
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ent-Rosane diterpenoids from Euphorbia milii showing an Epstein–Barr virus lytic replication assay†
Shao-Nan Liu‡
a,
Jiayuan Hu‡b,
Shen H. Tana,
Qian Wangb,
Jun Xu a,
Yan Wangc,
Yan Yuan*bd and
Qiong Gu*a
a,
Yan Wangc,
Yan Yuan*bd and
Qiong Gu*a
aResearch Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, People's Republic of China. E-mail: guqiong@mail.sysu.edu.cn; Fax: +86-20-39943077; Tel: +86-20-39943077
bThe Institute of Human Virology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong 510080, People's Republic of China. E-mail: yuan2@pobox.upenn.edu
cGuanghua School of Stomatology, Sun Yat-sen University, Guangdong Provincial Key Laboratory of Stomatology, Guangzhou 510080, People's Republic of China
dDepartment of Microbiology, School of Dental Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
First published on 4th October 2017
Abstract
The phytochemical investigation on the acetone extract of Euphorbia milii afforded thirteen new ent-rosane diterpenoids (1–13) through bioassay guided fractionation for evaluating its effect on Epstein–Barr virus (EBV) DNA lytic replication. Structures were determined by comprehensive spectroscopic analyses including 1D & 2D NMR techniques, chemical methods, and experimental and calculated electronic circular dichroism (ECD) data. The absolute configuration of euphominoid A (1) was established by single crystal X-ray diffraction analysis of its p-bromobenzoate derivative 1a. Compounds 1–3, and 10 displayed inhibitory activity with EC50 values ranging from 5.4 to 29.1 μM and selective index (SI) values varied from 4.5 to 9.3. Compound 2 showed the most potent inhibitory activity with an EC50 value of 5.4 μM comparing with the positive control (+)-rutamarin (EC50 = 5.4 μM). This is the first report of ent-rosane-type diterpenoids exhibiting significant inhibition of EBV lytic replication.
Introduction
Euphorbia milii Linn. (Euphorbiaceae) is a flowering plant mainly distributed in the southwestern region of China. The Euphorbia genus have provided approximately 400 characteristic diterpenoids possessing a broad range of biological activities.1–4 Several secondary metabolites isolated from the Euphorbia genus exhibited anti-viral activity against HIV, HSV, and Epstein–Barr virus (EBV).5–14 This plant has been extensively used as a detoxifying agent in traditional Chinese medical (TCM) treatments.15We have recently reported novel secondary metabolites from the E. milii,16 during a continuing program towards the discovery of anti-viral natural products.9,17,18 Herein, we report the detailed chemical investigation of the 80% acetone extract of the aerial parts of E. milii. This extract displayed inhibitory activity against EBV lytic replication with an EC50 value of 12.4 μg mL−1. Bioactivity-guided isolation led to the isolation and identification of thirteen new ent-rosane diterpenoids (1–13), some of which exhibited promising EBV lytic replication inhibitory activity.
Results and discussion
Compound 1, obtained as a white powder. Its HRESIMS spectrum showed the sodiated adduct ion at m/z 325.2132 [M + Na]+ corresponding to the molecular formula of C20H30O2. The IR signals at 3408 and 1695 cm−1 revealed the presence of a hydroxy group and a carbonyl group. The 1H NMR data of compound 1 (Table 1) exhibited characteristic signals of a vinyl group [δH 5.79 (1H, dd, J = 17.5, 10.7 Hz), 4.90 (1H, d, J = 17.5 Hz), and 4.82 (1H, d, J = 10.7 Hz)], a hydroxymethyl group [δH 3.59 (1H, d, J = 11.0 Hz), 3.65 (1H, d, J = 11.0 Hz)], and three methyl singlets (δH 0.91, 1.00, and 1.04). The 13C NMR and DEPT spectra of compound 1 showed 20 carbon resonances, comprising those of a carbonyl group at δC 216.5, two pairs of olefinic carbons (δC 129.7 and 141.3, 108.7 and 150.8), and three methyls (δC 16.9, 18.8, and 22.9). These data implied that the 2D structure of compound 1 is similar to that of engleromycenol,19 except for a carbonyl group in compound 1 in place of a methylene group at C-3 in engleromycenol. The HMBC cross-peaks of H3-18 with C-3, C-4, and C-5 (Fig. 1) confirmed a carbonyl group positioned at C-3. Thus, the 2D structure of compound 1 was established as shown.| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.43, m | 2.24, m | 1.92, m | 2.10, m | 2.35, m | 4.50, t (3.0) | |||
| 2.32, m | 1.99, m | ||||||||
| 2 | 2.45, m | 1.88, m | 1.75, m | 1.84, m | 2.69, dd (17.6, 5.5) | 1.81, m | 2.79, dd (17.8, 5.2) | 2.96, dd (17.3, 4.7) | 1.94, m |
| 1.71, m | 1.60, m | 2.42, dd (17.6, 11.3) | 1.64, m | 2.52, dd (17.8,11.0) | 2.46, dd (17.3, 8.2) | 1.76, m | |||
| 3 | 3.81, d (8.7) | 3.89, d (12.5) | 3.66, t (7.3) | 3.81, dd (11.3, 5.5) | 3.41, dd (11.6, 3.2) | 3.83, dd (11.0, 5.2) | 3.88, dd (8.2, 4.7) | 3.89, dd (3.2, 12.9) | |
| 6 | 2.06, m | 2.10, m | 2.07, m | 2.09, m | 2.35, m | 4.38, dd (4.2, 1.6) | |||
| 1.95, m | 1.92, m | 1.99, m | |||||||
| 7 | 1.42, m | 1.36, m | 1.38, m | 1.33, m | 1.36, m | 2.17, m | 2.24, m | 1.68, m | 2.23, m |
| 1.33, m | 1.32, m | 1.39, m | |||||||
| 8 | 1.54, m | 1.57, m | 1.53, m | 1.57, m | 1.47, m | 2.08, m | 2.07, m | 1.91, m | 2.19, m |
| 11 | 1.61, m | 1.62, m | 1.58, m | 1.57, m | 2.53, m | 1.69, m | 2.47, m | 2.50, m | 1.72, m |
| 1.30, m | 1.37, m | 1.30, m | 1.28, m | 1.12, m | 1.51, m | 1.25, m | 2.00, m | ||
| 12 | 1.54, m | 1.53, m | 1.53, m | 1.57, m | 1.61, m | 1.57, m | 1.64, m | 1.64, m | 1.40, m |
| 1.25, m | 1.29, m | 1.27, m | 1.28, m | 1.23, m | 1.35, m | 1.31, m | 1.28, m | 1.61, m | |
| 14 | 1.38, m | 1.36, m | 1.40, m | 1.39, m | 1.47, m | 1.40, m | 1.45, t (13.2) | 1.53, m | 1.18, m |
| 1.08, m | 1.06, m | 1.06, m | 1.06, m | 1.09, m | 1.13, m | 1.05, m | 1.10, m | 1.37, m | |
| 15 | 5.79, dd (17.5, 10.7) | 5.80, dd (17.5, 10.7) | 5.81, dd (17.5, 10.7) | 5.81, dd (17.5, 10.7) | 5.82, dd (17.5, 10.7) | 5.80, dd (17.5, 10.7) | 5.80, dd (17.5, 10.7) | 5.83, dd (17.5, 10.7) | 5.81, dd (17.5, 10.7) |
| 16 | 4.90, d (17.5) | 4.91, d (17.5) | 4.91, d (17.5) | 4.91, d (17.5) | 4.93, d (17.5) | 4.93, d (17.5, 1.3) | 4.90, d (17.5) | 4.94, d (17.5, 1.3) | 4.95, d (17.5, 1.2) |
| 4.82, d (10.7) | 4.83, d (10.7) | 4.84, d (10.7) | 4.84, d (10.7) | 4.85, d (10.7) | 4.87, d (10.7, 1.3) | 4.82, d (10.7) | 4.86, d (10.7, 1.3) | 4.90, d (10.7, 1.2) | |
| 17 | 1.00, s | 1.01, s | 1.01, s | 1.02, s | 1.02, s | 1.04, s | 1.37, s | 1.06, s | 1.06, s |
| 18 | 1.04, s | 1.02, s | 3.76, d (10.4) | 1.16, s | 1.21, s | 1.21, s | 1.29, s | 1.41, s | 1.15, s |
| 3.59, d (10.4) | |||||||||
| 19 | 3.59, d (11.0) | 3.56, d (10.9) | 1.01, s | 3.79, d (11.3) | 1.07, s | 1.29, s | 1.04, s | 1.19, s | 1.35, s |
| 3.65, d (11.0) | 3.51, d (10.9) | 3.53, d (11.3) | |||||||
| 20 | 0.91, s | 0.87, s | 0.85, s | 0.86, s | 1.07, s | 0.99, s | 1.19, s | 1.04, s | 0.99, s |
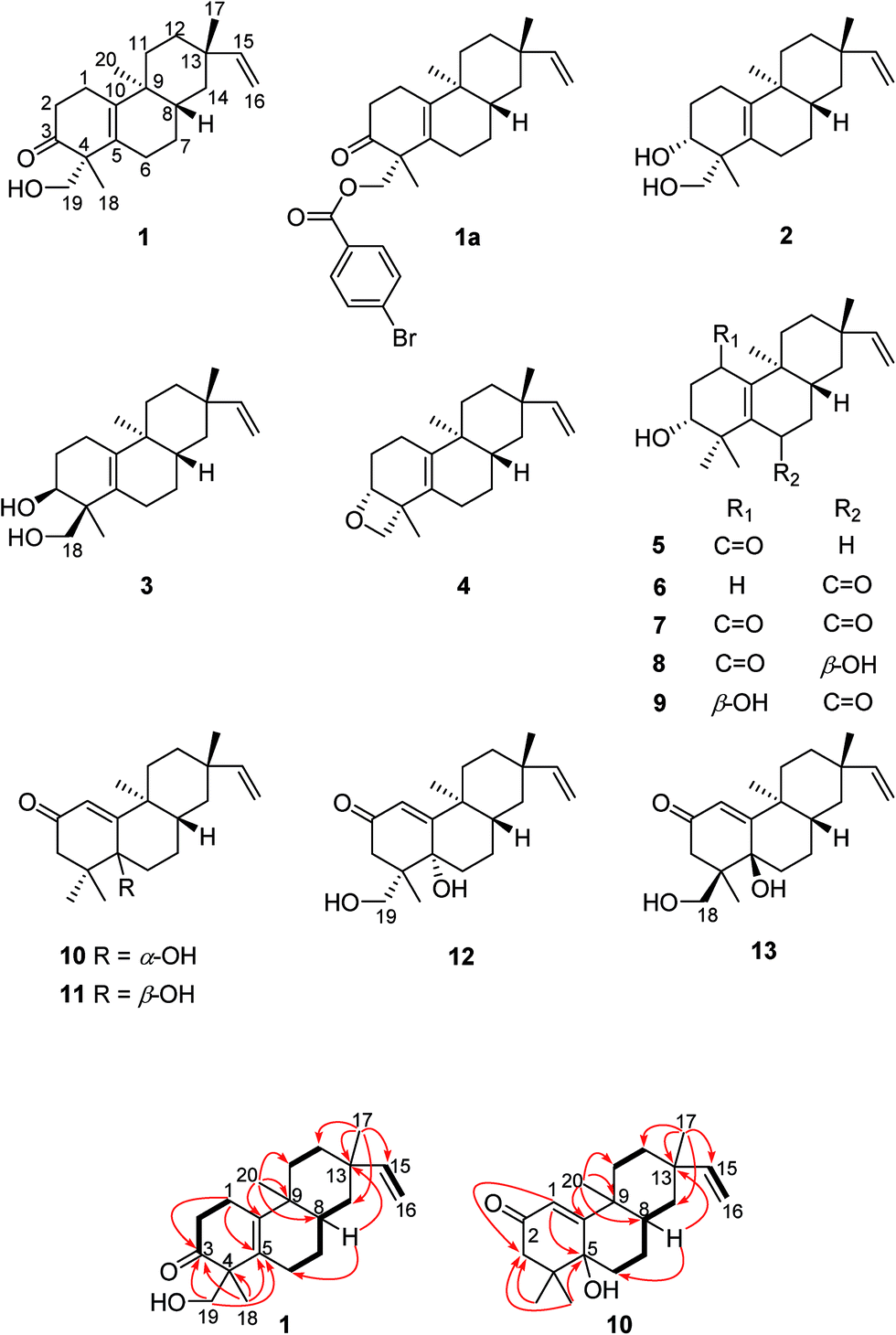 | ||
Fig. 1 Key 1H–1H COSY ( ) and HMBC ( ) and HMBC ( ) cross-peaks of compounds 1 and 10. ) cross-peaks of compounds 1 and 10. | ||
The relative configuration of compound 1 was deduced from the NOESY spectrum (Fig. S8, ESI†). The NOESY cross-peaks of H-8/H3-17, H-12a/H-15, and H-12a/H3-20 indicated an α-orientation of CH3-20 and β-orientations for H-8 and CH3-17. The absolute configuration of compound 1 was determined as 4R, 8S, 9S, and 13S by single crystal X-ray crystallographic diffraction analysis of its p-bromobenzoate derivative 1a with Cu Kα radiation (Fig. 2). Thus, the absolute configuration of compound 1 was defined as 4R, 8S, 9S, and 13S, and given the trivial name euphominoid A.
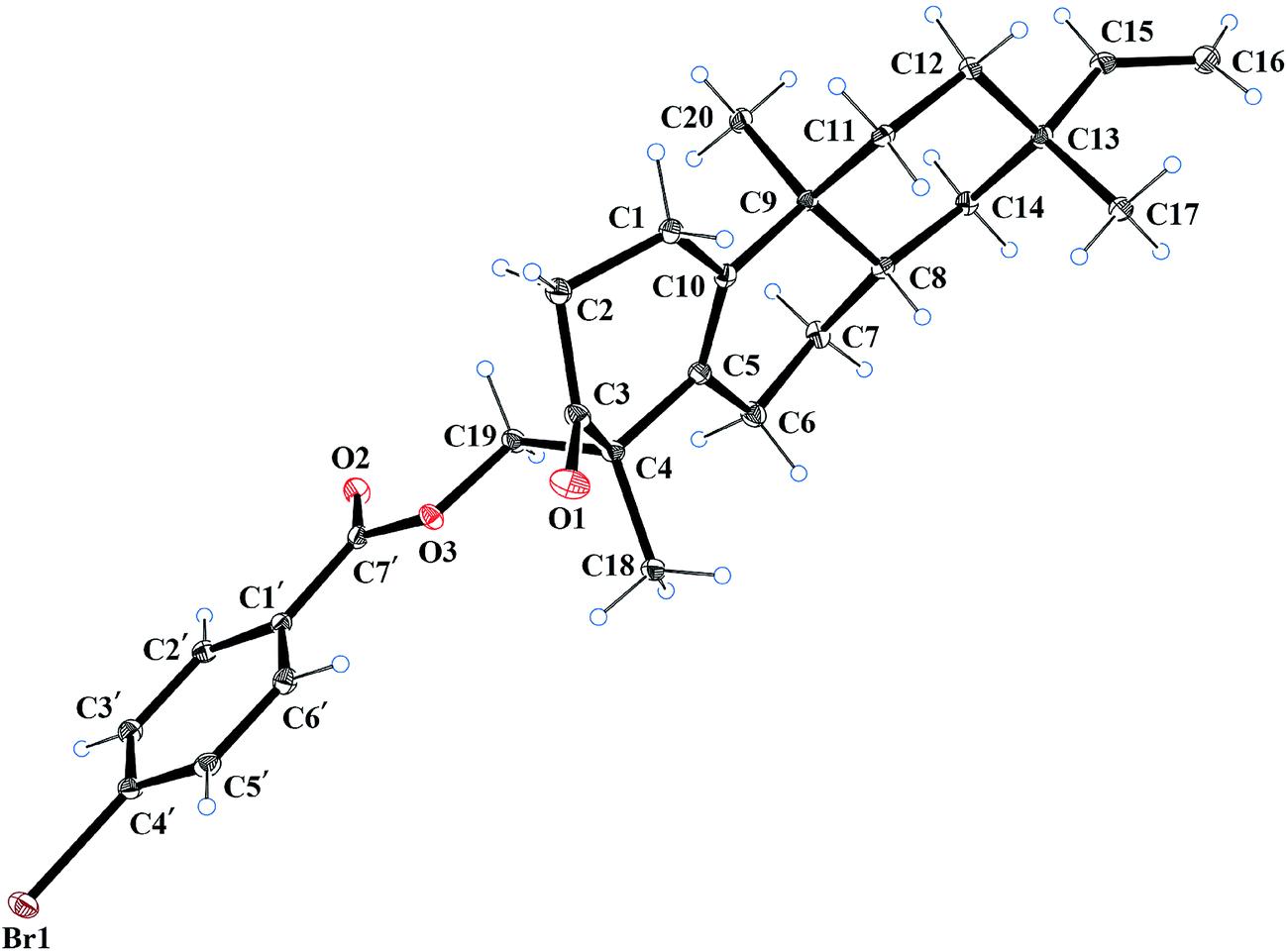 | ||
| Fig. 2 X-ray ORTEP plot of the molecular structure of compound 1a. | ||
Compounds 2 (euphominoid B) and 3 (euphominoid C) were isolated using a chiral RP-HPLC column and had the same molecular formula of C20H32O2. As shown in Table 1, the carbonyl signal at δC 216.5 in compound 1 was absent, while an oxygenated methine signal was observed at δC 72.4 for compound 2 and δC 73.2 for compound 3. These observations suggested the presence of a hydroxy group at C-3 in both compounds 2 and 3. For compound 2, the HMBC cross-peaks between H3-18 (δH 1.02, s)/H-19 (δH 3.56, d, J = 10.9 Hz; 3.51, d, J = 10.9 Hz) and the oxygenated methine carbon at δC 72.4 indicated that the hydroxy group was positioned at C-3. The corresponding cross-peaks observed in the HMBC spectrum obtained for compound 3 suggested similar results (Fig. S23, ESI†). By analysis of the NOESY spectra of compounds 2 and 3 (Fig. S16 and S24, ESI†), the cross-peaks of H-3/H3-18 in compound 2 and the cross-peaks of H-3/H3-19 in compound 3 revealed a cis-1,3-diol unit in 2 and 3. Thus, the relative configuration of compounds 2 and 3 were established as shown.
The molecular formula of compound 4 (euphominoid D) was identified as C20H30O from a protonated molecule at m/z 287.2355 [M + H]+ in the HRESIMS data, which indicated the loss of a water molecule from compounds 2 or 3. The NMR data of compound 4 were similar to those of compound 2 (Tables 1 and 2), except for a significant difference in the chemical shift of C-3 (δC 77.6 in compound 4 vs. 72.4 in compound 2). The downfield C-3 in 4 indicated the formation of an oxygen bridge between C-3 and C-19.20,21 The cross-peaks of H-3/H3-18 in the NOESY spectrum of compound 4 (Fig. S32, ESI†) indicated the same orientations of H-3 and CH3-18. Thus, the relative configuration of compound 4 was established as shown.
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 23.4 | 21.5 | 25.4 | 23.6 | 196.5 | 25.7 | 198.5 | 198.1 | 64.7 |
| 2 | 38.6 | 26.6 | 27.4 | 28.0 | 44.2 | 26.3 | 43.8 | 44.7 | 36.5 |
| 3 | 216.5 | 72.4 | 73.2 | 77.6 | 72.7 | 76.2 | 73.3 | 75.1 | 71.3 |
| 4 | 53.0 | 44.1 | 44.3 | 43.1 | 41.8 | 39.1 | 39.6 | 44.2 | 39.4 |
| 5 | 129.7 | 127.8 | 129.0 | 128.2 | 162.2 | 138.0 | 151.5 | 157.2 | 140.4 |
| 6 | 24.7 | 24.8 | 25.7 | 25.7 | 28.2 | 198.0 | 200.0 | 65.3 | 199.0 |
| 7 | 25.2 | 25.8 | 24.6 | 25.8 | 25.0 | 42.6 | 42.6 | 35.2 | 42.9 |
| 8 | 37.2 | 37.1 | 37.7 | 37.5 | 38.4 | 36.2 | 36.5 | 32.6 | 35.9 |
| 9 | 36.3 | 37.6 | 37.7 | 37.7 | 37.2 | 39.1 | 38.0 | 38.2 | 39.0 |
| 10 | 141.3 | 140.9 | 140.0 | 140.2 | 142.3 | 168.0 | 155.3 | 143.7 | 160.8 |
| 11 | 31.5 | 32.3 | 31.8 | 32.0 | 31.4 | 31.2 | 30.9 | 31.0 | 30.9 |
| 12 | 32.3 | 32.8 | 32.7 | 32.8 | 32.8 | 32.3 | 32.3 | 32.8 | 32.3 |
| 13 | 37.6 | 36.5 | 36.5 | 36.6 | 36.4 | 36.0 | 35.9 | 36.4 | 36.1 |
| 14 | 39.2 | 39.8 | 39.6 | 39.7 | 39.0 | 38.5 | 37.9 | 38.7 | 38.9 |
| 15 | 150.8 | 151.3 | 151.3 | 151.3 | 151.3 | 150.3 | 150.3 | 151.0 | 150.3 |
| 16 | 108.7 | 108.9 | 108.8 | 108.9 | 108.9 | 109.7 | 109.0 | 109.1 | 109.6 |
| 17 | 22.9 | 23.1 | 23.2 | 23.2 | 23.4 | 22.8 | 22.4 | 23.5 | 22.9 |
| 18 | 18.8 | 17.2 | 68.9 | 20.0 | 23.9 | 19.3 | 24.4 | 26.4 | 17.7 |
| 19 | 67.3 | 69.2 | 15.7 | 67.3 | 19.7 | 25.1 | 19.0 | 22.4 | 24.8 |
| 20 | 16.9 | 18.2 | 17.1 | 17.8 | 16.7 | 15.8 | 15.2 | 15.8 | 17.3 |
Biogenetically, compounds 2, 3, and 4 should possess the same absolute configurations as compound 1 at C-8, C-9, and C-13. Owing to the presence of the Δ5,10 double bond, no NOESY cross-peaks of H-3, H3-18, or H-19 with H-8, H3-17, or H3-20 were observed for compounds 2, 3, and 4. The absolute configurations of C-3 and C-4 in compounds 2, 3, and 4 could not be determined by comparison with those of compound 1. Thus chemical transformations (Fig. 3) were used to establish the absolute configurations at C-3 and C-4 for compounds 2 and 4. Compound 1 was reacted with NaBH4 in MeOH to yield compounds 2 and 4 in yields of 58 and 8%. Thus, the 3R absolute configurations in compounds 2 and 4 were defined through a combination of the 4R absolute configuration in compound 1 and the NOESY cross-peaks observed in compounds 2 and 4. The absolute configurations of compounds 2 and 4 were assigned as 3R, 4R, 8S, 9S, and 13S.
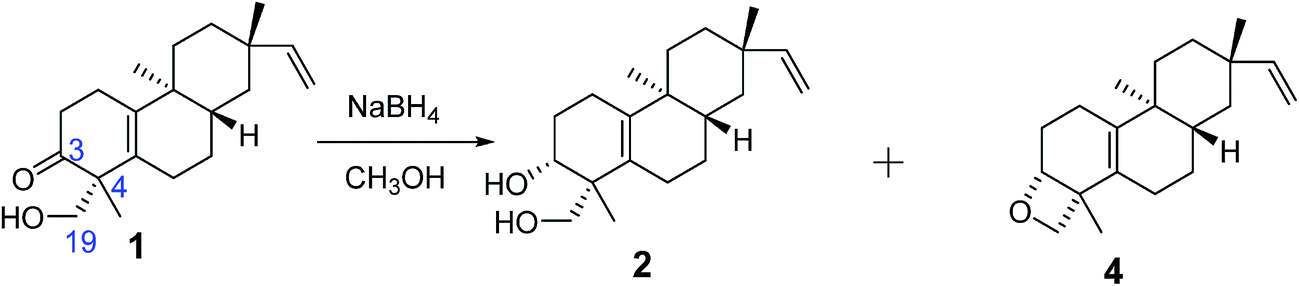 | ||
| Fig. 3 Chemical transformation of compounds 1, 2, and 4. | ||
Induced electronic circular dichroism spectrum (IECD) by [Mo2(OAc)4] was used to define the absolute configuration of the cis-1,3-diol unit.22,23 For the “semi-rigid” 1,3-diol moiety, only the syn-parallel orientation of the hydroxy groups allows for the formation a chiral complex with Mo2(OAc)4 leading to the Cotton effect (CE). The observed signal of the CE at 400 nm in the complex depends on the chirality of the 1,3-diol moiety. The strong positive CE band observed around 350 nm and a relatively weak negative CE band at 400 nm (Fig. 4) in the IECD spectra of compound 3 indicated that 3-OH and 18-CH2OH are co-facial. On the basis of the empirical sector rule for 1,3-diols, the negative CE at 400 nm observed in the IECD spectra of compound 3 and the sector rule (Fig. 4) indicated a 3S and 4S absolute configurations for compound 3. According to compound 1, the absolute configuration of compound 3 was assigned as 3S, 4S, 8S, 9S, and 13S.
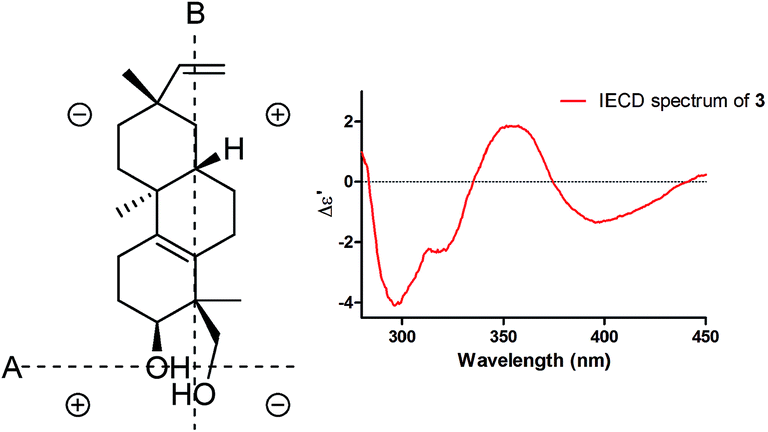 | ||
| Fig. 4 Sector rule (left) and IECD (right) of compound 3. | ||
Compound 5 (euphominoid E), was obtained as a colorless gum with a molecular formula of C20H30O3 based on the HRESIMS ion at m/z 301.2167 [M − H]−. A UV absorption maximum at 246 nm and IR signals at 1636 and 1579 cm−1, in combination with the observed 13C NMR chemical shifts of δC 196.5, 162.2, and 142.3 suggested the presence of an α,β-unsaturated carbonyl group. Detailed analysis of the 1D and 2D NMR data of compound 5 (Fig. S35–S40, ESI†) revealed an ent-rosane diterpenoid similar to compound 1. The HMBC cross-peaks from H3-18 (δH 1.21)/H3-19 (δH 1.07) to C-3 (δC 72.7), C-4 (δC 41.8), and C-5 (δC 162.2), and H3-20 (δH 1.07) to C-10 (δC 142.3) suggested the presence of a Δ5,10 double bond and a hydroxy group at C-3 (Fig. 5). A carbonyl group at C-1 was deduced from the HMBC cross-peak between H-3 and C-1 (Fig. 5). The absolute configuration of C-3 was determined using the Mosher's ester method.17 Treatment of compound 5 with (R)- or (S)-MTPA chloride in anhydrous dichloromethane, 4-dimethylaminopyridine, and triethylamine yielded the (S)- and (R)-MTPA ester derivatives, respectively. The 1H NMR chemical shift differences (ΔδS–R) between the 3R and 3S esters are shown in Fig. 5. The negative ΔδS–R values for H-6 and H3-18, and the positive values for H-2a, H-2b, and H-11a indicated the R configuration at C-3. In line with compound 1, the absolute configuration of compound 5 was determined as 3R, 8S, 9S, and 13S.
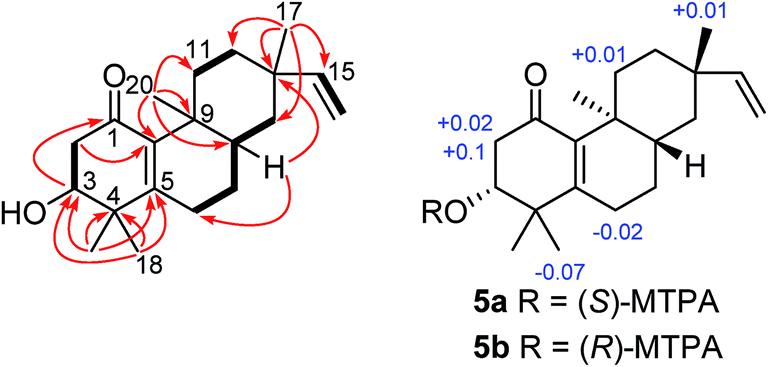 | ||
| Fig. 5 ΔδS–R values of MTPA esters of compound 5. | ||
The molecular formula of compound 6 (euphominoid F) was established as C20H30O2 based on the HRESIMS ion at m/z 303.2312 [M + H]+. The NMR data for this compound were similar to those of compound 5 (Tables 1 and 2), with the exception of the location of the carbonyl carbon. The carbonyl group at C-6 in compound 6 was deduced from the COSY cross-peaks of H-1 (δH 2.35, m)/H-2 (δH 1.81, m; 1.64, m) and H-2 (δH 1.81, m; 1.64, m)/H-3 (δH 3.41, dd, J = 11.6, 3.2 Hz), as well as an HMBC cross-peak between H-8 (δH 2.08, m) and C-6 (δC 198.0). The 2D structure of compound 6 was thus assigned as shown. Owing to the limited quantity obtained for compound 6, the computed ECD method was employed to determine its absolute configuration. The ECD spectra were calculated using the Gaussian 09 program at the TD-DFT-PBE1PBE/6-31++G(2d,2p) level in MeOH. This calculation for the 3R configuration was in good agreement with the experimental ECD data (Fig. 6). Thus, the absolute configuration of compound 6 was assigned as 3R, 8R, 9S, and 13S.
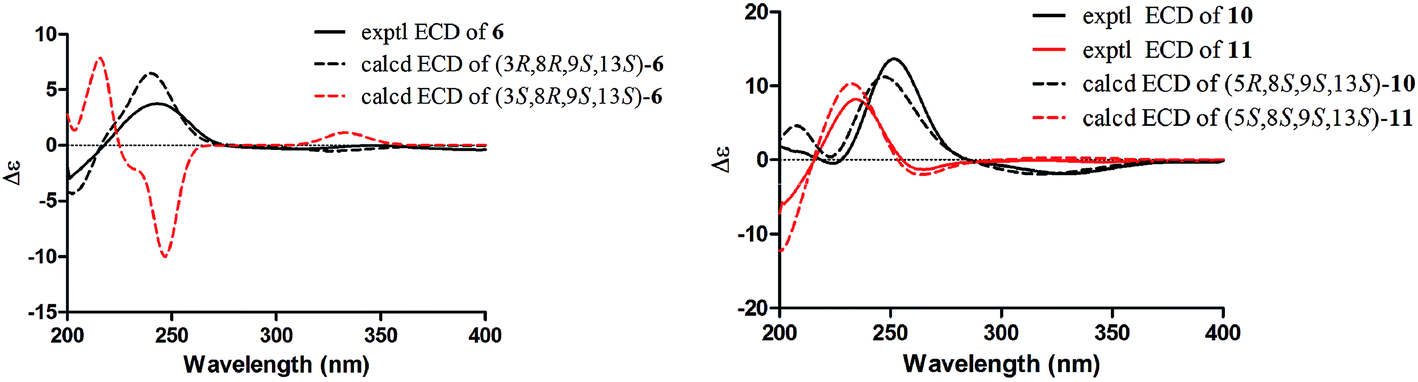 | ||
| Fig. 6 Calculated and experimental ECD spectra of compounds 6, 10 and 11. The calculated ECD spectra were computed at the PBE1PBE/6-31++G(2d,2p) level in MeOH. | ||
Compound 7 (euphominoid G) was found to have a molecular formula of C20H28O3 based on the HRESIMS (m/z 315.1954 [M − H]−). The structure of 7 was similar to compound 6, except for an additional carbonyl group at C-1, which was supported by the HMBC cross-peaks between H-3 (δH 3.83, dd, J = 11.0, 5.2 Hz) and C-1 (δC 198.5). Detailed analysis of the 2D NMR data (Fig. S53–S56, ESI†) confirmed the 2D structure of compound 7, and its absolute configuration was defined as 3R, 8R, 9S, and 13S through comparison of the calculated and experimental ECD data.
Compound 8 (euphominoid H) had the molecular formula C20H30O3 (HRESIMS). The NMR data (Tables 1 and 2) suggested a structure similar to that of compound 7. The HMBC cross-peaks of H-6 (δH 4.38, dd, J = 4.2, 1.6 Hz) with C-4 (δC 44.2), C-5 (δC 157.2), and C-8 (δC 32.6) indicated a hydroxy group at C-6 in compound 8. In the NOESY spectrum (Fig. S64, ESI†), the cross-peaks between H-6 and H3-20 suggested that H-6 and CH3-20 were co-facial and assigned as α-orientations. According to compound 7, the absolute configuration of compound 8 was defined as 3R, 6R, 8R, 9S, and 13S, which was confirmed by comparing the calculated and experimental ECD data.
Compound 9 (euphominoid I) was obtained as a colorless gum with a molecular formula of C20H30O3 based on the HRESIMS ion at m/z 319.2264 [M + H]+. Analysis of the 1D and 2D NMR data of compound 9 (Fig. S67–S72, ESI†) indicated a similar chemical structure to that of compound 8. The CHO–CH2–CHO fragment was deduced from the COSY cross-peaks of H-1 (δH 4.50, t, J = 3.0 Hz)/H-2 (δH 1.94, m; 1.76, m) and H-2 (δH 1.94, m; 1.76, m)/H-3 (δH 3.89, dd, J = 12.9, 3.2 Hz). The presence of a carbonyl group at C-6 was consistent with the HMBC cross-peaks of H-8/H-7 with C-6. In the NOESY spectrum (Fig. S72, ESI†), the cross-peaks of H-1/H3-20 indicated that the 1-OH group was β-oriented. Since there were insufficient signals observed in the NOESY spectrum attributed to H-3, comparison of its experimental and calculated ECD was used to identify the configuration of C-3 (Fig. S105, ESI†). Thus, according to compound 8, the absolute configuration of compound 9 was defined as 1R, 3R, 8R, 9S, and 13S.
Compound 10 (euphominoid J) was obtained as a colorless gum, and its molecular formula was determined as C20H30O2 from the HRESIMS ion at m/z 303.2307 [M + H]+. The NMR data of 10 (Table 3) were similar to those of ebractenoid J,24 except for an additional methyl group and the absence of a cyclopropyl ring. The HMBC cross-peaks from H3-18 (δH 1.06, s)/H3-19 (δH 1.02, s) to C-3 (δC 48.5), C-4 (δC 40.9), and C-5 (δC 75.0) indicated two methyl groups positioned at C-4 and a hydroxy group at C-5.
| Position | 10 | 11 | 12 | 13 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 5.95, s | 123.7 | 5.93, s | 123.2 | 5.98, s | 123.9 | 5.95, s | 123.4 |
| 2 | 200.5 | 200.7 | 200.8 | 201.2 | ||||
| 3 | 2.73, d (16.4) | 48.5 | 2.85, d (16.8) | 48.6 | 3.33, d (16.3) | 44.0 | 3.36, d (16.9) | 43.9 |
| 1.93, d(16.4) | 2.00, d (16.8) | 2.00, d (16.3) | 1.97, d (16.9) | |||||
| 4 | 40.9 | 41.3 | 43.4 | 43.9 | ||||
| 5 | 75.0 | 74.6 | 76.8 | 76.3 | ||||
| 6 | 1.92, m | 32.6 | 1.88, m | 23.2 | 2.09, m | 33.1 | 2.04, m | 23.9 |
| 1.82, m | 1.27, m | 1.83, m | 1.88, m | |||||
| 7 | 1.25, m | 24.3 | 1.88, m | 22.9 | 1.83, m | 24.2 | 1.87, m | 23.0 |
| 1.77, m | 1.30, m | 1.29, m | ||||||
| 8 | 1.47, m | 38.8 | 2.25, m | 29.7 | 1.48, m | 38.5 | 2.21, m | 29.6 |
| 9 | 40.0 | 38.3 | 40.0 | 38.2 | ||||
| 10 | 169.3 | 171.6 | 168.8 | 171.1 | ||||
| 11 | 1.71, m | 32.9 | 1.78, m | 34.2 | 1.73, m | 32.8 | 1.78, m | 34.1 |
| 1.53, m | 1.57, m | 1.53, m | 1.57, m | |||||
| 12 | 1.63, m | 32.3 | 1.57, m | 32.7 | 1.64, m | 32.2 | 1.57, m | 32.6 |
| 1.36, m | 1.36, m | 1.37, m | 1.36, m | |||||
| 13 | 36.2 | 36.3 | 36.2 | 36.2 | ||||
| 14 | 1.47, m | 39.4 | 1.27, m | 40.1 | 1.48, m | 39.4 | 1.25, m | 40.1 |
| 1.13, m | 1.15, m | 2.21, m | ||||||
| 15 | 5.81, dd (17.5, 10.7) | 150.6 | 5.81, dd (17.5, 10.7) | 150.7 | 5.82, dd (17.5, 10.7) | 150.6 | 5.81, dd (17.5, 10.7) | 150.7 |
| 16 | 4.94, dd (17.5, 1.0) | 109.0 | 4.93, dd (17.5, 1.2) | 109.4 | 4.94, dd (17.5, 1.3) | 109.4 | 4.95, dd (17.5, 1.3) | 109.3 |
| 4.87, dd (10.7, 1.0) | 4.87, dd (10.7, 1.2) | 4.88, dd (10.7, 1.3) | 4.88, dd (10.7, 1.3) | |||||
| 17 | 1.01, s | 22.9 | 0.99, s | 22.5 | 1.02, s | 22.9 | 1.00, s | 22.4 |
| 18 | 1.06, s | 25.4 | 1.09, s | 24.9 | 0.87, s | 21.4 | 4.05, d (11.4) | 69.3 |
| 3.38, d (11.4) | ||||||||
| 19 | 1.02, s | 23.4 | 0.97, s | 23.5 | 4.06, d (11.5) | 69.3 | 0.82, s | 20.9 |
| 3.35, d (11.5) | ||||||||
| 20 | 1.21, s | 19.2 | 1.09, s | 17.5 | 1.20, s | 19.6 | 1.00, s | 18.7 |
Interestingly, compound 11 (5-epi-euphominoid J), the C-5 epimer of 10, was also isolated. The 1H NMR spectra of compounds 10 and 11 were closely comparable, with an evident difference observed in the chemical shift of H-8 (δH 1.47 in compound 10 vs. 2.25 in compound 11), C-6 (δC 32.6 in compound 10 vs. 23.2 in compound 11), and C-8 (δC 32.6 in compound 10 vs. 23.2 in compound 11). Steric interactions existing between protons and their neighboring groups would result in a van-der-Waals effect that might lead to the deshielding of the protons.25 Thus, comparison of the 3D structures of compounds 10 and 11 indicated the existence of the van-der-Waals effect between 5β-OH and 8β-H in compound 11 (Fig. 7). The 1H NMR spectrum showed a discernible low field shift of the H-8 signal (δH 1.47 in compound 10 vs. 2.25 in compound 11), which indicated that the 5-OH in compound 11 is β-oriented whereas in compound 10 this hydroxyl group is α-oriented. The distance between the 5-OH and H-8 groups was calculated using the TDDFT method at the M06-2X/6-31+G(d,p) level. As shown in Fig. 7, the distance between these two groups in compound 11 was found to be smaller than that in compound 10. Thus, the absolute configuration of 10 was defined as 5R, 8S, 9S, and 13S, and compound 11 as 5S, 8S, 9S, and 13S, both of which were further confirmed by comparison of the experimental and calculated ECD spectra (Fig. 6).
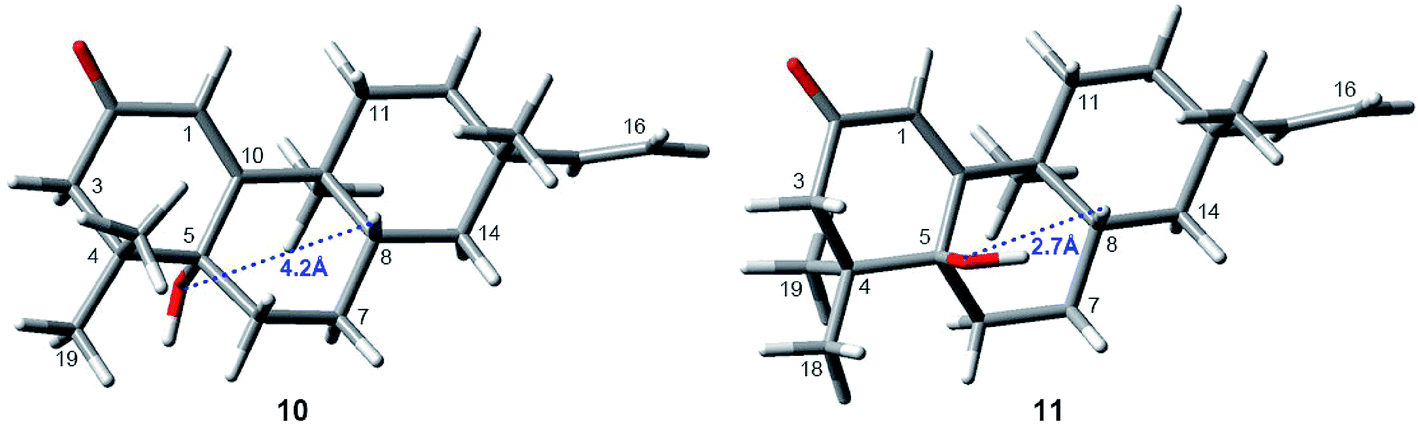 | ||
| Fig. 7 The distance between the 5-OH and 8-H groups within compounds 10 and 11. The calculated distance were computed at the M06-2X/6-31+G(d,p) level. | ||
Compound 12 (euphominoid K) had the molecular formula C20H30O3 based on the HRESIMS ion at m/z 317.2117 [M − H]−. The NMR data of compound 12 (Table 3) were similar to those of compound 10, except for a methyl group in compound 10 being replaced by a hydroxymethyl group (δC 69.3). The HMBC cross-peaks of H-19 (δH 4.06, d, J = 11.5 Hz; 3.35, d, J = 11.5 Hz) with C-3 (δC 44.0), C-4 (δC 43.4), C-5 (δC 76.8), and C-18 (δC 21.4) suggested that the hydroxymethyl group is located at C-4. The 5-OH group was found to be α-oriented based on comparison of the chemical shift of H-8 with that of compounds 10 and 11 (Table 3). Taking into consideration the van-der-Waals effect induced by the 5-OH, as well as comparison of the chemical shifts of H-6a (δH 2.09, m) and H-6b (δH 1.83, m), it was suggested that the H-6a and 5-OH groups are co-facial. The NOESY cross-peak of H-19/H-6a (Fig. S96, ESI†) revealed that the 19-CH2OH group was α-oriented. Thus, the absolute configuration of compound 12 was defined as 4S, 5S, 8S, 9S, and 13S, which was further confirmed by comparison of its experimental and calculated ECD spectra (Fig. S106, ESI†).
Compound 13 (euphominoid L) had the same molecular formula as that of compound 12, as well as similar 1H NMR data, with evident differences in the chemical shifts of H-8. Given that the chemical shifts of H-8 in compounds 13 and 11 were almost identical, and also taking into account the van-der-Waals effect, the 5-OH group in compound 13 was suggested to be β-oriented, as in compound 11. Therefore, the H-6a and 5-OH should be co-facial and have the same β-orientation. In the NOESY spectrum (Fig. S104, ESI†), the cross-peaks of H-18/H-6a indicated that the 18-CH2OH group was β-oriented. Thus, the absolute configuration of compound 13 was identified as 4R, 5R, 8S, 9S, and 13S.
Compounds 1–13 were evaluated for their potencies in the inhibition of EBV lytic DNA replication in P3HR-1 cells using previous methods.17,18 The tested compounds were initially assayed at 50 μM and (+)-rutamarin was used as a positive control.26 Compounds 1–3 and 10 exhibited greater than 50% inhibition of EBV DNA lytic replication at 50 μM. These four compounds were then subjected to further tests to determine their half-maximal antiviral effective concentration (EC50), half-maximal cytotoxic concentration (CC50), and selective index (SI) values (Table 4).
| Compound | EC50a | R2d | CC50b | R2d | SIc |
|---|---|---|---|---|---|
| a Inhibitory effects of the compounds against EBV lytic replication were tested and expressed as EC50 values (μM).b Cytotoxicities were measured after 2 days of compound treatment and expressed as CC50 values (μM).c Selective index (SI) = CC50/EC50.d Regression coefficients of the dose–response curves.e Positive control. | |||||
| 1 | 13.2 | 0.9008 | 59.6 | 0.7798 | 4.5 |
| 2 | 5.4 | 0.9711 | >50 | 0.8413 | >9.3 |
| 3 | 24.4 | 0.8706 | 113 | 0.7707 | 4.6 |
| 10 | 29.1 | 0.9121 | >200 | 0.7273 | >6.9 |
| (+)-Rutamarine | 5.4 | 0.879 | >150 | — | >39.6 |
Conclusions
Thirteen new ent-rosane diterpenoids were isolated from the air parts of E. milii. Four compounds 1–3 and 10 showed moderate inhibitory activity of EBV DNA lytic replication. Compound 2 exhibited significant inhibitory activity with EC50 values of 5.4 μM. Based on these results, preliminary SAR (structure–activity relationship) effects could be established. Thus, structurally, the 13 compounds tested were divided into two groups: in group 1, all of the compounds (1–4) possessed a Δ5,10 double bond, while the compounds in group 2 (5–13) contained an α,β-unsaturated carbonyl group. As shown in Table 4, compounds 1–3 showed medium to potent activity with EC50 values of 13.2, 5.4, and 24.4 μM, respectively. While compounds 5–13 found to be inactive against EBV lytic replication, except compound 10 showed medium activity (EC50 = 29.1 μM). These results suggested that the isolated olefinic scaffolds exhibited significant inhibitory activity than the α,β-unsaturated carbonyl group scaffolds against EBV lytic replication.Experimental section
General experimental procedures
Optical rotation was determined on a Perkin-Elmer 341 polarimeter. UV spectra were record on a Shimadzu UV2450 spectrophotometer. ECD data were collected on an Applied Photophysics Chirascan spectrometer. IR spectra were recorded from KBr pellets on a Bruker Tensor 37 infrared spectrophotometer. The 1H and 13C NMR spectra were measured on a Bruker AVANCE-400 NMR spectrometer operating at 400 MHz and 100 MHz, respectively, with TMS as the internal reference. The HRESIMS and ESIMS data were determined on a Shimadzu LCMS-IT-TOF mass spectrometer and an Agilent 1200 series LC-MS/MS system, respectively. Semipreparative chiral HPLC separation was carried out on an LC-20AT Shimadzu liquid chromatography system with a Phenomenex Lux cellulose-2 chiral-phase column (250 × 10 mm, 5 μm) and Agilent ZORBAX SB-C18 column (250 × 9.4 mm, 5 μm). X-ray data were collected using an Agilent Xcalibur (Onyx, Nova) diffractometer. TLC silica gel plates were purchased from Marine Chemical Ltd., Qingdao, People's Republic of China. RP18 reversed-phase silica gel (Fuji, 40-75 μm), MCI gel (CHP20P, 75-150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan), silica gel (200–300 Mesh, Marine Chemical Ltd.), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) were used for column chromatography (CC).Plant material
The aerial parts of E. milii (3.0 kg) were collected in Baoshan District, Yunnan Province, People's Republic of China, on September 19, 2014, and identified by Dr Chunyan Han from the Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (XG-2014001) was deposited at the School of Pharmacy Sciences, Sun Yat-sen University.Extraction and isolation
The air-dried aerial parts of E. milii (3.0 kg) was powdered and extracted with 80% acetone in water (3 × 20 L) at room temperature for 2 days. The solvent was removed under reduced pressure to give a crude extract (400 g), which was suspended in H2O and extracted successively with EtOAc (3 × 3 L). The EtOAc-soluble fraction (180 g) was chromatographed over a silica gel column, eluting with a step gradient of cyclohexane–EtOAc to give fractions A–C. Partition B (50 g) was loaded onto a silica gel column and eluted with a gradient of CH2Cl2–EtOAc from 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to afford six fractions (B1–B6). Fraction B2 was separated on a silica gel column, eluting with cyclohexane–EtOAc (40:1), and further purified by preparative HPLC using 80% CH3CN–H2O as the solvent to afford compounds 1 (500 mg) and 6 (4 mg). Compounds 10 (10 mg) and 11 (10 mg) were isolated from fraction B3 by semi-preparative HPLC, eluting with MeOH–H2O (88:12). Fraction B4 (16 g) was loaded onto a silica gel column and eluted with cyclohexane–EtOAc (40:1) to afford subfractions B4a–B4c, which were sequentially purified by preparative HPLC, eluting with CH3CN–H2O (85%, 75%, and 65%) to afford compounds 2 (400 mg), 3 (15 mg), 4 (10 mg), 5 (4 mg), 7 (4 mg), 8 (6 mg), 9 (4 mg), 12 (8 mg), and 13 (14 mg).
ε) 247 (3.71) nm; ECD (MeOH) λmax (Δε) 246 (+7.72), 326 (−0.75) nm; IR (KBr) νmax 3407, 2932, 1636, 1579, 1314 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 301.2167 [M − H]− (calcd for C20H29O2, 301.2173).ε) 248 (3.77) nm; ECD (MeOH) λmax (Δε) 244 (+3.74) nm; IR (KBr) νmax 3446, 2928, 1644, 1582, 1372, 1067 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 303.2312 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 206 (3.22), 261 (3.55) nm; ECD (MeOH) λmax (Δε) 211 (−2.82), 262 (+3.33) nm; IR (KBr) νmax 3390, 2922, 1677, 1656, 1465, 1051 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 315.1954 [M − H]− (calcd for C20H28O3Na, 315.1966).ε) 204 (2.71), 244 (3.00) nm; ECD (MeOH) λmax (Δε) 202 (−1.19), 247 (+2.20), 232 (−0.57) nm; IR (KBr) νmax 3394, 2924, 1651, 1464, 1044, 802 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 319.2276 [M + H]+ (calcd for C20H31O3, 319.2268).ε) 203 (3.05), 244 (3.45) nm; ECD (MeOH) λmax (Δε) 204 (−0.50), 243 (+0.65), 352 (+0.36) nm; IR (KBr) νmax 3390, 2923, 1650, 1380, 1044 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 319.2264 [M + H]+ (calcd for C20H31O3, 319.2268).ε) 234 (4.03) nm; ECD (MeOH) λmax (Δε) 251 (+13.64), 330 (−1.89) nm; IR (KBr) νmax 3483, 2968, 1664, 1610, 1317 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 303.2307 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 237 (3.98) nm; ECD (MeOH) λmax (Δε) 234 (+8.21), 265 (−1.31) nm; IR (KBr) νmax 3387, 2936, 1655, 1611, 1467, 1316 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 303.2308 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 208 (3.74), 231 (3.79) nm; ECD (MeOH) λmax (Δε) 201 (+6.17), 248 (+4.87) nm; IR (KBr) νmax 3427, 2964, 1657, 1094 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 317.2117 [M − H]− (calcd for C20H29O3, 319.2122).ε) 237 (3.79) nm; ECD (MeOH) λmax (Δε) 201 (−1.56), 222 (−1.24), 234 (+2.23) nm; IR (KBr) νmax 3475, 2935, 1654, 1046 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 317.2116 [M − H]− (calcd for C20H29O3, 319.2122).
:1 to afford six fractions (B1–B6). Fraction B2 was separated on a silica gel column, eluting with cyclohexane–EtOAc (40:1), and further purified by preparative HPLC using 80% CH3CN–H2O as the solvent to afford compounds 1 (500 mg) and 6 (4 mg). Compounds 10 (10 mg) and 11 (10 mg) were isolated from fraction B3 by semi-preparative HPLC, eluting with MeOH–H2O (88:12). Fraction B4 (16 g) was loaded onto a silica gel column and eluted with cyclohexane–EtOAc (40:1) to afford subfractions B4a–B4c, which were sequentially purified by preparative HPLC, eluting with CH3CN–H2O (85%, 75%, and 65%) to afford compounds 2 (400 mg), 3 (15 mg), 4 (10 mg), 5 (4 mg), 7 (4 mg), 8 (6 mg), 9 (4 mg), 12 (8 mg), and 13 (14 mg).
ε) 247 (3.71) nm; ECD (MeOH) λmax (Δε) 246 (+7.72), 326 (−0.75) nm; IR (KBr) νmax 3407, 2932, 1636, 1579, 1314 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 301.2167 [M − H]− (calcd for C20H29O2, 301.2173).ε) 248 (3.77) nm; ECD (MeOH) λmax (Δε) 244 (+3.74) nm; IR (KBr) νmax 3446, 2928, 1644, 1582, 1372, 1067 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 303.2312 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 206 (3.22), 261 (3.55) nm; ECD (MeOH) λmax (Δε) 211 (−2.82), 262 (+3.33) nm; IR (KBr) νmax 3390, 2922, 1677, 1656, 1465, 1051 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 315.1954 [M − H]− (calcd for C20H28O3Na, 315.1966).ε) 204 (2.71), 244 (3.00) nm; ECD (MeOH) λmax (Δε) 202 (−1.19), 247 (+2.20), 232 (−0.57) nm; IR (KBr) νmax 3394, 2924, 1651, 1464, 1044, 802 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 319.2276 [M + H]+ (calcd for C20H31O3, 319.2268).ε) 203 (3.05), 244 (3.45) nm; ECD (MeOH) λmax (Δε) 204 (−0.50), 243 (+0.65), 352 (+0.36) nm; IR (KBr) νmax 3390, 2923, 1650, 1380, 1044 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Tables 1 and 2; HRESIMS m/z 319.2264 [M + H]+ (calcd for C20H31O3, 319.2268).ε) 234 (4.03) nm; ECD (MeOH) λmax (Δε) 251 (+13.64), 330 (−1.89) nm; IR (KBr) νmax 3483, 2968, 1664, 1610, 1317 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 303.2307 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 237 (3.98) nm; ECD (MeOH) λmax (Δε) 234 (+8.21), 265 (−1.31) nm; IR (KBr) νmax 3387, 2936, 1655, 1611, 1467, 1316 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 303.2308 [M + H]+ (calcd for C20H31O2, 303.2319).ε) 208 (3.74), 231 (3.79) nm; ECD (MeOH) λmax (Δε) 201 (+6.17), 248 (+4.87) nm; IR (KBr) νmax 3427, 2964, 1657, 1094 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 317.2117 [M − H]− (calcd for C20H29O3, 319.2122).ε) 237 (3.79) nm; ECD (MeOH) λmax (Δε) 201 (−1.56), 222 (−1.24), 234 (+2.23) nm; IR (KBr) νmax 3475, 2935, 1654, 1046 cm−1; 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 3; HRESIMS m/z 317.2116 [M − H]− (calcd for C20H29O3, 319.2122).Preparation of the p-bromobenzoyl derivative of compound 1
To a stirred solution of compound 1 (50 mg, 0.16 mmol) in dry dichloromethane (10 mL) were added p-bromobenzoyl chloride (100 mg, 0.45 mmol), 4-dimethylaminopyridine (10 mg, 0.08 mmol), and trimethylamine (0.1 mL) at room temperature. When compound 1 was completely consumed (monitored by TLC), the reaction mixture was extracted with diluted hydrochloric acid and concentrated under reduced pressure to give a white solid residue, which was further purified by silica gel chromatography using dichloromethane as solvent to yield compound 1a (60 mg, 77%). Compound 1a was recrystallized from ethyl acetate as colorless crystals. 1H NMR (400 MHz, CDCl3): δH 7.78 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 5.80 (dd, J = 10.7, 17.5 Hz, 1H), 4.88 (m, 2H), 4.53 (d, J = 10.9 Hz, 1H), 4.35 (d, J = 10.9 Hz, 1H), 2.53 (m, 2H), 2.39 (m, 2H), 1.68–1.48 (m, 3H), 1.48–1.25 (m, 5H), 1.19 (s, 3H), 1.02 (s, 3H), 1.09 (m, 1H), 0.81 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δC 213.7, 165.2, 151.0, 141.7, 131.9, 131.1, 129.3, 128.9, 128.3, 109.0, 68.8, 51.3, 39.4, 38.3, 38.0, 37.5, 36.5, 32.6, 31.8, 25.4, 23.6, 23.2, 19.3, 17.1 ppm.X-ray crystallographic study of compound 1a
C27H33BrO3, M = 485.46, monoclinic, 0.4 × 0.3 × 0.2 mm3, space group P21, a = 5.86059 (7) Å, b = 12.67519 (17) Å, c = 15.73654 (19) Å, α = 90°, β = 94.4504 (11)°, γ = 90°, V = 1165.45 (3) Å3, Z = 2, Dcalc = 1.3833 g cm−3, F000 = 508.0, Xcalibur, Onyx, Nova, Cu Kα radiation, λ = 1.54184 Å, T = 100 K, 2θmax = 144.24°, 21844 reflections collected, 4565 unique (Rint = 0.0358). Final GooF = 1.035, R1 = 0.0257, wR2 = 0.0704, R indices based on 4565 reflections with I > 2 sigma(I) (refinement on F2), 291 parameters, 1 restraint. Lp and absorption corrections applied, μ = 2.602 mm−1. Flack parameter = 0.003 (10). Crystallographic data for the structure of 1a have been deposited at the Cambridge Crystallographic Data Centre under the reference number CCDC 1568169.†
Conversion of 1 to 2 and 4
To a solution of 1 (50 mg, 0.17 mmol) in MeOH (5 mL), NaBH4 (6.3 mg, 0.34 mmol) was added at 0 °C. The reaction was stirred for 10 h at room temperature until all starting material was consumed. The solvent was removed under reduced pressure. The residue was purified by preparative TLC (cyclohexane/EtOAc, 2/1) to afford compounds 2 (30 mg, 58% yield) and 4 (4 mg, 8% yield).Determination of the absolute configuration of the cis-1,3-diol unit within compound 3
The IECD spectrum of compound 3 (0.50 mM in a [Mo2(OAc)4] (0.10 mM) DMSO solution for 0 and 30 min) were measured on an Applied Photophysics Chirascan spectrometer in a 1 cm pathlength between 450 nm and 280 nm. The IECD spectrum of compound 3 was subtracted from the IECD spectrum of those complexes at 0 min, and the absolute configuration of the cis-1,3-diol moiety of compound 3 was assessed following sector rule. [Mo2(OAc)4] (Strem) and DMSO (Sigma-Aldrich, anhydrous ≥99.9%) were commercially available and used without further purification.Preparation of (R)- and (S)-MTPA esters of compound 5
To a solution of compound 5 (3 mg), 4-dimethylaminopyridine (DMAP, 10 mg) and triethylamine (0.025 mL) in anhydrous dichloromethane (1 mL) was added an excess of (R)- or (S)-MTPA chloride (20 μL), and the reaction was stirred for 1 h at room temperature. The reaction mixture was then purified by preparative silica gel TLC using cyclohexane–EtOAc (2:1) as the developing solvent.
1H NMR data of (R)-MTPA ester of compound 5 (400 MHz, CDCl3): δH 7.53–7.40 (5H, m, Ar–H), 5.82 (1H, dd, J = 10.7, 17.5 Hz, H-15), 5.17 (1H, dd, J = 5.6, 10.6 Hz, H-3), 4.93 (1H, dd, J = 1.4, 17.5 Hz, H-16a), 4.86 (1H, dd, J = 1.4, 10.7 Hz, H-16b), 2.88 (1H, dd, J = 5.6, 17.6 Hz, H-2a), 2.48 (1H, m, H-11a), 2.46 (1H, dd, J = 10.6, 17.6 Hz, H-2b), 2.34 (2H, m, H-6), 1.16 (3H, s, H-18), 1.07 (3H, s, H-19 or H-20), 1.04 (3H, s, H-20 or H-19), 1.02 (3H, s, H-17).
1H NMR data of (S)-MTPA ester of compound 5 (400 MHz, CDCl3): δH 7.53–7.40 (5H, m, Ar–H), 5.82 (1H, dd, J = 10.7, 17.5 Hz, H-15), 5.21 (1H, dd, J = 5.5, 10.5 Hz, H-3), 4.93 (1H, dd, J = 1.3, 17.5 Hz, H-16a), 4.86 (1H, dd, J = 1.3, 10.7 Hz, H-16b), 2.90 (1H, dd, J = 5.5, 17.5 Hz, H-2a), 2.56 (1H, dd, J = 10.5, 17.5 Hz, H-2b), 2.49 (1H, dt, J = 3.6, 13.3 Hz, H-11a), 2.32 (2H, m, H-6), 1.09 (3H, s, H-18), 1.06 (3H, s, H-19 or H-20), 1.04 (3H, s, H-20 or H-19), 1.03 (3H, s, H-17).
ECD computational calculations for compounds 5–13
The ECD spectra were calculated using Gaussian 09 and analyzed using the GUI GaussView (version 5.0) at the density functional theory (DFT) and time-dependent DFT (TD-DFT) levels. Conformational analysis of these compounds was performed using the Discovery Studio 3.5 (Accelrys Inc., San Diego, CA) software package. The minimum energy conformations were fully optimized at the B3LYP/6-31G(d) level in the gas phase to obtain accurate conformers. The ECD spectra were simulated at the PBE1PBE/6-31++G(2d,2p) level in MeOH. The calculated ECD curve was generated using SpecDis with σ = 0.2 eV.Configuration computational calculations for compounds 10–13
The structures of compounds 10–13 were optimized at the M06-2X/6-31+G(d,p) in the gas phase using Gaussian 09. The distance between hydroxyl and H-8 was measured using Gaussian 09 and analyzed using GUIs GaussView (version 5.0), and the spectra were given in Fig. S155 and S156.†Bioassay of extracts and compounds isolated from E. milii on the inhibition of EBV DNA lytic replication
The anti-EBV activities were assayed according to a method reported in the literature.17,18 P3HR-1 cells were cultured in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco-BRL), streptomycin (100 μg mL−1), and penicillin (100 units per mL). The crude extracts and isolated compounds were dissolved in dimethyl sulfoxide (DMSO), and the final DMSO concentration in the culture medium was less than 0.5%. P3HR-1 cells were induced with 12-O-tetradecanoylphorbol-13-acetate (TPA) (20 ng mL−1) and sodium butyrate (0.3 mM) for EBV lytic replication. After 3 h, the induced cells were treated with the extracts or compounds over varying concentrations. After 48 h, the P3HR-1 cells were harvested, and the total DNA in the cells was purified using the HiPure Tissue DNA Kit according to the manufacturer's protocol (Magen). The EBV genomic copy number was quantified by real-time PCR on a Roche 480 LightCycler using the LightCycler FastStart DNA MasterPlus SYBR Green Kit with primers for the detection of EBNA1 (sense: 5′-CATTGAGTCGTCTCCCCTTTGGAAT-3′; antisense: 5′-TCATAACAAGGTCCTTAATCGCATC-3′). The intracellular viral genomic DNA in each sample was normalized to GAPDH using the primers directed to GAPDH (sense: 5′-ACATCATCCCTGCCTCTAC-3′; antisense: 5′-TCAAAGGTGGAGGAGTGG-3′). The EC50 value of each compound was determined from a dose–response curve of the EBV DNA contents from TPA/butyrate-induced and compound-treated cells. The EC50 values for each compound were calculated according to previous methods17,18 using GraphPad Prism software.Cytotoxicity assays
The cell viability of P3HR-1 cells, after treatment with either the compounds or only DMSO, was tested by counting the trypan blue-stained cells using a light microscope after two days of treatment. The cell viability was defined relative to the control cells (non-drug-treated). The CC50 value was obtained from the dose–response curves using GraphPad Prism software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by the Guangdong Province Frontier and Key Technology Innovation Program (2015B010109004), the College Key Training Program for Excellent Young Teacher (17ykzd13), the Guangdong Innovative Research Team Program (2009010058), and the National Natural Science Foundation of China (Nos. 81471138, 81573310, 81371793, 81530069).References
- Q. W. Shi, X. H. Su and H. Kiyota, Chem. Rev., 2008, 108, 4295–4327 CrossRef CAS PubMed.
- A. Vasas and J. Hohmann, Chem. Rev., 2014, 114, 8579–8612 CrossRef CAS PubMed.
- H. B. Wang, X. Y. Wang, L. P. Liu, G. W. Qin and T. G Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed.
- A. Vasas, D. Rédei, D. Csupor, J. Molnár and J. Hohmann, Eur. J. Org. Chem., 2012, 2012, 5115–5130 CrossRef CAS.
- Y. Ito, M. Kawanishi, T. Harayama and S. Takabayashi, Cancer Lett., 1981, 12, 175–180 CrossRef CAS PubMed.
- C. M. Yang, H. Y. Cheng, T. C. Lin, L. C. Chiang and C. C. Lin, Clin. Exp. Pharmacol. Physiol., 2005, 32, 346–349 CrossRef CAS PubMed.
- L. Avila, M. Perez, G. Sanchez-Duffhues, R. Hernandez-Galan, E. Munoz, F. Cabezas, W. Quinones, F. Torres and F. Echeverri, Phytochemistry, 2010, 71, 243–248 CrossRef CAS PubMed.
- R. J. Gulakowski, J. B. McMahon, R. W. Buckheit Jr, K. R. Gustafson and M. R. Boyd, Antiviral Res., 1997, 33, 87–97 CrossRef CAS PubMed.
- C. Jiang, P. Luo, Y. Zhao, J. Hong, S. L. Morris-Natschke, J. Xu, C. H. Chen, K. H. Lee and Q. Gu, J. Nat. Prod., 2016, 79, 578–583 CrossRef CAS PubMed.
- H. Y. Cheng, T. C. Lin, C. M. Yang, K. C. Wang, L. T. Lin and C. C. Lin, J. Antimicrob. Chemother., 2004, 53, 577–583 CrossRef CAS PubMed.
- I. Mucsi, J. Molnar, J. Hohmann and D. Redei, Planta Med., 2001, 67, 672–674 CrossRef CAS PubMed.
- T. Akihisa, E. M. Kithsiri Wijeratne, H. Tokuda, F. Enjo, M. Toriumi, Y. Kimura, K. Koike, T. Nikaido, Y. Tezuka and H. Nishino, J. Nat. Prod., 2002, 65, 158–162 CrossRef CAS.
- A. M. Madureira, J. R. Ascenso, L. Valdeira, A. Duarte, J. P. Frade, G. Freitas and M. J. Ferreira, Nat. Prod. Res., 2003, 17, 375–380 CrossRef CAS PubMed.
- M. J. Ahn, C. Y. Kim, J. S. Lee, T. G. Kim, S. H. Kim, C. K. Lee, B. B. Lee, C. G. Shin, H. Huh and J. Kim, Planta Med., 2002, 68, 457–459 CrossRef CAS PubMed.
- W. Li, X. Cai, Z. H. Hu and Y. J. Wang, Chinese Wild Plant Resources, 2008, 27, 1–4 Search PubMed.
- S. N. Liu, D. Huang, S. L. Morris-Natschke, H. Ma, Z. H. Liu, N. P. Seeram, J. Xu, K. H. Lee and Q. Gu, Org. Lett., 2016, 18, 6132–6135 CrossRef CAS PubMed.
- H. Cui, B. Xu, T. Wu, J. Xu, Y. Yuan and Q. Gu, J. Nat. Prod., 2014, 77, 100–110 CrossRef CAS PubMed.
- T. Wu, Q. Wang, C. Jiang, S. L. Morris-Natschke, H. Cui, Y. Wang, Y. Yan, J. Xu, K. H. Lee and Q. Gu, J. Nat. Prod., 2015, 78, 500–509 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, F. Wang, Z. H. Li, Z. J. Dong and J. K. Liu, Nat. Prod. Bioprospect., 2015, 5, 69–75 CrossRef CAS PubMed.
- I. Jantan and P. G. Waterman, Phytochemistry, 1994, 37(5), 1477–1479 CrossRef CAS.
- L. Anwar, E. Mai, M. Ninomiya, S. Ibrahim, D. P. Putra, K. Tanaka and M. Koketsu, Med. Chem. Res., 2017 DOI:10.1007/s00044-017-1937-3.
- J. Frelek, G. Snatzke and W. J. Szczepek, Fresenius. J. Anal. Chem., 1993, 345, 683–687 CrossRef CAS.
- J. Frelek, W. Szczepek and W. Voelter, J. Prakt. Chem., 1997, 339, 135–139 CrossRef CAS.
- Z. G. Liu, Z. L. Li, J. Bai, D. L. Meng, N. Li, Y. H. Pei, F. Zhao and H. M. Hua, J. Nat. Prod., 2014, 77, 792–799 CrossRef CAS PubMed.
- H. Günther, NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry, Wiley-VCH, Germany, 3rd edn, 2013 Search PubMed.
- T. Wu, Y. Wang and Y. Yuan, Antiviral Res., 2014, 107, 95–101 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: IR, MS, and 1D and 2D NMR data for compounds 1–13. CCDC 1568169. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08877a |
| ‡ Shaonan Liu and Jiayuan Hu contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |