pseudo-axially oriented aromatic ring on the methyl groups of 1a. The chemical shifts of C-7/C-7′ observed at δC 82.9, and of C-8/C-8′ at δC 41.6, provided further evidences for the cis configurations between the methyl and aryl groups of 1a (Tables 1 and S1†). All the 1H and 13C NMR data indicated that the lignan 1a is all-cis isomer of 2a (grandisin)35 thus, the structure was determined as rel-(7R,8S,7′S,8′R)-3,4,5,3′,4′,5′-hexamethoxy-7,7′-epoxylignan.
Compound 1b was isolated as pale-yellow oil by preparative TLC of a methanolic extract of the leaves of P. solmsianum with 99.2% purity indicated by HPLC analysis. The molecular formula of 1b was determined as C23H28O7 according to the quasi-molecular ion [M + H]+ observed in the HR-ESIMS at m/z 417.1915.
Analysis of the 1H and 13C NMR spectra of 1b indicated the structure of an all-cis tetrahydrofuran lignan similarly to 1a (Table 1; Fig. S5 and S6†). The major differences were associated to the replacement of two methoxy by one methylenedioxy group, which account for the difference of 16 Da between the molecular ions of 1a and 1b. The chemical shift of the equivalent aromatic hydrogens of the 3,4,5-trimethoxyphenyl ring (δH 6.65, s, 2H) was similar to that of 1a, while the hydrogens of the second aromatic ring appeared as two broad singlets at δH 6.63 and 6.61 and were assigned to the two aromatic hydrogens of the 3-methoxy-4,5-methylenedioxyphenyl ring. The HMBC contour plot (Fig. S8†) displayed correlations between the oxymethine protons (H-7/H-7′) to C-6/C-6′ and C-2/C-2′, and between the H-7/H-7′ hydrogens to signals of C-9/C-9′ (Fig. 2). All NMR data, including HSQC (Fig. S7†), confirmed the assignments made and thus 1b was determined as rel-(7R,8S,7′S,8′R)-3′,4′-methylenedioxy-3,4,5,5′-tetramethoxy-7,7′-epoxylignan.
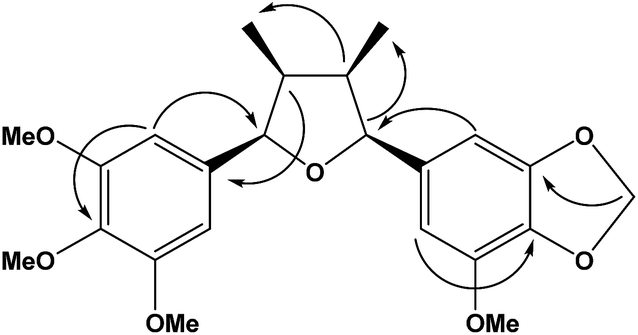 | ||
| Fig. 2 Important 1H–13C observed in the HMBC correlations of lignan 1b. | ||
Compound 2b has been reported previously as rel-(7R,8S,7′S,8′)-3′,4′-methylenedioxy-3,4,5,5′-tetramethoxy-7,7′-epoxylignan.35
The compound 3a, determined as (−)-epigrandisin based on the spectrometric data analysis, is a lignan formerly isolated from the stem bark of Aglaia leptantha but characterized in a mixture with (−)-grandisin (2a) with an estimated [α]21D value of −15 ± 10.15 (−)-Epigrandisin, without configuration assigned, was also reported from leaves of Rhaphidophora decursiva.10
Determination of relative configurations of lignans 1a, 1b, 2b and 3a
The relative configurations of the THF lignans 1a, 1b, 2b and 3a were determined by comparison of ECD spectra with that of (−)-grandisin (2a) (Fig. 3), in which its configuration had been previously determined by X-ray analysis.7 Lignan 1a is optically inactive and no Cotton effect was observed in its ECD curve. The ECD curve of lignan 2b was very similar to that of (−)-grandisin (2a) with a negative Cotton effect at 233 nm, indicating the trans configuration between the aromatic rings. In contrast, the lignans 1b and 3a showed a positive Cotton effect at 220 nm in the ECD curve, i.e. opposite to that of 2a, hence the cis configuration between the aromatic rings were assigned (Fig. 3).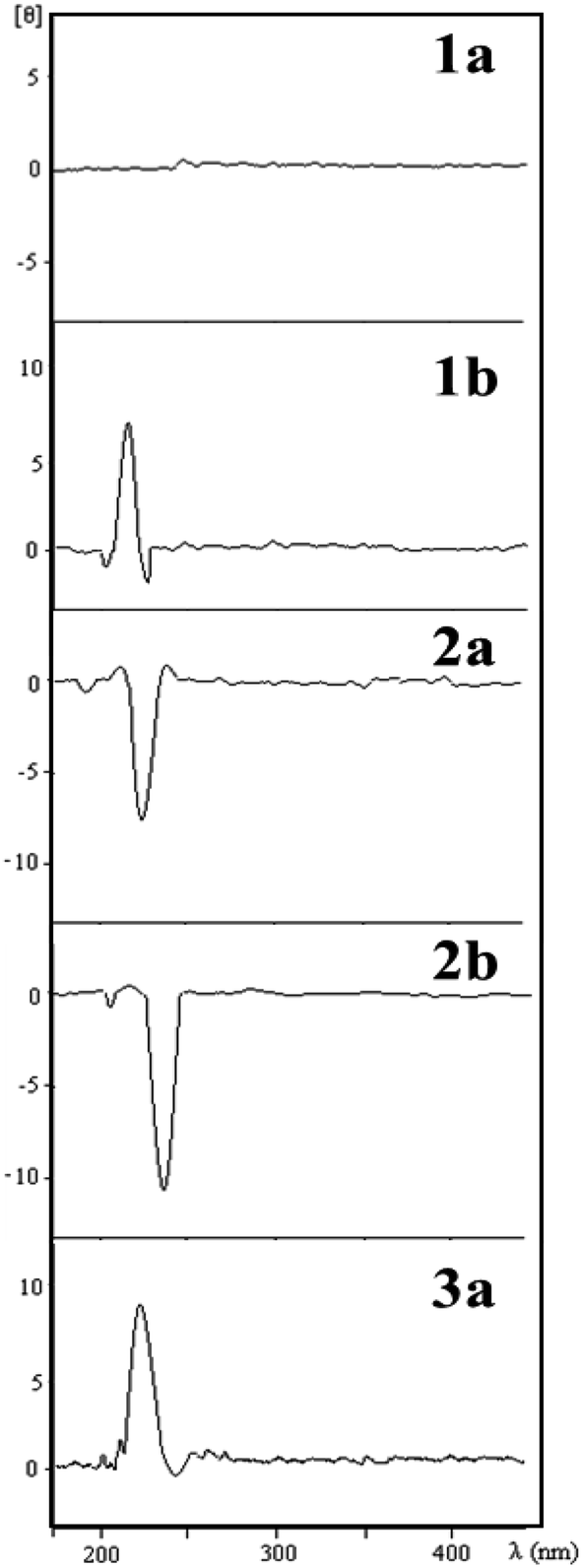 | ||
| Fig. 3 ECD curves of tetrahydrofuran lignans from P. solmsianum. | ||
Energetics of the lignans in gas phase
Optimization of the tetrahydrofuran lignans (gas phase) structures was performed with the aid of Gaussian 03 (revision B.05) software,37 using DFT at the B3LYP level with the 6-31G(d,p) basis set. Becke's three parameter adiabatic connection approach is well established to provide structural parameters for large molecules including the intramolecular and intermolecular hydrogen bondings.38,39 Harmonic frequencies were calculated in order to confirm the equilibrium geometries corresponding to minima energy and zero point energy (ZPE). Correction term was added to the electronic energies of the calculated structures to estimate differences in the relative energies between the lignans 1a, 2a and 3a (Table S2†).In order to predict the energy and the solvent effect on lignans stability in polar and apolar environments, an single point energy calculation was performed, with the PCM (B3LYP) SCRF models using the 6-31G(d,p) basis set. The tight SCF convergence criteria were specified for all jobs and the predicted energy and solvent effects in cyclohexane and water media were calculated (ESI, Table S3†).
The calculations at the B3LYP/6-31G(d,p) level indicated lignan 2a as the most stable among the lignans 1a and 3a in gas phase, cyclohexane and water. An exception was observed for lignans 2a and 3a, which have similar electronic energy in water medium (Tables S2 and S3†). The calculations showed that inclusion of solvent effects does not change the overall relative results obtained in the gas phase. Furthermore, the stabilities increased with the solvent polarity, and only the change in the energy produced in the high polar medium (i.e., water) was more effective in stabilizing the conformers as compared to the gas phase or apolar solvent (i.e., cyclohexane).
Gas phase optimization geometry of the THF lignans
A remarkable feature in the analysis of the geometrical parameters of 1a, 1b, 2a, 2b and 3a lignans is the stabilization resulting from intramolecular C–H⋯O hydrogen bonding. Energy calculations predict that the all-trans THF lignan 2a would be 0.42 kcal mol−1 more stable than epigrandisin 3a, and 6.5 kcal mol−1 more stable than the all-cis THF lignan 1a. Such sequence of stability is somewhat compatible with the rare natural occurrence of all-cis THF lignan isomer. In fact, there are only two previous report for it.32,33 It is also noteworthy that the methyl group (C9′) attached to the THF ring lies close to the aromatic rings in the all-cis lignan 1a, revealing one further possible hydrogen bonding contributing to an unfavourable gauche conformation around the C8′ bond (Fig. S9†). Accordingly, the calculated angle at C8–C8′–C7′ is 3° smaller than the corresponding C7–C8–C8′ angle at the THF ring, a feature also observed in 1b. Additionally, the phenyl rings in 1a lie closer to the THF ring compared with the all-trans isomer, resulting in smaller dihedral angles, which is less favourable.The results concerning structural changes associated to the phenyl rings suggested that the geometrical equilibrium structures of 1b and 2b are dependent on intramolecular hydrogen bonding. On the other hand, the geometrical energy minimization of 2b taking into account both hydrogens but not hydrogen bonding (a difference of ∼1 kcal mol−1), showed that these additional hydrogen bonds are not energetically significant. It is also worth to note that in these geometry optimizations, the methoxy groups attached to each of the phenyl ring do not necessary lie close to the ring plane, but assumed the most favourable hydrogen bonding with conformers differing from those of crystallographic data.14
The calculated equilibrium geometries for lignans 1a–2a are shown in Fig. S9 and S10 (ESI†), and the main geometrical parameters, obtained after minimized energies using the B3LYP method, are presented in Tables S4–S7 (ESI†).
Relative energies of the THF isomers without methoxy groups in the phenyl rings
In order to compare the behavior of the THF lignan isomers, a singular qualitative calculation was made for the ten possible tetrahydrofuran isomers (four diastereomeric pairs and two meso forms) (Table S8†) with a lower level of theory as B3LYP/3-21G*. The methoxy groups were not included in the calculation in order to minimize the effect of the hydrogen bondings in the stabilities and thus to determine only the effect of configuration in the stabilities of THF rings. The electronic energy of the optimized structures indicates that the all trans and the all cis structures are the most and the least stable among the ten stereoisomers, respectively (Table S8†). With this finding, we can predict that the configuration and the degree of methoxylation contribute to the stabilization of the THFs lignans.Conclusions
The structures and absolute configurations of two novel THF lignans (1a, 1b), isolated from leaves of the species P. solmsianum, and of two known lignans (2b, 3a),10,15,35 were determined. In addition, the theoretical electronic calculations provided a new data set for the structures of the THF lignans in gas phase showing that the configuration of the tetrahydrofuran ring and intramolecular hydrogen bonding are related to the geometrical equilibrium energy profile of the conformers. The calculated energies in the gas phase and in solvents (water and cyclohexane) indicated that lignan 2a is the most stable among the isomeric lignans 1a–3a. Moreover, the calculated data for 1b and 2b could explain the theoretical equilibrium geometries of these lignans since the configuration of the THF ring and intramolecular hydrogen bonding originated from the methoxy groups in the phenyl ring, compared to the methylenedioxy moiety, contributed to the stabilization of the THF lignan structures.Experimental
Plant material
Leaves of Piper solmsianum C. DC were collected at the Instituto de Biociências (IB), Universidade de São Paulo (SP, Brazil) and at the Horto Florestal (HF), Instituto Florestal do Estado de Sao Paulo (SP, Brazil) in January 2003 (Permit ICMBio-15780-1). Plants were identified by Dr Elsie Franklin Guimarães (Instituto de Pesquisas Jardim Botânico do Rio de Janeiro, RJ, Brazil), and voucher specimens were deposited at the herbarium of the Instituto de Pesquisas Jardim Botânico do Rio de Janeiro (IB specimen – Kato-0367; HF specimen – Kato-0175).Extraction and isolation of lignan 1a
Dried leaves (3.2 g) of P. solmsianum (IB specimens) were milled and the powder extracted with CH2Cl2 for 48 h. The extract was concentrated under vacuum to yield 300 mg of a residue that was submitted to CC over C18 column (7 g, 15 × 2.5 cm i.d.). Elution with mixtures of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH containing increasing amounts of MeOH (from 50 to 100%) gave 10 fractions. Fractions 3 and 4 (eluted with 70% MeOH) were pooled, extracted with CH2Cl2 (3 × 100 mL), the organic fractions dried over anhydrous Na2SO4 and concentrated under vacuum to yield 1a (6 mg). Data for compound 1a: pale white powder; [α]21D 0 (c 1 × 10−4 mol L−1, CH2Cl2); UV λmax 225 nm (c 3.0 × 10−2 mol L−1 MeOH); CD θ220 nm + 6.6 (c 2.5 × 10−3 mol L−1 MeOH); IR νfilmmax cm−1: 2965, 2930, 2837, 1593, 1127, 1095, 843; 1H and 13C NMR: see Table 1; EI/MS m/z (rel. int): 432 [M]+ (31), 236 (100), 221 (70), 205 (83); HR-ESIMS [M + H]+ calcd for C24H33O7 433.2221, found 433.2232. Anal. calcd for C24H32O7: C, 66.66; H, 7.40; O, 25.92. Found C, 66.57; H, 7.25; O 25.88.
:MeOH containing increasing amounts of MeOH (from 50 to 100%) gave 10 fractions. Fractions 3 and 4 (eluted with 70% MeOH) were pooled, extracted with CH2Cl2 (3 × 100 mL), the organic fractions dried over anhydrous Na2SO4 and concentrated under vacuum to yield 1a (6 mg). Data for compound 1a: pale white powder; [α]21D 0 (c 1 × 10−4 mol L−1, CH2Cl2); UV λmax 225 nm (c 3.0 × 10−2 mol L−1 MeOH); CD θ220 nm + 6.6 (c 2.5 × 10−3 mol L−1 MeOH); IR νfilmmax cm−1: 2965, 2930, 2837, 1593, 1127, 1095, 843; 1H and 13C NMR: see Table 1; EI/MS m/z (rel. int): 432 [M]+ (31), 236 (100), 221 (70), 205 (83); HR-ESIMS [M + H]+ calcd for C24H33O7 433.2221, found 433.2232. Anal. calcd for C24H32O7: C, 66.66; H, 7.40; O, 25.92. Found C, 66.57; H, 7.25; O 25.88.
Extraction and isolation of lignans 1b, 2b and 3a
Dried leaves (6 g) of P. solmsianum (HF specimens) were milled and the powder extracted with methanol for 48 h. The extract was concentrated under vacuum to yield 400 mg of a residue that was suspended in 150 mL of MeOH:H2O (4:1), filtered over a bed of Celite, and extracted with CH2Cl2 (3 × 100 mL). The CH2Cl2 fraction was dried over anhydrous Na2SO4, concentrated under vacuum to yield 70 mg of residue that was submitted to preparative TLC developed twice with hexanes:EtOAc (85:15) to afford 1b (10 mg), 2b (3 mg) and 3a (5 mg). Data for compound 1b: pale yellow oil; [α]21D + 80.7 (c 1 × 10−4 mol L−1, CH2Cl2); UV λmax 225 nm (c 3.0 × 10−2 mol L−1 MeOH); CD θ220 nm + 6.6 (c 2.5 × 10−3 mol L−1 MeOH); IR νfilmmax cm−1: 2965, 2935, 1634, 1507, 1233, 1228, 1095, 843; 1H and 13C NMR: see Table 1; EI/MS m/z (rel. int): 416 [M]+ (14), 220 (100), 205 (85), 236 (43), 208 (21); HR-ESIMS [M + H]+ calcd for C23H28O7 417.1907, found 417.1915. Data for compound 2b: pale white powder; [α]21D − 62.2 (c 1 × 10−4 mol L−1, CH2Cl2); UV λmax 240 nm (c 5.0 × 10−2 mol L−1 MeOH); CD θ233 nm − 10.5 (c 3.0 × 10−3 mol L−1 MeOH); IR νfilmmax cm−1: 2965, 2935, 1634, 1507, 1233, 1228, 1095, 843; 1H and 13C NMR;36 EI/MS m/z (rel. int): 432 [M]+ (12), 205 (100), 2221 (85), 236 (90), 208 (30); HR-ESIMS [M + H]+ calcd for C24H33O7 433.2221 found 433.2234. Data for compound 3a: pale yellow oil; [α]21D − 71.4 (c 1 × 10−4 mol L−1, CH2Cl2); 1H and 13C NMR.15
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by grants provided by FAPESP (09/51850-9, 14/50316-7), CNPq, and PRP-USP (Pró-Reitoria de Pesquisas da USP). MJK and HVL are grateful to CNPq for research fellowships, and CSR acknowledges FAPESP for a scholarship. The authors are indebted to the Laboratório de Computação Cientifica Avançada (LCCA), Universidade de São Paulo (SP, Brazil) for computer facilities.References
- R. S. Ward, Nat. Prod. Rep., 1999, 16, 75–96 RSC.
- J. Y. Pan, S. L. Chen, M. H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC.
- P. K. Agrawal and R. S. Thakur, Magn. Reson. Chem., 1985, 23, 389–418 CrossRef CAS.
- D. A. Whiting, Nat. Prod. Rep., 1985, 2, 191–211 RSC.
- L. B. Davin, M. Jourdes, A. M. Patten, K. W. Kim, D. G. Vassão and N. G. Lewis, Nat. Prod. Rep., 2008, 25, 1015–1090 RSC.
- T. Biftu and R. Stevenson, Phytother. Res., 1987, 1, 97–106 CrossRef CAS.
- D. Holloway and F. Scheinmann, Phytochemistry, 1974, 13, 1233–1236 CrossRef CAS.
- J. M. Barbosa, M. S. da Silva, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1989, 28, 2209–2211 CrossRef.
- J. M. Saad, E. Soepadamo, X. P. Fang, J. L. McLaughlin and P. E. Fanwick, J. Nat. Prod., 1991, 54, 1681–1683 CrossRef CAS.
- H. J. Zhang, P. A. Tamez, V. D. Hoang, G. T. Tan, N. Van Hung, L. T. Xuan, L. M. Huong, N. M. Cuong, D. T. Thao, D. D. Soejarto, H. H. S. Fong and J. M. Pezzuto, J. Nat. Prod., 2001, 64, 772–777 CrossRef CAS.
- C. S. Ramos, S. A. Vanin and M. J. Kato, Phytochemistry, 2008, 69, 2157–2161 CrossRef CAS PubMed.
- N. P. Lopes, M. J. Kato and M. Yoshida, Phytochemistry, 1999, 51, 29–33 CrossRef CAS.
- M. Kuroyanagi, K. Yoshida, A. Yamamoto and M. Miwa, Chem. Pharm. Bull., 2000, 48, 832–837 CrossRef CAS PubMed.
- H. K. Fun, K. Sivakumar, B. C. Yip, A. H. Othman and I. M. Said, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 414–416 CrossRef.
- H. Greger, T. Pacher, S. Vajrodaya, M. Bacher and O. Hofer, J. Nat. Prod., 2000, 63, 616–620 CrossRef CAS.
- Y. Qin, C. L. Yin and Z. H. Cheng, Molecules, 2013, 18, 13636–13644 CrossRef CAS PubMed.
- T. Biftu, J. C. Chabala, J. Acton, T. Beattie, D. Brooker, R. Bugianesi, M. N. Chang, P. C. Chiang, N. Gamble, N. Girotra, D. Graham, C. H. Kuo, M. Ponpipom, S. Sahoo, T. Y. Shen, K. L. Thompson, S. S. Yang, S. B. Hwang and T. Doebber, Prostaglandins, 1988, 35, 846 CrossRef.
- R. C. C. Martins, L. R. Latorre, P. Sartorelli and M. J. Kato, Phytochemistry, 2000, 55, 843–846 CrossRef CAS PubMed.
- L. D. Ferreira, D. R. Callejon, A. Engemann, B. Cramer, H. U. Humpf, V. P. de Barros, M. D. Assis, D. B. da Silva, S. de Albuquerque, L. T. Okano, M. J. Kato and N. P. Lopes, Planta Med., 2012, 78, 1939–1941 CrossRef CAS PubMed.
- L. G. Felippe, D. C. Baldoqui, M. J. Kato, V. D. Bolzani, E. F. Guimarães, R. M. B. Cicarelli and M. Furlan, Phytochemistry, 2008, 69, 445–450 CrossRef CAS PubMed.
- T. J. Schmidt, Phytochem. Lett., 2012, 5, 632 CrossRef CAS.
- M. M. O. Cabral, J. A. Alencar, A. E. Guimarães and M. J. Kato, J. Am. Mosq. Control Assoc., 2009, 25, 103–105 CrossRef CAS PubMed.
- A. Leite, M. J. Kato, R. O. A. Soares, A. E. Guimarães, J. R. Santos-Mallet and M. M. O. Cabral, Rev. Bras. Farmacogn., 2012, 22, 517–521 CrossRef CAS.
- J. J. Chen, E. T. Chou, C. Y. Duh, S. Z. Yang and I. S. Chen, Planta Med., 2006, 72, 351–357 CAS.
- H.-W. Liu, X.-Z. Yu, D. Padula, G. Pescitelli, Z.-W. Lin, F. Wang, K. Ding, M. Lei and J.-M. Gao, Eur. J. Med. Chem., 2013, 59, 265–273 CrossRef CAS PubMed.
- J. L. Wu, N. Li, T. Hasegawa, J. Sakai, S. Kakuta, W. X. Tang, S. Oka, M. Kiuchi, H. Ogura, T. Kataoka, A. Tomida, T. Tsuruo and M. Ando, J. Nat. Prod., 2005, 68, 1656–1660 CrossRef CAS PubMed.
- M.-M. Bai, W. Shi, J.-M. Tian, M. Lei, Y. H. Kim, J. H. Kim, Y. N. Sun and J.-M. Gao, J. Agric. Food Chem., 2015, 63, 2198–2205 CrossRef CAS PubMed.
- H. S. Kim, C. M. Wooten, Y. Park and J. Y. Hong, Org. Lett., 2007, 9, 3965–3968 CrossRef CAS PubMed.
- K. Nihei, K. Konno, L. S. C. Bernardes, N. P. Lopes, S. Albuquerque, I. de Carvalho, M. T. Pupo, R. C. C. Martins and M. J. Kato, ARKIVOC, 2004, 112–126 CAS.
- L. S. C. Bernardes, M. J. Kato, S. Albuquerque and I. Carvalho, Bioorg. Med. Chem., 2006, 14, 7075–7082 CrossRef CAS PubMed.
- H. Nishiwaki, K. Nakayama, Y. Shuto and S. Yamauchi, J. Agric. Food Chem., 2014, 62, 651–659 CrossRef CAS PubMed.
- A. M. Rimando, J. M. Pezzuto, N. R. Farnsworth, T. Santisuk, V. Reutrakul and K. Kawanishi, J. Nat. Prod., 1994, 57, 896–904 CrossRef CAS.
- T. J. Schmidt and J. Heilmann, Planta Med., 2000, 66, 749–751 CrossRef CAS PubMed.
- J. G. Blears and R. D. Haworth, J. Chem. Soc., 1958, 1985–1987 RSC.
- R. C. C. Martins, J. H. G. Lago, S. Albuquerque and M. J. Kato, Phytochemistry, 2003, 64, 667–670 CrossRef CAS PubMed.
- S. F. Fonseca, L. E. S. Barata, E. A. Ruveda and P. M. Baker, Can. J. Chem., 1979, 57, 441–443 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, 03-Revision B.05, Gaussian Inc., Pittsburgh, 2003 Search PubMed.
- H. Matsuura, H. Yoshida, M. Hieda, S. Yamanaka, T. Harada, K. Shin-ya and K. Ohno, J. Am. Chem. Soc., 2003, 125, 13910–13911 CrossRef CAS PubMed.
- P. Gilli, V. Bertolasi, L. Pretto, L. Antonov and G. Gilli, J. Am. Chem. Soc., 2005, 127, 4943–4953 CrossRef CAS PubMed.
- S. Hanessian, G. J. Reddy and N. Chahal, Org. Lett., 2006, 8, 5477–5480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 1a and 1b; Fig. S1–S11, Tables S1–S7. See DOI: 10.1039/c7ra09262h |
| This journal is © The Royal Society of Chemistry 2017 |