
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-switchable polymer-hybrid silver nanoparticles and their gas-tunable catalytic activity†
Zanru Guo *a,
Hongjian Gua,
Wei Maa,
Qiang Chena,
Zhanfeng He*b,
Jiali Zhanga,
Yongxin Liua,
Longzhen Zhenga and
Yujun Feng*c
*a,
Hongjian Gua,
Wei Maa,
Qiang Chena,
Zhanfeng He*b,
Jiali Zhanga,
Yongxin Liua,
Longzhen Zhenga and
Yujun Feng*c
aDepartment of Polymer Materials and Chemical Engineering, School of Materials Science and Engineering, East China Jiaotong University, Nanchang, Jiangxi 330013, P. R. China. E-mail: guozanru@ecjtu.edu.cn
bState Key Laboratory of Oil and Gas Reservoir Geology, Exploitation Southwest Petroleum University, Chengdu 610500, P. R. China. E-mail: he_zhanfeng2008@126.com
cPolymer Research Institute, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: yjfeng@scu.edu.cn
First published on 26th October 2017
Abstract
The design of controllable or “signal-triggered” metal nanoparticles is one of the emerging trends in nanotechnology and advanced materials. CO2-switchable polymer-hybrid silver nanoparticles (AgNPs) were prepared by a one-pot reaction reducing AgNO3 and trithioester terminated PDEAEMA with sodium borohydride (NaBH4). The hybrids showed a long-term stability, and their size and size distribution can be easily modulated by tuning the molar ratio of polymers to AgNO3. The hybrids not only exhibit hydrophobic–hydrophilic transitions in immiscible mixed solvents, but also undergo a switchable dispersion/aggregation states upon alternately treating with CO2 and N2. Moreover, this smart hybrid was preliminarily used as catalyst for the reduction of 4-nitrophenol. The catalytic activity of the hybrids can be switched and monotonously tuned by varying the flow rate of CO2 purged into the reaction system, which may open a new avenue for tailoring the catalytic activity of metal nanoparticles toward a given reaction.
Introduction
In recent years, metal nanoparticles (NPs) such as Au and Ag, have garnered much interest, especially in the field of catalysts, owing to their unusual physicochemical properties that are quite different from those of the bulk solids or atomic state.1–5 However, metal NPs are biased toward self-aggregation to form big particles due to their high surface energy,6 which not only severely hinders their long-term storage and processing but also reduces their catalytic activity.7–9 To counter this problem, several supported agents such as surfactants,10 solid surface,11 polyelectrolyte brushes,12 hollow capsules,13 dendrimers14 and hydrogels15 have been exploited to make metal NPs dispersed or individualized. Recently, metal NPs were studied for application in “smart” catalysts with switchable, tunable and reusable catalytic properties.9,16–23 To be successfully applied, metal NPs should be not only dispersed but also sensitive to external stimulus.9,16–23 Functionalization of metal NPs with stimuli-responsive polymers represents one of the choices to satisfy such requirements.Up to date, several kinds of stimuli-responsive polymers have been used to functionalize metal NPs to form “smart” catalysts. For example, temperature-responsive polymeric hydrogel,24 micelle,25,26 microgel,27 “yolk–shell” structure,28 and polymers29 were employed to support metal NPs, forming temperature switchable or tunable catalysts. Similarly, metal NPs were immobilized in pH-responsive polymeric hydrogel,30,31 microsphere,32 and micelle33 to get pH-responsive catalytic systems. Besides, light-controllable catalyst was obtained by the combination of a temperature-responsive polymer with metal nanoparticles which can convert light into heat through light irradiation.9 Though the use of temperature as a trigger for metal “smart” catalysts can switch reaction and modulate reaction rate, changing reaction temperature would result in some side reaction or affect reaction rate,9 which lead to a nonmonotone correlation with temperature. As for pH trigger based on acids and bases, it is hard to tune reaction rate as reaction usually happened in a fix pH range.23,31 Moreover, the use of acids and bases for tuning pH may contaminate or modify the final products.9 Therefore, to switch catalytic activity and modulate catalytic rate, it is desirable to develop a “green” and simple trigger for metal “smart” catalysts.
Very recently, we and others have employed CO2 and inert gas such as nitrogen to switch between the hydrophobic and hydrophilic property of polymers with amidine34,35 or amino groups,36–40 and then control the morphology of nanomaterials34,40 or its self-assemblies.36,39 As an abundant, economical, nontoxic, biocompatible, and renewable resource, CO2 trigger just bubble gas and leave no contamination during the stimulate process,34–39 which may satisfy the requirements of metal “smart” catalysts. Zhao and coworkers41 pioneered the fabrication of CO2-switchable gold nanoparticles (AuNPs) by functionalizing AuNPs with CO2-switchable polymers, and found the obtained AuNPs hybrids can be dispersed–re-dispersed in and separated from aqueous solution by CO2 and N2 bubbling. Besides, the hybrids exhibit high catalytic activity, easier separation and better reusability for 4-nitrophenol reduction. Yuan et al.42 embedded AuNPs into the shell of CO2-responsive magnetic hybrid nanospheres, and switch their catalytic activity through the access of swollen or collapsed of CO2 sensitive shell. Although these metal hybrid catalysts exhibit CO2-responsive properties, the studies were focused on gold metal, and its reusability and switchability. To the best of our knowledge, there have been few reports of other CO2-responsive metal catalyst and the gas-modulated catalytic activity. Compared with AuNPs and other metal nanoparticles (like Pt, Pd),43,44 silver nanoparticles (AgNPs) can be prepared more readily and inexpensively and also exhibits similar catalytic properties, which may have broad application prospects in catalysis.45 As such, the appeal for CO2-switchable AgNPs with gas-tunable catalytic activity for the general development of smart catalysts remains high.
In this report, we used an reversible addition–fragmentation transfer polymerization (RAFT) technique to synthesize a CO2-responsive polymer poly(2-(diethylamino)-ethyl methacrylate) (PDEAEMA), since RAFT polymerization has the advantage to prepare the thiol terminated polymer because the chain transfer agents always contain the dithioester or trithioester group that can be easily reduced to thiol terminated group.46–48 Resorting to reducing agent sodium borohydride (NaBH4), AgNO3 and trithioester end group of PDEAEMA were reduced to silver nanoparticles (AgNPs) and thiol moiety, respectively; then PDEAEMA adsorbed onto the surface of AgNPs via strong Ag–sulfur interaction, forming PDEAEMA–AgNPs (Ag–P) hybrids (Scheme 1). The hydrophilic–hydrophobic properties and dispersibility of the AgNPs were examined by bubbling CO2 or N2. The hybrids were applied as a catalyst in a model catalytic reduction of 4-nitrophenol, as it is one of the most refractory pollutants that can occur in industrial waste waters.42,48 Besides, the catalytic activity at different CO2 flow rate was preliminarily discerned, which may open a new avenue for tailoring the catalytic activity of metal nanoparticles toward a given reaction.
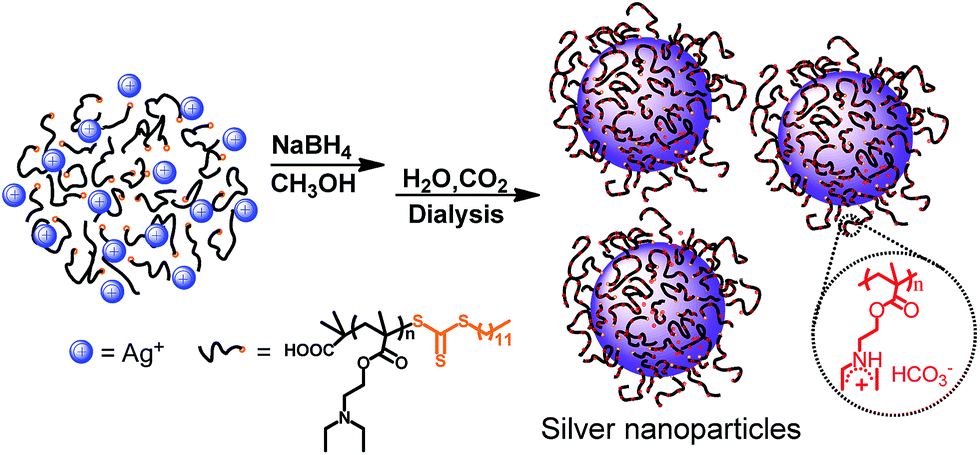 | ||
| Scheme 1 Schematic representation of the formation of polymer-hybrid silver nanoparticles. | ||
Experimental
Materials
2-Diethylaminoethyl methacrylate (DEAEMA, ≥99.5%), silver nitrate (AgNO3, ≥99.8%), and sodium borohydride (NaBH4, ≥98%) were purchased from Aladdin co., Ltd. 2,2′-Azoisobutyronitrile (AIBN) (Aldrich, 98%) was recrystallized from diethylether prior to use. The chain transfer agent (CTA), S-1-dodecyl-S′-(α,α′-dimethyl-α′′-aceticacid)trithiocarbonate, was synthesized following a previously reported procedure.49 All the chemicals were used as received, and all aqueous solutions were prepared with deionized water.Preparation of PDEAEMA
PDEAEMA with trithioester terminal group was prepared via RAFT polymerization. DEAEMA (6.0 g, 0.032 mol), AIBN (21 mg, 0.128 mmol), and CTA (0.23 g, 0.64 mmol) were added into a three-necked flask, followed by the addition of 30 mL of THF. After bubbling with nitrogen for at least 30 min, the reaction was heated to 70 °C for 24 h. Then the polymerization was quenched by immerging the reaction flask into liquid nitrogen for about 5 minutes. The product mixture was diluted by 10 mL THF, and the final product was gained via precipitation in cold n-hexane (−78 °C, ca. 500 mL) followed by filtration over a G4 frit. The obtained product was redissolved in 20 mL THF and reprecipitated in cold n-hexane (−78 °C, ca. 500 mL) again. The resultant solid was collected by filtration and dried overnight in a vacuum oven for 24 h to give PDEAEMA. Yield: 5.45 g (∼85%).Preparation of silver nanoparticles
Different molar ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24, 1:12 and 1:6) of PDEAEMA to AgNO3 were investigated, and PDEAEMA-stabilized silver nanoparticles (Ag–P) dispersions were prepared by the reduction of AgNO3 using NaBH4 in methanol solution. The ratio of polymer to silver (1:24) is as an example: 0.192 g of PDEAEMA was dissolved in 50 mL of methanol. 0.081 g of AgNO3 was dissolved in 2 mL of deionized water, and the solution of AgNO3 was added into the PDEAEMA solution drop by drop under vigorous stirring (1200 rpm). After stirring for 30 minutes, 0.359 g of NaBH4 solid was slowly added to the mixed solution with stirring. The appearance of the mixture immediately changed from light yellow to dark brown. After stirring for 24 h, the mixture was concentrated to ca. 10 mL by rotary evaporator. Then 2 mL of water was added with bubbling CO2 (30 mL min−1), which does not have solid precipitation. After bubbling for 30 minutes, another 2 mL of water was added. The above procedure was repeated until 10 mL of deionized water was added. Then the mixture was dialyzed against distilled water for one week to remove methanol, sodium borate and other unreacted impurities. The distilled water was changed every 4 h during the dialysing. Finally, the polymer-hybrid silver nanoparticles dispersion was obtained.
:24, 1:12 and 1:6) of PDEAEMA to AgNO3 were investigated, and PDEAEMA-stabilized silver nanoparticles (Ag–P) dispersions were prepared by the reduction of AgNO3 using NaBH4 in methanol solution. The ratio of polymer to silver (1:24) is as an example: 0.192 g of PDEAEMA was dissolved in 50 mL of methanol. 0.081 g of AgNO3 was dissolved in 2 mL of deionized water, and the solution of AgNO3 was added into the PDEAEMA solution drop by drop under vigorous stirring (1200 rpm). After stirring for 30 minutes, 0.359 g of NaBH4 solid was slowly added to the mixed solution with stirring. The appearance of the mixture immediately changed from light yellow to dark brown. After stirring for 24 h, the mixture was concentrated to ca. 10 mL by rotary evaporator. Then 2 mL of water was added with bubbling CO2 (30 mL min−1), which does not have solid precipitation. After bubbling for 30 minutes, another 2 mL of water was added. The above procedure was repeated until 10 mL of deionized water was added. Then the mixture was dialyzed against distilled water for one week to remove methanol, sodium borate and other unreacted impurities. The distilled water was changed every 4 h during the dialysing. Finally, the polymer-hybrid silver nanoparticles dispersion was obtained.
Catalytic reduction of 4-nitrophenol
Referring to previous methods,55 1 mL of 2.5 mM 4-nitrophenol and 1 mL of 250 mM NaBH4 solution was diluted by 23 mL of distilled water in a 50 mL beaker, the mixture was stirred for 5 min at room temperature. Then, the mixture was transferred to a test tube and 0.01 mL of 5 mg mL−1 silver nanoparticle dispersion was added, the mixture was taken out for UV-vis adsorption monitoring immediately. Based on the strength of the peak at 400 nm, the initial concentration (C0) of 4-nitrophenolate ions was calculated. At a certain reaction time, 3 mL of the mixture was taken out for UV-vis adsorption monitoring, and the concentration (C) was calculated. The correlation of ln(C/C0) versus the reduction time t was estimated to be linear, and the slope was estimated as the apparent reaction rate constant (kapp). kapp is the average values calculated from three runs of a certain measurement. Following the above steps, the flow rate of CO2 was controlled at 10 mL min−1, 15 mL min−1, 20 mL min−1, 25 mL min−1, 30 mL min−1, 40 mL min−1 and 50 mL min−1, respectively, to carry out catalytic reaction. With the progress of the reaction, the color of the solution gradually fades from yellow to colorless. Noteworthy, after bubbling CO2 for a minute, the pH of the mixture will be less than 6.7. At the same time, there was a peak at 317 nm, the peak of 4-nitrophenol, indicating that NaBH4 was consumed by CO2 and the catalytic reaction was not complete. Therefore, a certain amount of NaBH4 should be added. At the end of the reaction, silver nanoparticles were separated by bubbling N2.Characterization
UNICO UV-4802 double-beam spectrophotometer (UV-vis spectroscopy) was used to observe the location and migration of silver nanoparticle peaks. The full width at half-maximum (FWHM) values of the UV-vis spectra were measured by previous methods.57 In the catalytic reduction of 4-nitrophenol, UV-visible spectroscopy can be used to determine its reduction.Infrared spectra were registered on a Nicolet MX-1E FTIR (USA) spectrophotometer in the scanning range of 4000–400 cm−1 using KBr pellet method.
The 1H NMR of the polymer was measured using a Nuclear Magnetic Resonance Spectrometer (AV CORP300, Bruker, Germany). The polymer was purified according to its solubility in CDCl3, and the molecular structure of the polymer was determined from the corresponding chemical shift and integral area in the 1H NMR.
The molecular weight and the molecular weight distribution of the polymers were determined using a gel permeation chromatography (GPC) system equipped with a Waters 515 pump and a 2410 detector. The column temperature was set at 25 °C and the THF was used as the mobile phase. Polystyrene (PSt) was used as the reference material.
Thermal gravimetric analysis (TGA, 299-F1, NETZSCH, Germany) was used to test the polymer modified silver nanoparticles in the polymer content. The sample was heated to a temperature of 800 °C at a rate of 10 °C min−1 in a nitrogen atmosphere (flow rate 20 mL min−1). The dialyzed dark brown concentrated mixture was purged with N2 for 1 h and centrifuged at 10000 rpm for 5 minutes, the upper layer was discarded, washed with deionized water and centrifuged. After three times, the mixture was suction filtered and the solid sample was vacuum dried at 50 °C for 24 hours prior to TGA and XRD characterizations.
The zeta potential values of silver colloid were measured with zetameter ZetaPALS (Brookhaven, USA). Each test was carried out for five times and the average values were taken as the final results.
X-ray diffraction (XRD) patterns of AgNPs hybrids was recorded using a RigakuDMAX2200 with Ni-filtered Cu Kα radiation over a scanning range of 30 to 80° at an X-ray power of 40 kV and 40 mA.
The conductivity of Ag–P1 dispersion was measured with a DDS-11A conductometer (Chengdu Fangzhou Instrument) at 25 °C, and the average values were calculated from three runs of a certain measurement.
Transmission electron microscope (Holland, Philips Company, Tecnai12) was used to observe the particle size distribution of silver nanoparticles. The size distributions of each sample were determined at least 1000 particles from photographs of the TEM images by image analysis software (Nano Measurer). As for negative staining TEM, the sample was stained with phosphotungstic acid for TEM observation.
Results and discussion
Synthesis and characterization
To bestow the CO2-switchable property on AgNPs, CO2-responsive polymer was needed. Based on previous reports, polymer with amidine34,35 or tertiary amine groups36–40 exhibits CO2-responsive characteristic. However, as for amidine-based polymer, heating is usually needed to expel the captured CO2.34,35 In contrast, polymer containing tertiary amine groups could realize reversible switchability in the same condition upon alternate treatment of CO2 and N2.36–40 PDEAEMA was a typical tertiary amine type CO2-responsive polymer.36,37 Thus PDEAEMA was synthesized through RAFT polymerization, as RAFT not only is a powerful and versatile controlled radical polymerization technique, enables precise control over the MW, MW distribution, but also introduce trithioester group to the end of polymer.49,50 Detailed preparation and characterization procedures can be found in ESI.†To form CO2-switchable AgNPs, the silver nanoparticles were prepared by reducing silver nitrate (AgNO3) with NaBH4 in the presence of the PDEAEMA. Meanwhile, trithioester end group of PDEAEMA was reduced to thiol group with NaBH4,46–49 providing the necessary for chemical attaching PDEAEMA on the surface of AgNPs (Scheme 1). With such one-pot protocol, a series of AgNPs–PDEAEMA (Ag–P) dispersion were prepared by varying the molar ratios (1:6, 1:12 and 1:24) of PDEAEMA to AgNO3, as shown in Table 1. For characterization and storage, AgNPs hybrids were transferred from methanol to aqueous environment by bubbling CO2. And three dark brown dispersion were finally obtained (insert, Fig. 1), indicative of the formation of AgNPs.51
| Samplea | [PDEAEMA]:[AgNO3]b |
% PDEAEMA/% Agc | Average diameter of Ag–NPsd, nm |
|---|---|---|---|
| a Ag–P refers to PDEAEMA polymer-stabilized silver nanoparticles.b Molar ratios of PDEAEMA to AgNO3.c Weight percentage, determined by TGA.d Estimated from TEM images. | |||
| Ag–P1 | 1:6 |
90/10 | 8.51 ± 2.8 |
| Ag–P2 | 1:12 |
84/16 | 10.06 ± 3.7 |
| Ag–P3 | 1:24 |
71/29 | 14.16 ± 6.6 |
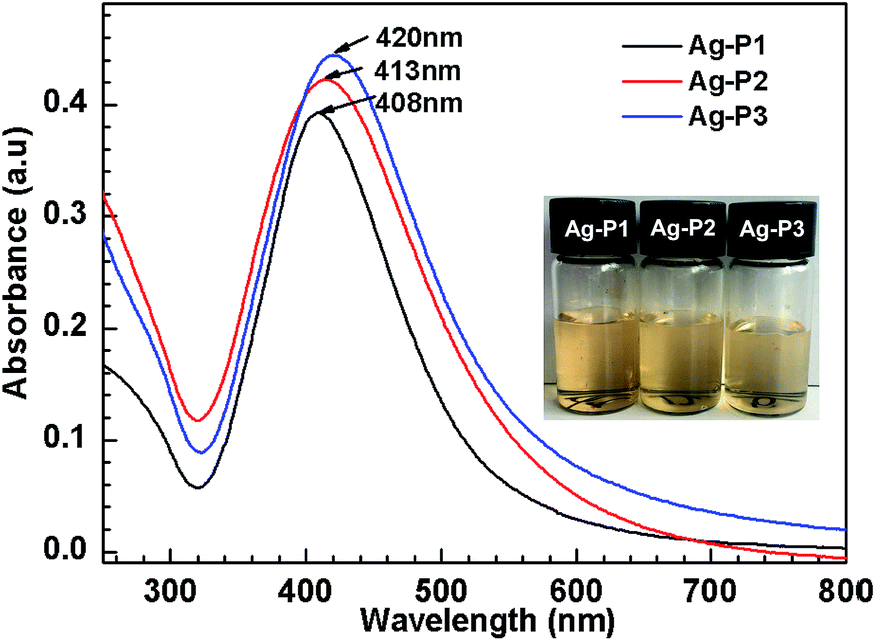 | ||
| Fig. 1 UV-vis spectra of Ag–P1, Ag–P2 and Ag–P3 in water after bubbling CO2 at 25 °C. The inset images of the hybrids solution refer to corresponding samples. The concentration of hybrids is 0.06 mg mL−1. | ||
To characterize the formation of AgNPs hybrids, UV-vis spectroscopy which is known for the sensitivity to the size, size distribution and morphology of metal nanoparticles51,52 was employed. As shown in Fig. 1, single absorption peak is found in the region of 320–600 nm, resulting from intense surface plasmon resonances (SPR) of the obtained AgNPs.53 It is noteworthy that the λmax gradually increases with increasing the silver contents of Ag-polymer dispersion. Usually, λmax of AgNPs are biased to shift to longer wavelengths with increasing nanoparticle size,52 suggesting that the AgNPs size increases slightly upon increasing the silver contents. This may arise from the increased collision frequency due to the formation of more Ag atoms.46,54 Furthermore, the full width at half-maximum (FWHM) could be calculated from the UV-vis spectra, since FWHM is useful to evaluate the polydispersity of AgNPs.46,56 The FWHM value of Ag–P1 is 105 nm, which are similar to or slightly smaller than those previously reported for AgNPs.46,52,55 Correlating with symmetric absorption peaks, this implies that the size of Ag–P1 is uniform.56 Compared with Ag–P1, the FWHM for Ag–P2 (118 nm), Ag–P3 (124 nm) become wider, suggesting that the polydispersity of the hybrids increased with increasing the silver ratio.
In order to get more direct information on the size, size distribution and morphology of AgNPs, TEM observations were performed. Fig. 2 shows TEM images and size distribution histograms of three AgNPs. One can find that the AgNPs hybrids display good dispersion and a spherical shape. With total 1000 particles counted by an image analysis software (Nano Measure) on number distribution, we found Ag–P1 have an average size of 8.51 ± 2.8 nm with a narrow distribution. With increasing the silver ratio, the size and size distribution become bigger. The sizes of the other two AgNPs are 10.06 ± 3.7 nm and 14.16 ± 6.6 nm, respectively. The TEM results are in good agreement with those of the UV-vis spectra analysis. Thus the size and size distribution of as obtained AgNPs can be easily modulated by varying the molar ratio of polymers to AgNO3. In addition, XRD was carried out to confirm the structure of AgNPs. As illustrated in Fig. S3,† their XRD pattern of AgNPs shows characteristic diffraction peaks for metallic silver [111], [200], [220] and [311] facets, indicative of the formation of pure Ag.46,55,57,58
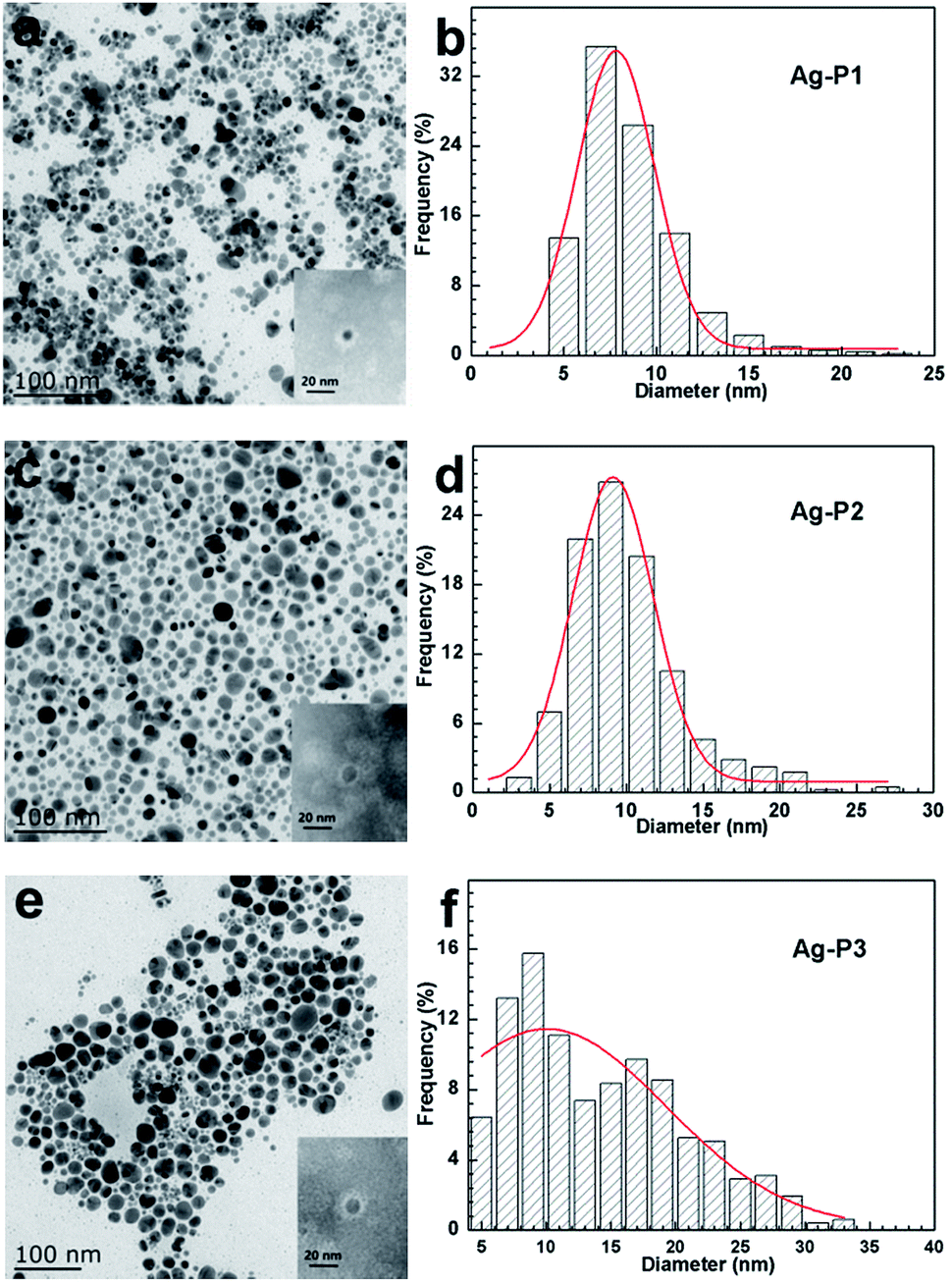 | ||
| Fig. 2 TEM images of Ag–P1 (a), Ag–P2 (c) and Ag–P3 (e) in water and particle size distribution histograms of silver particles evaluated from the corresponding TEM images (b, d and f). The inset TEM images of the sample negatively staining with phosphotungstic acid. | ||
As stated above, the AgNPs were clearly observed by TEM. However, the grafted PDEAEMA were not observed under TEM observation, which may attribute to the polymer with lower atomic mass.55 To confirm that PDEAEMA was grafted onto the surface of AgNPs, FT-IR spectroscopy was employed. As given in Fig. S4,† The IR spectra of the nanoparticles and the PDEAEMA are similar to one another, indicating that the polymer molecules have indeed grafted onto AgNPs. However, a remarkable difference in the peak intensity is found between the peaks in polymer and the hybrids. Those peaks correspond to the stretching mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S (1062 cm−1) and –CH2–S– (725 cm−1).49 After reaction, CS was disappeared because the trithioester group was reduced to thio group. The decreased intensity for –CH2–S– is believed to be that the thiol end group of the polymer on the nanoparticle form a relatively close packed thiol layer and molecular motion is constrained,56 which suggesting that polymer attached to AgNPs surface through a chemical bond between S ions and Ag atoms.
S (1062 cm−1) and –CH2–S– (725 cm−1).49 After reaction, CS was disappeared because the trithioester group was reduced to thio group. The decreased intensity for –CH2–S– is believed to be that the thiol end group of the polymer on the nanoparticle form a relatively close packed thiol layer and molecular motion is constrained,56 which suggesting that polymer attached to AgNPs surface through a chemical bond between S ions and Ag atoms.
To determine the relative amount of PDEAEMA on AgNPs, thermal gravimetric analysis (TGA) measurement was carried out. From the TGA curve given in Fig. 3, the weight percentage of PDEAEMA in the Ag–P1, Ag–P2, Ag–P3 hybrids were ca. 90 wt%, 84 wt%, 71 wt%, respectively, from which we could calculate that one AgNPs was wrapped by roughly 2000 polymer chains (see ESI†). To visualize the polymer on nanoparticles, AgNPs samples treated with negative staining technique was used for TEM observation, which provides reverse-contrast negative electron optical images for the unstained component.55 One can find that black AgNPs dot surrounded by the brighter polymer part, showing typical cocoon-like morphology (inset, Fig. 2). The thickness of observed PDEAEMA layer is about 6–10 nm, indicating that the polymers were attached to AgNPs. Compared with Ag–P2 and Ag–P3, Ag–P1 has smaller size and narrower size distribution. Therefore, in the following experiments, we will mainly focus on Ag–P1.
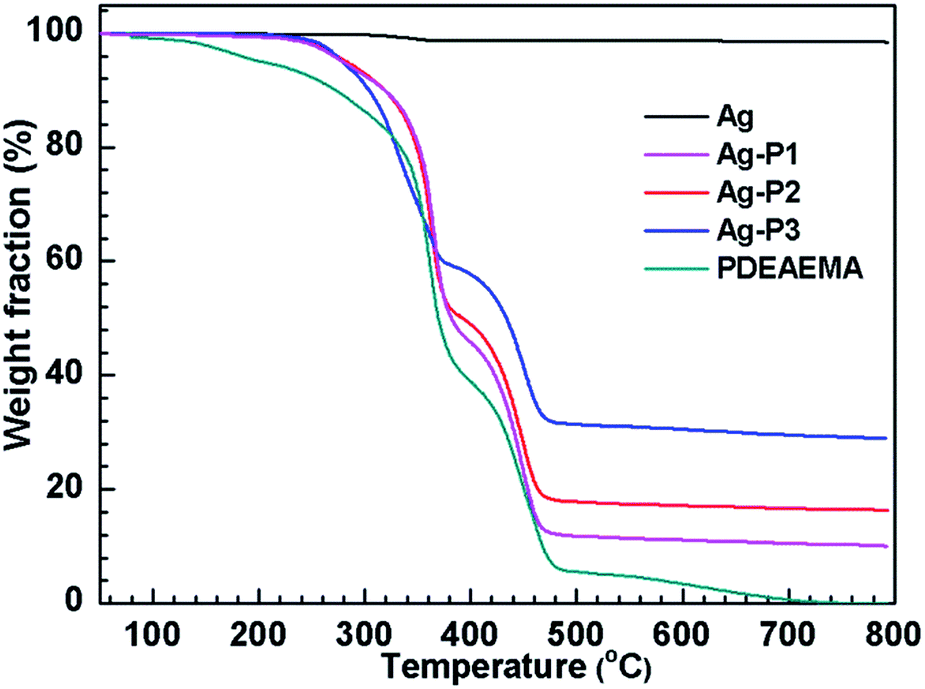 | ||
| Fig. 3 TGA for Ag, Ag–P1, Ag–P2, Ag–P3 and PDEAEMA. | ||
In addition, the stability of AgNPs in aqueous environment is important for their application.55,56 To detect the stability of the PDEAEMA-protected AgNPs in water, we measured the absorption spectra of one of the AgNPs hybrids systems (Ag–P1) with the same concentration at different times. As shown from Fig. S5,† there is no obvious difference in the shape, position, and symmetry of the absorption peak during 12 months, indicative of the long-term stability of the hybrid.
CO2-switchable behavior of AgNPs hybrids
:1, v/v) in the presence and in the absence of CO2, respectively. In the former case, the dispersion separates into two phases; the upper layer is brown while the lower one is transparent, indicating that Ag–P1 reside in the top water layer (inset, Fig. 4a). However, when N2 is bubbled into the mixing solution to expel CO2, the upper water phase turns into transparent, suggesting that the AgNPs hybrids move from the aqueous phase to the organic one, i.e., the Ag–P1 hybrids transform from hydrophilic to hydrophobic, and thus dissolve in the organic layer (inset, Fig. 4b).
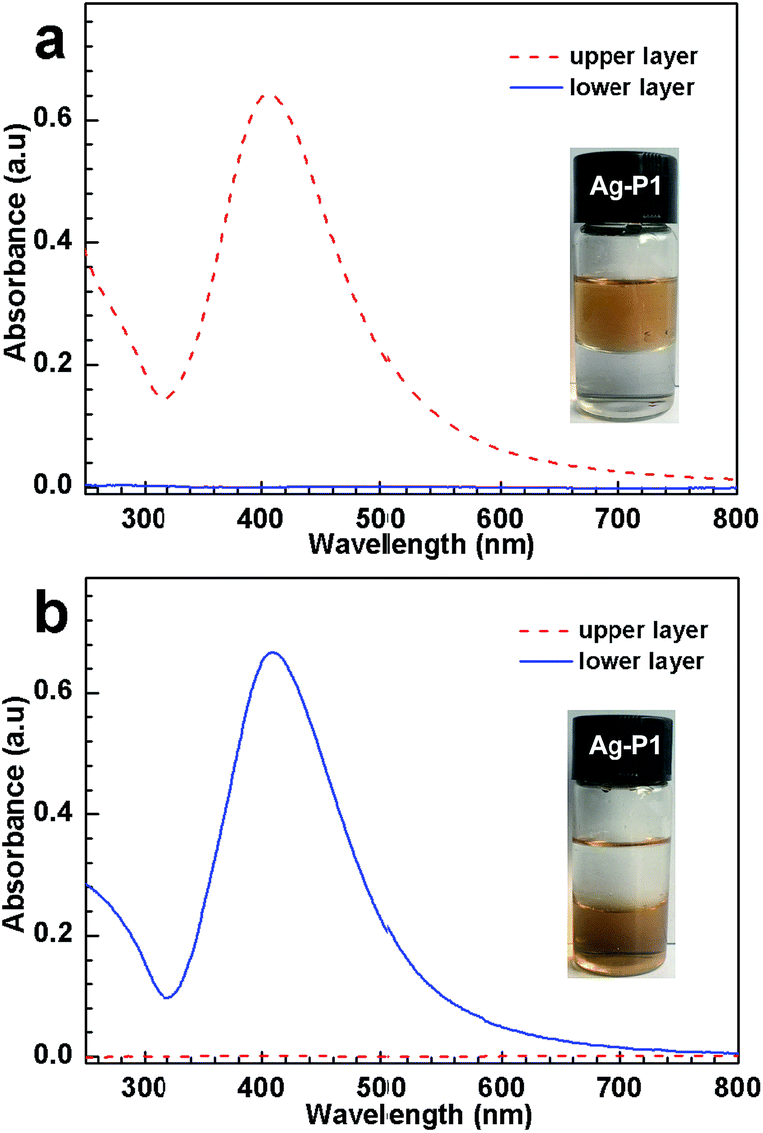 | ||
| Fig. 4 Hydrophobic–hydrophilic transition of Ag–P1 hybrids (0.08 mg mL−1, 25 °C) in DCM/water (1:1, v/v) monitored by the UV-vis spectrum: (a) treated with CO2 and (b) bubbling N2. | ||
During this process, UV-vis spectroscopy was used to investigate the dispersion state in the mixed solvent. As the hybrids was treated with CO2, the upper aqueous solution exhibited SPR at 408 nm (Fig. 4a), indicating that Ag–P1 hybrids were dispersed in the water phase. In contrast, the lower DCM solution shows no obvious signals in the range of 250–800 nm, suggesting no Ag–P1 presented in lower organic layer. When N2 was bubbled into the biphasic solution, the UV-vis spectra of the two phases was reversed, that is, the lower DCM phase exhibits strong SPR peak (Fig. 4b), while no signals appeared in the upper aqueous solution. These UV-vis spectra clearly show that Ag–P1 experiences a hydrophobic–hydrophilic transition. Based on these macroscopic results, it is noteworthy that Ag–P1 can switch between aqueous media and organic solvents, which is convenient for separation/collection.
To further reveal the dispersion state of the hybrids, UV-vis spectroscopy, was employed to monitor the variation of absorbance of hybrid suspension after bubbling and removing CO2, respectively. As shown in Fig. 5a, a strong SPR absorbance was observed after bubbling CO2. With bubbling N2, on the other hand, the absorbance gradually decreased, and concomitantly the SPR peak showed blue shift (from 408 nm to 425 nm), indicative of aggregation and precipitation.52,56 In addition, the FWHM values of the spectra increase with time of bubbling N2 (Fig. 5c), implying the formation of AgNPs aggregates with larger size and broad size distribution. In order to get more direct information on the aggregation state of AgNPs hybrids in water, TEM were performed. An obvious aggregation state of the AgNPs hybrids can be observed (Fig. S6†), which is in good agreement with the abovementioned results. When the dispersion was treated with CO2 again, the SPR peak and its FWHM reinstated (Fig. 5b and c), indicating that AgNPs hybrids were re-dispersed again.
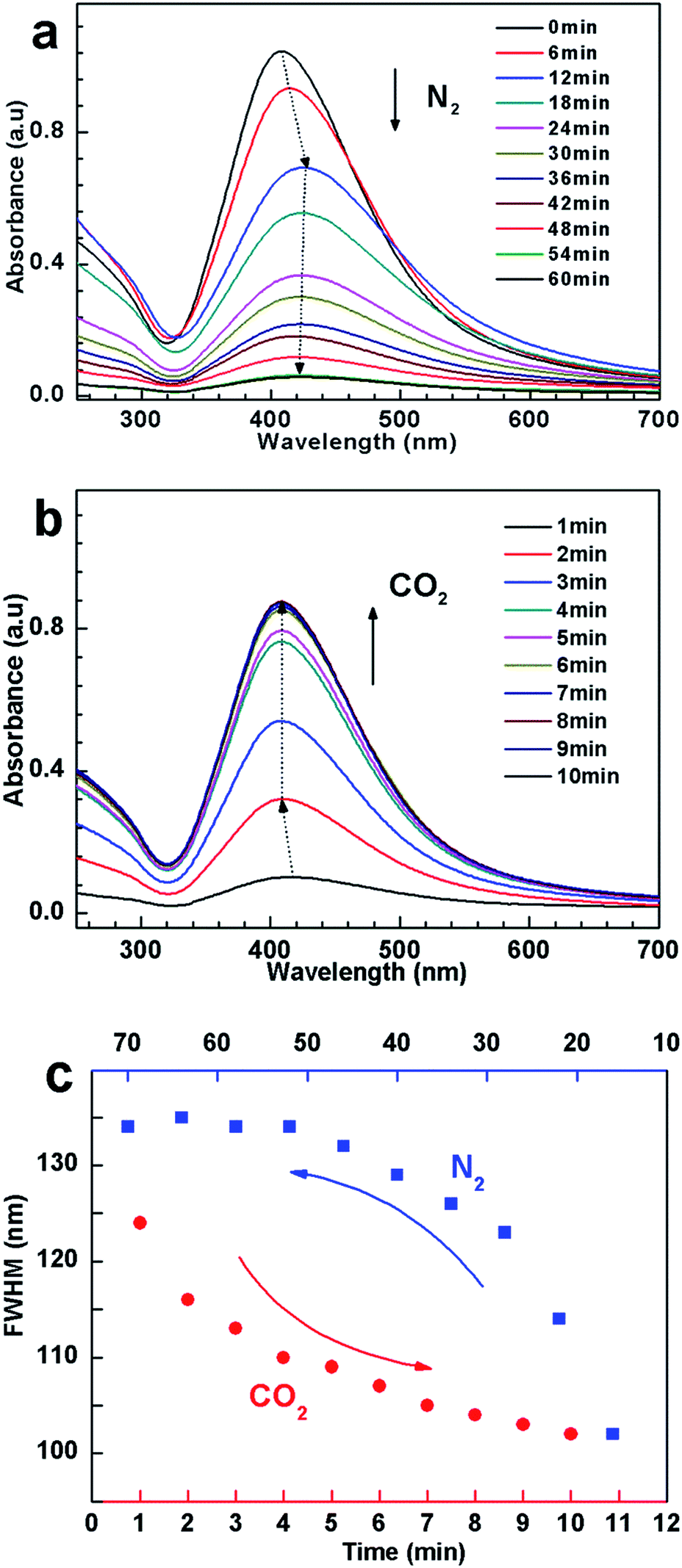 | ||
| Fig. 5 UV-vis spectra changes of 0.1 mg mL−1 Ag–P1 aqueous dispersion with (a) N2 bubbling (120 mL min−1) and (b) CO2 bubbling (100 mL min−1). (c) The cyclic changes of FWHM values with N2 and CO2 bubbling, which was calculated from UV-vis spectra. | ||
To elucidate the dispersed/aggregated transition, electrical conductivity measurements were performed to monitor the change of conductivity for the suspension when cyclically bubbling CO2 and N2 (Fig. 6a). When CO2 was introduced into dispersion, the conductivity sharply rises from about ca. 5.8 to ∼130 μS cm−1, with a net enhancement of about 124 μS cm−1, which must be due to the tertiary amine groups in PDEAEMA reacting with CO2 in water to form charged ammonium bicarbonate.37–42 When CO2 was displaced by N2, the conductivity recovered to its original value, and this reversible change in conductivity could be repeated several times, which amply demonstrate that the response of the suspension to CO2 was fully reversible and reproducible.34
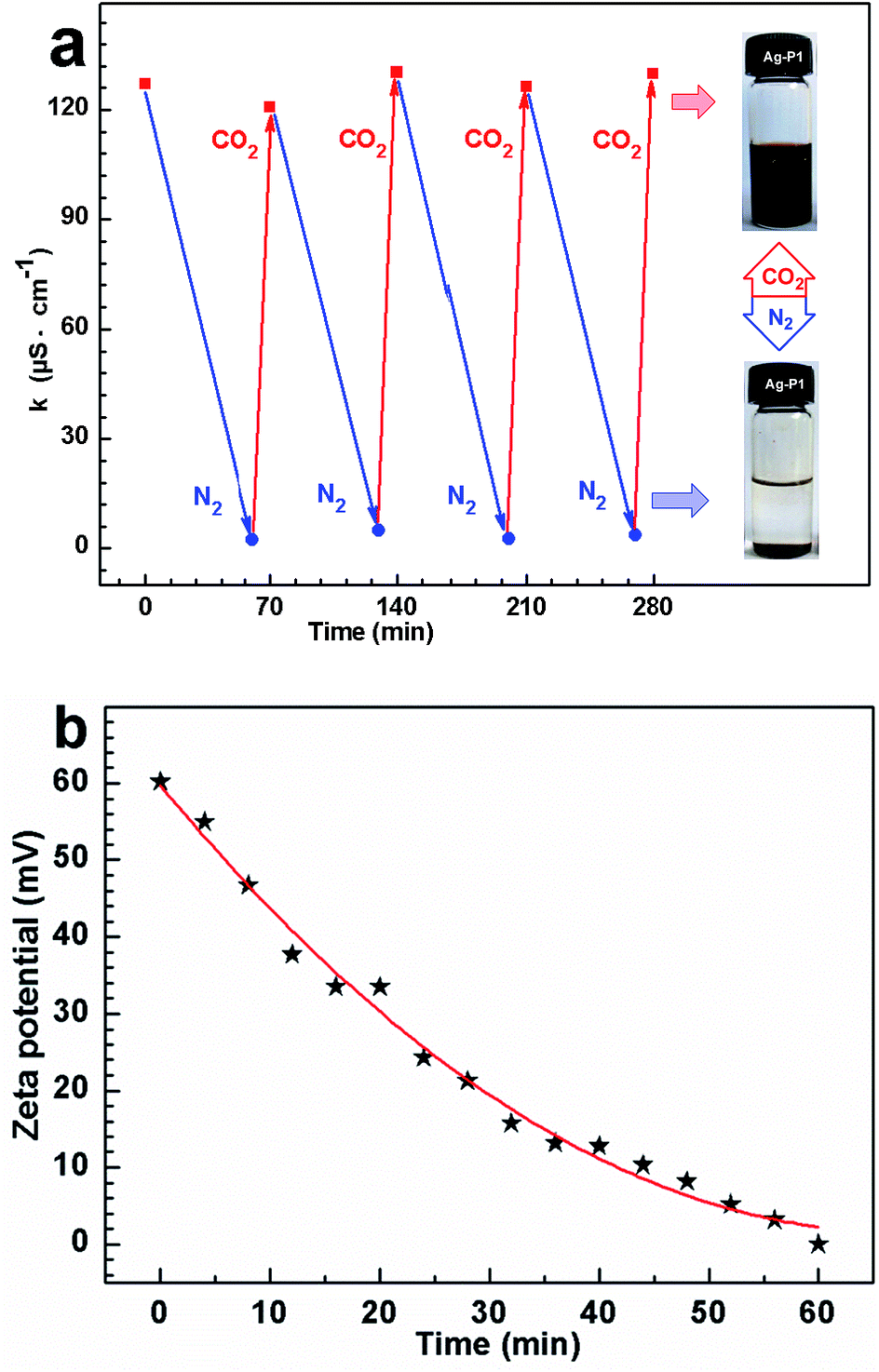 | ||
| Fig. 6 (a) Cyclic changes in conductivity of Ag–P1 aqueous dispersion measured at 25 °C with treatment of CO2 and N2, where the concentrations of Ag–P1 is 2.23 mg mL−1. (b) The zeta potential of Ag–P1 aqueous dispersion at different time after bubbling N2 at 25 °C. | ||
To further confirm that ionization happened on the cocoon of the hybrids, zeta potential of the silver colloidal solution has been measured. The zeta potential for the particle treated with CO2 reached +60.2 mV, as exhibited in Fig. 6b, supporting the formation of positive ammonium ions of the surface coated polymers. After removing CO2 by purging N2, the zeta potential decreases with increasing in the time of purging N2, and finally reduced to +0.66 mV (Fig. 6b), suggesting that positive ammonium ions of the polymer cocoon were mostly deprotonated due to the deportation of the CO2.
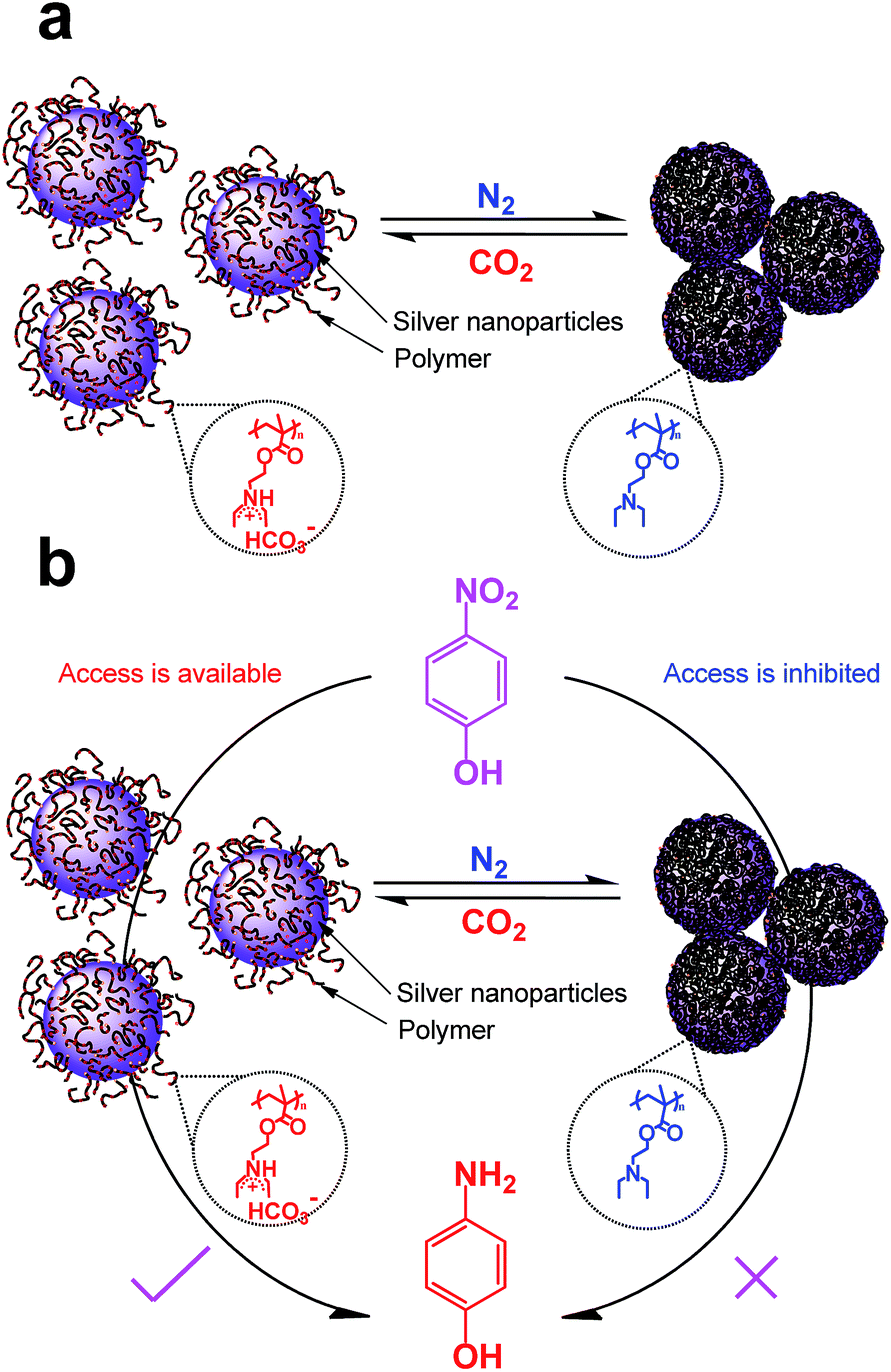 | ||
| Scheme 2 Schematic illustration of the AgNPs hybrids response to the stimulus of CO2 (a), and switch and tune the catalytic activity for reduction of 4-nitrophenol (b). | ||
The reversible dispersion/aggregation states of AgNPs hybrids controlled by CO2 in water could also be understood based on the CO2-responsive behaviour of PDEAEMA. As shown in Scheme 2a, the tertiary amine groups of PDEAEMA coated on AgNPs were protonated when reacted with CO2 in water, leading an extended conformation of the polymer. Thus the interchain electrostatic repulsion and the steric hindrance among the AgNPs protected by the charged PDEAEMA should allow the long-term dispersing in water. After the removal of CO2, the electrostatic repulsion of PDEAEMA disappears owing to an opposite deprotonation effect,36,37 thus diminishing the polymer–polymer electrostatic repulsions and increasing the interaction among polymer chains,34 resulting in larger particles, and precipitate from aqueous solution.
Application in catalysis
As the AgNPs hybrids exhibit sensitive to CO2-switchable behaviour, it is interesting to see if the hybrids can be used as a gas-tunable catalysis. Here, in order to evaluate the catalytic activity of the as-obtained AgNPs hybrids, the reduction of 4-nitrophenol, to 4-aminophenol by NaBH4 was used as model reaction. Because 4-nitrophenol is one of the most refractory pollutants in industrial wastewaters while 4-aminophenol is a commercially important intermediate for the manufacture of analgesic and antipyretic drugs.59 In addition, the reactant 4-nitrophenol could transfer into 4-nitrophenolate ion at high pH and shows characteristic peak at 400 nm; and the product 4-aminophenol gives a typical absorption at 300 nm, which is easily monitored by UV-vis measurement.9,55,59Fig. S7a† shows the time-dependent UV spectra for the reduction of 4-nitrophenol in presence of CO2-treated AgNPs hybrids. One can find that the peak height at 400 nm exhibit a slight decreases within 40 min (Fig. S7a†), indicating a very low conversion of 4-nitrophenol. Based on the absorption at 400 nm values, linear correlation between ln(C/C0) (C is the concentration at a certain reaction time and C0 is the initial concentration of 4-nitrophenolate ions) versus reaction time was obtained (Fig. S7a†), indicating that such a catalytic reduction follows a pseudo-first-order law. The apparent reaction rate constant (kapp) was calculated from linear and is only 1.15 × 10−5 s−1. This result confronts earlier observations that CO2-bubbled AuNPs protected with PDEAEMA still exhibits a high catalytic activity for 4-nitrophenol.41,42 This may arise from that the density of PDEAEMA grafted on AgNPs hybrids, ca. 2000 polymer chains in one particle as aforedescribed, is higher than that of reported ones, though the previous papers did not provide this data.41,42 When CO2-treated AgNPs hybrids was introduce into the solution, the charged ammonium bicarbonates would be deprotonated because the basic NaBH4 reacted with carbonic acid formed by CO2 and water (i.e., NaBH4 + H+ + 3H2O = Na+ + 4H2↑ + H3BO3). Thus PDEAEMA cocoon became hydrophobic and wrapped AgNPs tightly, which inhibited access to the catalytic sites of AgNPs (Scheme 2b).
To open the access for 4-nitrophenol, we tried to bubble CO2 into the mixing solution. Firstly, we wonder whether 4-nitrophenol could be reduced by CO2. CO2 was bubbled into the mixing solution of 4-nitrophenol and NaBH4 (without AgNPs hybrids). After bubbling CO2 for 50 min, the peak height at 400 nm has no change (Fig. S8, ESI†), suggesting that 4-nitrophenol could not be reduced by CO2 in absent of AgNPs hybrids. In contrast, CO2 was purged into the mixture in presence of Ag–P1 hybrids. When the flow rate of CO2 is 10 mL min−1, it was found that the absorption at 400 nm decreased slowly within the first 18 min, and then dropped fast to nearly zero (Fig. S7b†). Meanwhile, the peak of 4-aminophenol at 300 nm was observed,42,55 implying that 4-nitrophenol was reduced to 4-aminophenol. It clearly shows an induction time before the conversion of reactants into products takes place (Fig. 7a and S7b†). After deducting the induction time (ti), the kapp is 1.57 × 10−3 s−1 and bigger than that of without bubbling CO2. This result suggests that access to the encapsulated AgNPs was opened, since polymer chains contacted with CO2 and extended in solution. When the flow rate increased, the induction time decreased and disappeared at 25 mL min−1 (Fig. 7a). And concomitantly the kapp increased with the flow rate, and reach to ca. 3.6 × 10−3 s−1 at 50 mL min−1 (Fig. S7h†), which is close to gold nanoparticle reaction rate constant.42 The kapp as function of CO2 flow rate was presented in Fig. 7b. Two linear regimes in the kapp curve are found: a monotonous linear increase in kapp is evidenced while CO2 flow within 30 mL min−1, after which it is slight change.
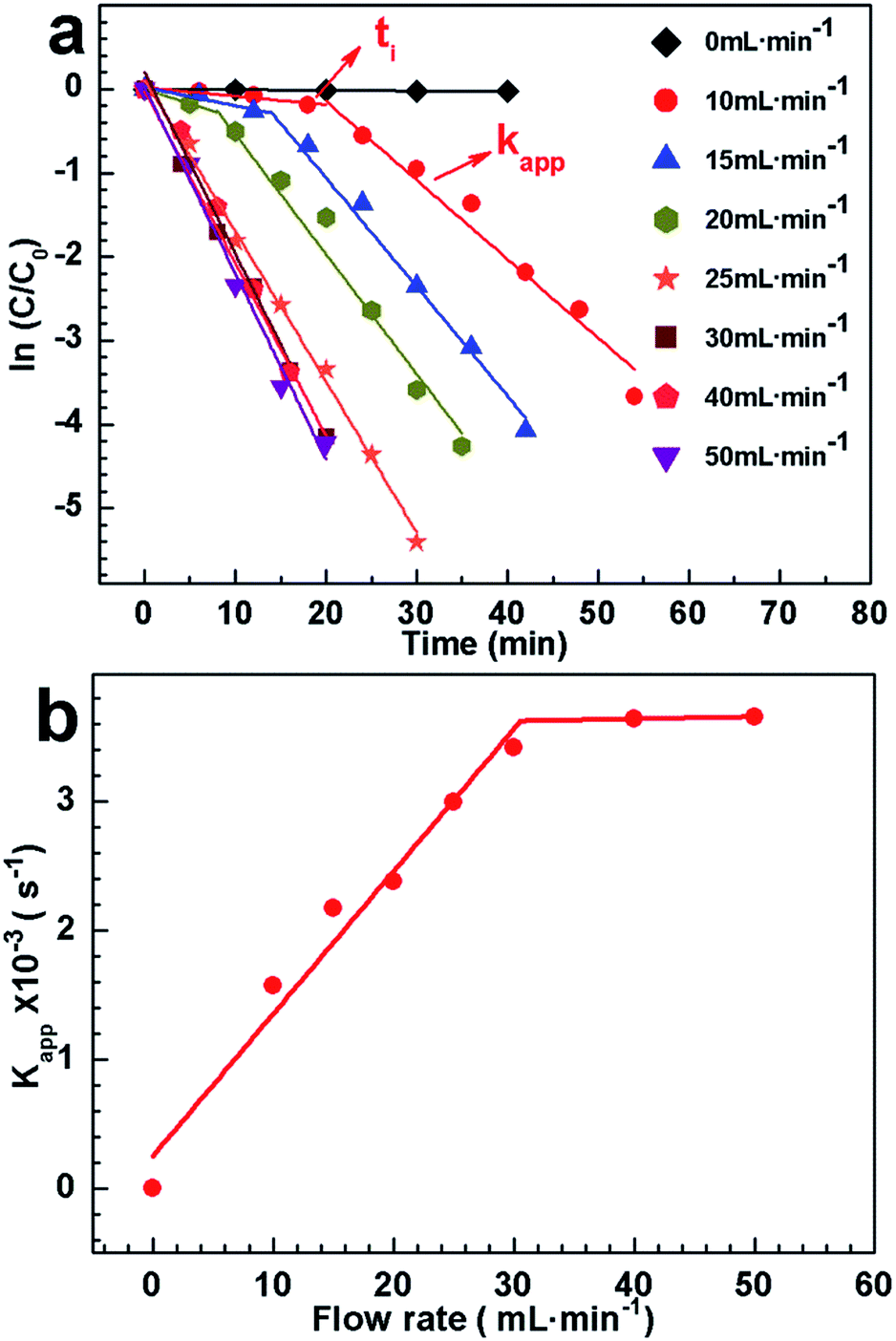 | ||
| Fig. 7 (a) The plots of ln(C/C0) versus the time t at different flow rate of CO2 stimuli. (b) The variation of the apparent reaction rate constant (kapp) at different CO2 flow rate. The concentrations of 4-NP and Ag–P1 are 0.0139 mg mL−1 and 2 × 10−3 mg mL−1, respectively. | ||
To elucidate this variation, pH value of the solution under bubbling CO2 was tested, since the degree of protonation increases with decreasing of pH.37 As exhibited in Fig. S9,† the pH decreased with bubbling CO2 and maintained at ca. 7 for catalysis. Note that if the pH was less than 6.7, the peak at 317 nm ascribed to 4-nitrophenol would appear, which would affect the catalytic reaction.60 During the reaction, additional NaBH4 should be added to compensate that of consumed by CO2 and keep pH stable. Besides, it clearly shows that the time for decreasing pH to ca. 7 increases with low flow rate, which may cause the induction time. As there was no obvious difference on pH for catalysis at different flow rate, the hybrid should show a similar protonation and has similar kapp. A possible explanation for increasing kapp is that the frequency for PDEAEMA cocoon contacting with CO2 increased under the high CO2 flow rate, which is favourable to opening the access to AgNPs. Such preliminary findings, particularly the linearity found in the flow rate below 30 mL min−1, imply that the catalytic activity of Ag–P1 hybrids can be switched and monotonously tuned by varying the flow rate of CO2 purged into the reaction system.
Conclusions
In summary, we have prepared CO2-switchable AgNPs hybrids by one-pot reducing AgNO3 and trithioester terminated PDEAEMA with NaBH4. Apart from the long-term stability, the size and size distribution of AgNPs hybrids can be easily modulated by varying the molar ratio of polymers to AgNO3. The hybrids not only exhibit hydrophobic–hydrophilic transitions in immiscible mixed solvents, but also undergo a switchable dispersion/aggregation states upon alternate treated with CO2 and N2. In addition, we have demonstrated that the catalytic activity of the hybrids for the reduction of 4-nitrophenol can be switched and monotonously tuned by varying the flow rate of CO2 purged into the reaction system, which may open a new avenue for tailoring the catalytic activity of metal nanoparticles toward a given reaction. The strategies described in this work can also be used to functionalize other nanoparticles in a quick and easy way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported by Open Fund (PLN1508) of State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation (Southwest Petroleum University) and National Natural Science Foundation of China (51563009, 21465011, 21464006, 21464005). W. M. thanks the financial support from the Innovation Fund Designated for Graduate Students of Jiangxi Province (YC2016-S265).Notes and references
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- H. Song, Acc. Chem. Res., 2015, 48, 491–499 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- Y. Xu, L. Chen, X. Wang, W. Yao and Q. Zhang, Nanoscale, 2015, 7, 10559–10583 RSC.
- J. Zhou, J. Ralston, R. Sedev and D. A. Beattie, J. Colloid Interface Sci., 2009, 331, 251–262 CrossRef CAS PubMed.
- A. Shahzad, W.-S. Kim and T. Yu, RSC Adv., 2015, 5, 28652–28661 RSC.
- L. Xie, M. Chen and L. Wu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4919–4926 CrossRef CAS.
- J.-T. Zhang, G. Wei, T. F. Keller, H. Gallagher, C. Stötzel, F. A. Müller, M. Gottschaldt, U. S. Schubert and K. D. Jandt, Macromol. Mater. Eng., 2010, 295, 1049–1057 CrossRef CAS.
- S. Li, D. Lin, J. Zhou and L. Zha, J. Phys. Chem. C, 2016, 7, 343–349 Search PubMed.
- K. J. Rao and S. Paria, ACS Sustainable Chem. Eng., 2015, 3, 483–491 CrossRef CAS.
- K. Layek, M. L. Kantam, M. Shirai, D. Nishio-Hamane, T. Sasaki and H. Maheswaran, Green Chem., 2012, 14, 3164–3174 RSC.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- T. Yao, T. Cui, H. Wang, L. Xu, F. Cui and J. Wu, Nanoscale, 2014, 6, 7666–7674 RSC.
- R. Rajesh, S. S. Kumar and R. Venkatesan, New J. Chem., 2014, 38, 1551–1558 RSC.
- E. Kumacheva, Nat. Mater., 2012, 11, 665–666 CrossRef CAS PubMed.
- S. Cao, J. Chen, Y. Ge, L. Fang, Y. Zhang and A. P. Turner, Chem. Commun., 2014, 50, 118–120 RSC.
- G. Marcelo, M. Lópezgonzález, F. Mendicuti, M. P. Tarazona and M. Valiente, Macromolecules, 2014, 47, 6028–6036 CrossRef CAS.
- J. Zhang, M. Zhang, K. Tang, F. Verpoort and T. Sun, Small, 2013, 10, 32–46 CrossRef PubMed.
- A. Döring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- Y. Zhou, M. Zhu and S. Li, J. Mater. Chem. A, 2014, 2, 6834–6839 CAS.
- D. A. Burnat, R. Kontic, L. Holzer, P. Steiger, D. Ferri and A. Heel, J. Mater. Chem. A, 2016, 4, 11939–11948 CAS.
- Z. Liu, X. Tong, J. Liu and S. Xue, Catal. Sci. Technol., 2016, 6, 1214–1221 CAS.
- R. Begum, K. Naseem and Z. H. Farooqi, J. Sol-Gel Sci. Technol., 2016, 77, 497–515 CrossRef CAS.
- X. Jiang, D. A. Xiong, Y. An, P. Zheng, W. Zhang and L. Shi, J. Polym. Sci., Part A: Polym. Chem., 2010, 45, 2812–2819 CrossRef.
- J. Bigot, B. Charleux, G. Cooke, D. Fournier and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- J. Zhang, S. Xu and E. Kumacheva, Adv. Mater., 2005, 17, 2336–2340 CrossRef CAS.
- Z. Chen, Z. M. Cui, C. Y. Cao, W. D. He, L. Jiang and W. G. Song, Langmuir, 2012, 28, 13452–13458 CrossRef CAS PubMed.
- S. Li, G. Yi and A. P. F. Turner, Adv. Funct. Mater., 2011, 21, 1194–1200 CrossRef CAS.
- M. Z. And and W. Zhang, J. Phys. Chem. C, 2008, 112, 6245–6252 Search PubMed.
- Z. Chen, L. Xu, Y. Liang and M. Zhao, Adv. Mater., 2010, 22, 1488–1492 CrossRef CAS PubMed.
- C. Xiao, S. Chen, L. Zhang, S. Zhou and W. Wu, Chem. Commun., 2012, 48, 11751–11753 RSC.
- X. Chen, Y. An, D. Zhao, Z. He, Y. Zhang, J. Cheng and L. Shi, Langmuir, 2008, 24, 8198–8204 CrossRef CAS PubMed.
- Z. Guo, Y. Feng, S. He, M. Qu, H. Chen, H. Liu, Y. Wu and Y. Wang, Adv. Mater., 2013, 25, 584–590 CrossRef CAS PubMed.
- Z. Guo, Y. Feng, Y. Wang, J. Wang, Y. Wu and Y. Zhang, Chem. Commun., 2011, 47, 9348–9350 RSC.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed. Engl., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2013, 1, 57–61 CrossRef.
- P. G. Jessop, S. M. Mercer and D. J. Heldebrant, Energy Environ. Sci., 2012, 5, 7240–7253 CAS.
- H. Liu, Z. Guo, S. He, H. Yin, C. Fei and Y. Feng, Polym. Chem., 2014, 5, 4756–4763 RSC.
- M. Ying, K. Promthaveepong and L. Nan, Anal. Chem., 2016, 88, 8289–8293 CrossRef PubMed.
- J. Zhang, D. Han, H. Zhang, M. Chaker, Y. Zhao and D. Ma, Chem. Commun., 2012, 48, 11510–11512 RSC.
- A. Feng, Y. Wang, L. Peng, X. Wang and J. Yuan, RSC Adv., 2016, 6, 97030–97035 RSC.
- H. Song, R. M. Rioux, J. D. Hoefelmeyer, R. Komor, K. Niesz, M. Grass, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3027–3037 CrossRef CAS PubMed.
- G. J. Leong, A. Ebnonnasir, M. C. Schulze, M. B. Strand, C. Ngo, D. Maloney, S. L. Frisco, H. N. Dinh, B. Pivovar and G. H. Gilmer, Nanoscale, 2014, 6, 11364–11371 RSC.
- E. Shahbazali, V. Hessel, T. Noël and Q. Wang, Nanotechnol. Rev., 2014, 3, 65–86 CAS.
- Y. Wu, S. He, Z. Guo and Y. Feng, Polym. Sci., 2013, 55, 634–642 CAS.
- Y. Sun, Y. Liu, G. Zhao, X. Zhou, J. Gao and Q. Zhang, J. Mater. Sci., 2008, 43, 4625–4630 CrossRef CAS.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499–3504 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- R. Brause, H. Möltgen and K. Kleinermanns, Appl. Phys. B, 2002, 75, 711–716 CrossRef CAS.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC.
- D.-H. Chen and Y.-W. Huang, J. Colloid Interface Sci., 2002, 255, 299–302 CrossRef CAS PubMed.
- S. He, H. Chen, Z. Guo, B. Wang, C. Tang and Y. Feng, Colloids Surf., A, 2013, 429, 98–105 CrossRef CAS.
- S. He, J. Yao, P. Jiang, D. Shi, H. Zhang, S. Xie, A. S. Pang and H. Gao, Langmuir, 2001, 17, 1571–1575 CrossRef CAS.
- J. Yang, H. Yin, J. Jia and Y. Wei, Langmuir, 2011, 27, 5047–5053 CrossRef CAS PubMed.
- R. Shankar, L. Groven, A. Amert, K. W. Whites and J. J. Kellar, J. Mater. Chem., 2011, 21, 10871–10877 RSC.
- Y. Lu, Y. Mei, M. Ballauff and M. Drechsler, J. Phys. Chem. B, 2006, 110, 3930–3937 CrossRef CAS PubMed.
- L. A. Shah, A. Haleem, M. Sayed and M. Siddiq, J. Environ. Chem. Eng., 2016, 4, 3492–3497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and GPC of polymers, IR spectra, XRD data and TEM images of hybrids, the data of catalysis and pH variation during the catalytic process and some calculations. See DOI: 10.1039/c7ra09233d |
| This journal is © The Royal Society of Chemistry 2017 |