
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc bimetallics supported by a xanthene-bridged dinucleating ligand: synthesis, characterization, and lactide polymerization studies†
Thilini S. Hollingswortha,
Ryan L. Hollingswortha,
Tomer Rosen*b and
Stanislav Groysman *a
*a
aDepartment of Chemistry, Wayne State University, 5101 Cass Ave, Detroit, MI 48202, USA. E-mail: groysman@chem.wayne.edu
bSchool of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel-Aviv University, Tel-Aviv 69978, Israel. E-mail: tomer.rsn@gmail.com
First published on 29th August 2017
Abstract
A new, potentially dinucleating xanthene-bridged bis(iminophenolate) ligand L (L = 6,6′-((1E,1′E)-((2,7-di-tert-butyl-9,9-dimethyl-9H-xanthene-4,5-diyl)bis(azanylylidene))bis(methanylylidene))bis(2,4-di-tert-butylphenol)) has been synthesized and its coordination chemistry with zinc precursors featuring alkoxide, chloride, and ethyl leaving groups has been investigated. The reaction of a zinc precursor bearing two bulky alkoxides, Zn(Cl)(μ2-OR)2Li(THF) (OR = di-tert-butyl-phenylmethoxide), formed a mononuclear complex Zn(L) that was isolated as an H-bond adduct with HOR, Zn(L)·HOR. In contrast, the reaction of L (or its lithium salt) with diethylzinc (or zinc chloride) led to the formation of the corresponding dinuclear complexes Zn2(L)(Et)2 and Zn2(L)(μ2-Cl)4Li2(OEt2)2. X-ray crystallography revealed syn-parallel geometry for Zn2(L)(Et)2 (Zn⋯Zn distance of 4.5 Å) and anti-parallel geometry for Zn2(L)(μ2-Cl)4Li2(OEt2)2 (Zn⋯Zn distance of 6.7 Å). Zn2(L)(Et)2 was found to be somewhat unstable, demonstrating decomposition into Zn(L) and ZnEt2; this decomposition can be reversed by the addition of excess ZnEt2. Treatment of Zn2(L)(Et)2 with benzyl alcohol (BnOH) in deuterated benzene, toluene, or dichloromethane resulted in the formation of Zn2(L)(OBn)2, which was characterized by 1H and 13C NMR spectroscopy. Zn2(L)(OBn)2 was found to be active in the ring-opening polymerization of rac-lactide to afford heterotactically inclined PLA.
Introduction
Bimetallic cooperativity is a common theme in biological catalysis,1 and is currently an active area of research in chemical catalysis.2,3 Among other fields, bimetallic cooperativity has been postulated in polymerization of lactide and other cyclic esters. Lactide is a cyclic diester that is derived from renewable sources.4 The corresponding polymer, poly(lactic acid), features many characteristic properties similar to those of polyolefins, but offers an important advantage of biodegradability.5 Lactide polymerization is generally catalyzed by strongly Lewis-acidic metals, in particular zinc.6,7 The structure of the metal catalyst affects the properties of the resulting poly(lactic acid), including tacticity, molecular weight, and molecular weight distribution. Several recent reports demonstrated advantageous reactivity of predesigned bimetallics8–16 that may exhibit higher catalytic activity, higher molecular weights, or improved tacticity, in comparison with related mononuclear species. The observed synergism of predesigned bimetallics is corroborated by the interesting observation that bimetallic alkoxide-bridged active sites may be preferential even for precursors lacking dinucleating ligands.17 Thus, Mehrkhodavandi and coworkers have recently demonstrated that a di-indium complex, bridged by a monodentate alkoxide (Fig. 1), retains its dinuclear structure in solution and functions as a bimetallic polymerization catalyst.17 | ||
| Fig. 1 Selected recently reported bimetallic pre-catalysts for lactide polymerization.9–11,17 | ||
As the reactivity of bimetallic catalyst depends on the metal–metal separation, there is a growing interest in bimetallic catalysts where a fixed metal–metal distance is enforced by a sufficiently rigid dinucleating ligand.2 There is a significant number of bimetallic lactide polymerization catalysts stabilized by dinucleating ligands, including Salen derivatives,13 2,6-diaminophenolate derivatives,8,9b,16 2,2′-phenolate derivatives,10 and other systems (for selected examples see Fig. 1). Our group investigates cooperative reactivity of bimetallics supported by dinucleating ligands exhibiting various degrees of rigidity.18 We, and others, have recently begun investigating potentially dinucleating ligands based on 4,5-diamino xanthene linkers.19,20 Xanthene linker positions two metals sufficiently close to each other to allow cooperative reactivity. Yet, the open-chain nature of xanthene-bridged bis(iminopyridine) ligands, coupled with single C4–N and C5–N bonds, allow the two chelating units sufficient freedom of movement versus each other, which may be beneficial if varying metal–metal distances are required to satisfy different bimetallic reaction intermediates. Following our recent report on cooperative alkyne cyclotrimerization catalyzed by xanthene-based bis(iminopyridine)dinickel complex,19b we turned to investigate the reactivity of xanthene-based di-zinc complexes in lactide polymerization. We hypothesized that neutral bis(iminopyridine) ligands would not be suitable for the relatively hard dicationic zinc(II), and thus decided to replace neutral iminopyridine with monoanionic iminophenolate. The presence of a single monoanionic chelate at each zinc centre enables coordination of an additional monoanionic monodentate ligand (alkoxide or amide), as required for an active lactide polymerization catalyst. Herein we describe coordination chemistry of the new xanthene-bridged bis(iminophenolate) ligand with zinc, and present the reactivity of the resulting bimetallic complexes in lactide polymerization.
Results and discussion
Ligand synthesis
Dinucleating ligand L was obtained by condensation of previously reported 2,7-di-tert-butyl-9,9-dimethyl-4,5-diaminoxanthene20 with 3,5-di-tert-butyl-2-hydroxybenzaldehyde in methanol. Pure ligand was obtained by recrystallization from benzene/methanol as bright yellow crystals in 76% yield. NMR data for L is consistent with the single species in solution featuring effective C2v symmetry as indicated by one peak for both xanthene methyl groups, and one peak for both xanthene tert-butyl peaks. Interestingly, two different types of crystals were obtained by recrystallization of analytically- and NMR-pure sample of L from hexanes (see ESI for details†). The major difference between the structures (Lsyn and Lanti) is in the relative disposition of the chelating units – iminophenolates. In Lsyn, the phenolates are on the same side of xanthene linker, which leads to their syn-parallel arrangement. In Lanti, the phenolates are on two different sides of the xanthene linker, in the anti-parallel arrangement. One noteworthy feature of the structures is the flexibility of the xanthene linker. While the linker appears to be perfectly flat in Lsyn, it is slightly puckered in Lanti.Complex synthesis and characterization
Metal-alkoxide complexes constitute the most commonly used form of precatalysts for lactide polymerization. We have previously described series of divalent 3d metal complexes of the M(OR)2(THF)2 form (OR = OCtBu2Ph), whose monomeric structure was made possible by the use of excessively bulky alkoxides.21,22 One could postulate that a zinc complex of similar form could provide an ideal precursor for the reaction with L, as: (1) phenols are significantly more acidic than alcohols; (2) a bulky alkoxide should render zinc centres in a hypothetical “Zn2(L)(OR)2” complex coordinatively unsaturated and therefore highly reactive toward monomer insertion. Thus, our initial route towards bimetallic zinc complexes with L targeted synthesis of a “Zn(OR)2(THF)2” species first. The addition of two equivalents of ROH22d to diethylzinc failed to produce the expected zinc-alkoxide product. Intriguingly, no reaction was observed by 1H NMR spectroscopy, suggesting that our alkoxide ligand may be more basic than alkyl (ethyl) ligand in the zinc coordination environment. Salt metathesis reaction between zinc chloride and two equivalents of ROLi produced Zn(Cl)(μ2-OR)2Li(THF) (1), which was isolated as colourless crystals from hexanes in 75% yield. The synthesis and structure of 1 are given in Fig. 2, along with the corresponding bond distances and angles. 1 demonstrates distorted trigonal planar zinc centre ligated by two OR ligands and one chloride.23 Notably, the chloride is terminal while the alkoxides are bridging. In contrast, in the corresponding nickel(II) structure (Ni(OR)(μ2-Cl)(μ2-OR)Li(THF)2), the chloride was bridging while one of the alkoxides was terminal.24 The bridging nature of both alkoxides in Zn(Cl)(μ2-OR)2Li(THF) supports the notion of the increased alkoxide basicity when coordinated to zinc.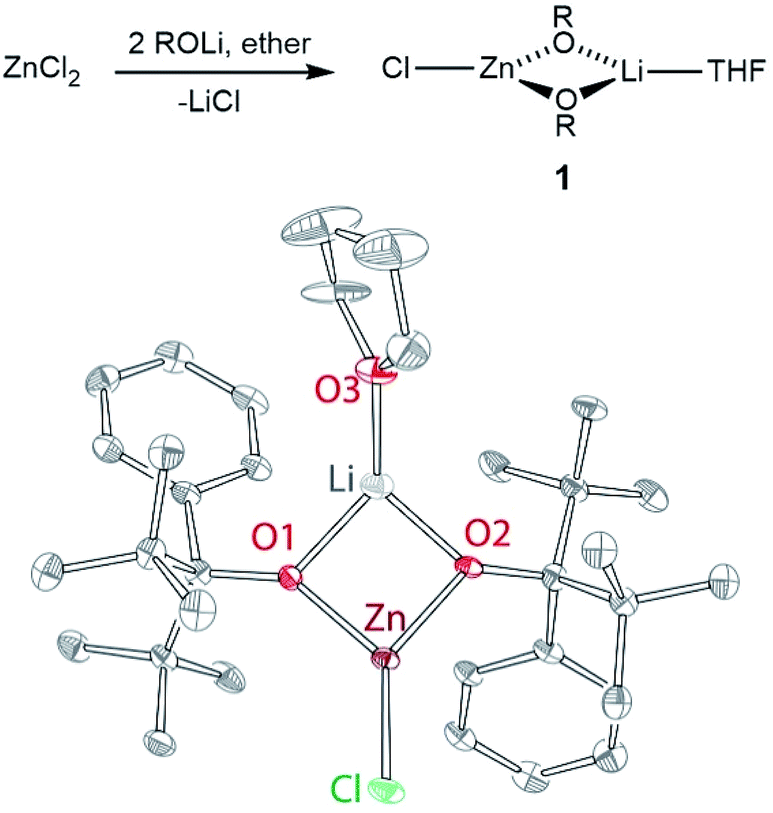 | ||
| Fig. 2 Top: synthesis of compound 1; OR = OCtBu2Ph. Bottom: X-ray structure of 1, 50% probability ellipsoids. Selected bond distances (Å) and angles (°): Zn Cl 2.155(1), Zn O1 1.902(3), Zn O2 1.914(3), O1 Zn Cl 130.5(1), O2 Zn Cl 137.7(1), O1 Zn O2 89.6(1). | ||
To abstract LiCl from 1, it was treated with TlPF6 or AgPF6. In both cases, an immediate reaction was observed, as indicated by the formation of TlCl (or AgCl) precipitate. However, while 1H NMR spectrum of the isolated product suggested formation of “Zn(OR)2” species, it was also accompanied by the formation of free alcohol, ROH. Our numerous attempts to isolate pure “Zn(OR)2” by recrystallization led to product mixtures containing varying amounts of the protonated ligand ROH. We note that while this behaviour stands in contrast to the Cr(II), Mn(II), Fe(II), and Co(II), for which M(OR)2(THF)2 could be isolated in good yields following similar protocols,24,22a Ni(II) and Cu(II) also failed to lead to isolable M(OR)2(THF)2 complexes.24
As we could not isolate pure “Zn(OR)2”, we investigated the reactivity of L with two equivalents of complex 1, or with “Zn(OR)2” prepared in situ. Slow addition of L to the cold (−35 °C) THF solution containing two equivalents of complex 1 led to the clean formation of mononuclear complex 2 (Scheme 1). Similarly, addition of L to the mixture of 1 and two equivalents of TlPF6 led to the formation of complex 2. It is hypothesized that the inability of L to function as a dinucleating ligand in this case results from the basicity of zinc-alkoxide precursors. It is also possible that the steric bulk of the Zn-OR fragment, occupying one of the L sites, prevents the reaction of the second site with another equivalent of 1, triggering instead intramolecular protonolysis. In support of this hypothesis, complex 2 was obtained as an ROH adduct, 2·HOR, both in solution and in solid state. Solid-state structure of 2 (Fig. 3) indicates hydrogen bonding between one of the L phenolates and the hydrogen atom of ROH (2.13 Å).
 | ||
| Scheme 1 Coordination chemistry of L with zinc described in this manuscript. Complex in square brackets (6) was not characterized by X-ray crystallography, and its structure is proposed based on NMR spectroscopy. | ||
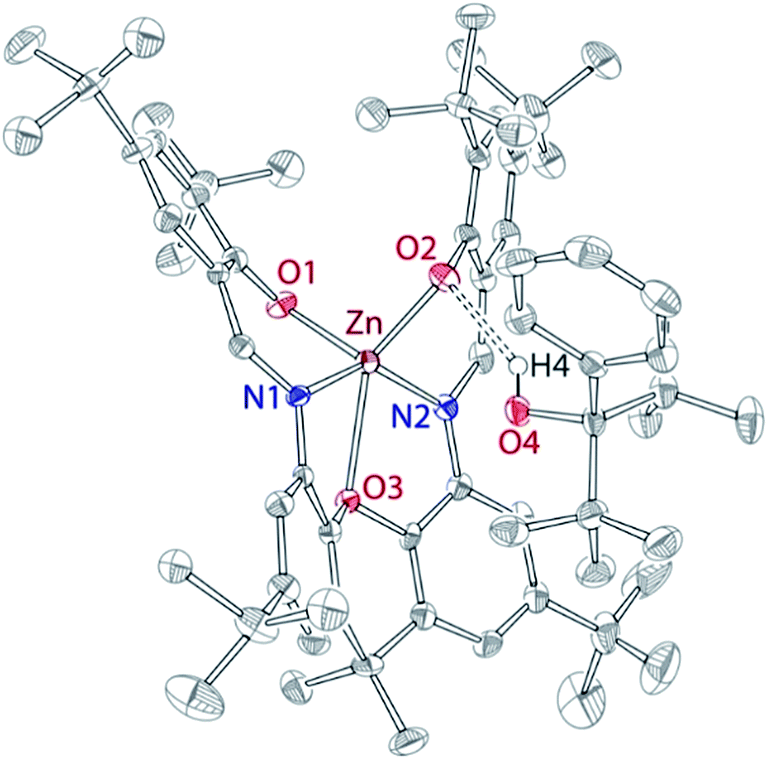 | ||
| Fig. 3 X-ray structure of 2·HOR, 50% probability ellipsoids. H atoms (except for H4), co-crystallized solvent, and alternative conformation of tBu groups are omitted for clarity. Selected bond distances (Å): Zn O1 1.938(3), Zn O2 1.979(3), Zn N1 2.007(3), Zn N2 2.037(3), Zn O3 2.396(2), O2⋯H4 2.132. | ||
As the direct reaction of a zinc alkoxide precursor did not yield the desired dinuclear bis(alkoxide) complex, salt metathesis route of deprotonated L was pursued. Treatment of L with approximately two equivalents of tBuOLi produced compound 3, which was obtained as yellow microcrystalline solid by recrystallization from hexanes. X-ray quality crystals of 3 were obtained from ether, and the solid-state structure is presented in Fig. 4. Compound 3 is a dilithium salt of L that incorporates an additional equivalent of LiOtBu to form the [Li3O3] cluster, in which lithiums are bridged by the μ3-OtBu ligand. In the solid-state structure, all lithium sites are different, two being tetra-coordinate and the third tri-coordinate (Fig. 4). One of the tetra-coordinate lithium sites is ligated by an ether molecule. The 1H NMR spectrum of 3 (obtained from hexanes) is in general agreement with this structure, with the major difference being coordinated THF in place of ether observed in the solid-state structure. A singlet for the [OtBu] group is observed at 1.34 ppm. The two iminophenoxide chelates are different in solid state but equilibrate in solution, giving rise to five different aromatic signals, two different signals for the phenoxide tBu groups, and one signal for the xanthene tBu groups. In contrast, xanthene methyls give rise to two different signals (1.77 and 1.58 ppm), breaking the effective xanthene plane symmetry, due to the Li3O3 cluster bridging the two iminophenoxide sites. The spectrum shows varying amounts of coordinated THF (one to two equivalents), whose labile coordination is likely responsible for the observed higher symmetry in solution. The attempts to use less than two equivalents of tBuOLi led to the isolation of the same structure, obtained in lower yield.
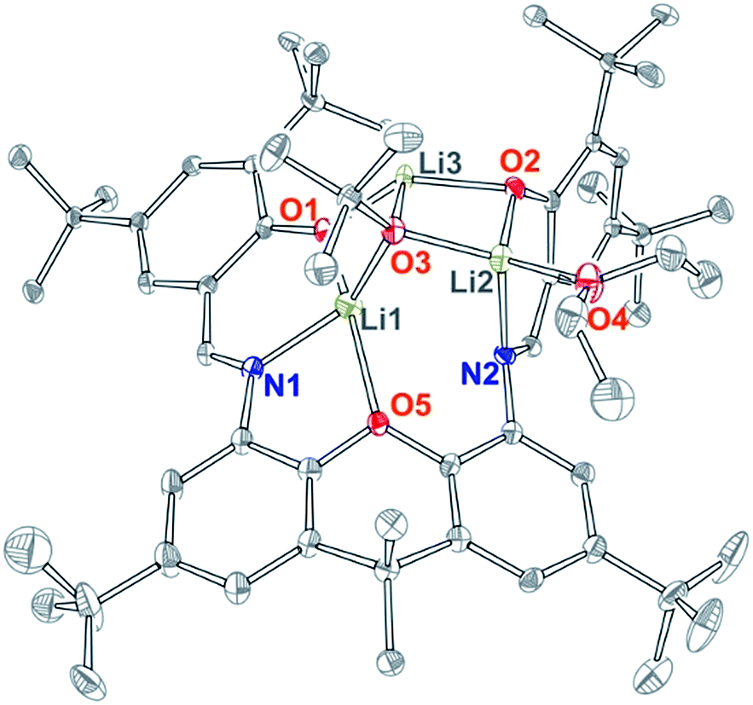 | ||
| Fig. 4 X-ray structure of 3, 30% probability ellipsoids. H atoms, co-crystallized solvent, and alternative conformation of tBu groups are omitted for clarity. | ||
Addition of 3 to two equivalents of zinc chloride led to the formation of the dinuclear complex Zn2Li2(L)(Cl4)(OEt2)4 (4). Complex 4 was isolated as yellow blocks from a hexane/ether mixture in 43% yield. The complex was characterized by proton NMR spectroscopy, X-ray crystallography and elemental analysis. 1H NMR spectrum of 4 contains broad resonances, suggesting a dynamic process. The X-ray structure of 4 is given in Fig. 5 below. The structure reveals that 4 is a di-zinc complex, where each zinc displays a pseudotetrahedral coordination environment including two chlorides, in addition to the iminophenolate chelate. The structure incorporates lithium ions that are linked to the complex through two chloride bridges each; pseudotetrahedral geometry of the lithium sites is completed by two coordinated ether molecules. Most significantly, the structure reveals anti-parallel arrangement of the two zinc metals coordinated by L, with Zn–Zn distance of 6.7 Å. While the anti-parallel arrangement is expected to be unfavourable for catalysis, it is likely due to the steric pressure around each zinc centre. We postulated that the removal of the incorporated lithium ions might release steric pressure and enable syn geometry of the zinc centres. To remove LiCl, 4 was treated with TlPF6 or AgPF6. In both cases, complex mixtures of products formed, from which we were not able to isolate any zinc-containing products. We have also attempted a direct reaction between 4 and two equivalents of LiOR, which failed to lead to a well-defined zinc-containing product.
 | ||
| Fig. 5 X-ray structure (50% probability ellipsoids) of 4. Selected bond distances (Å) and angles (°): Zn1 O1 1.900(3), Zn1 N1 1.989(4), Zn1 Cl1 2.269(1), Zn1 Cl2 2.268(1), Zn2 O2 1.902(3), Zn2 N2 1.987(4), Zn2 Cl3 2.262(1), Zn2 Cl4 2.271(1), O1 Zn1 N1 96.4(1), O2 Zn2 N2 96.9(1) Cl1 Zn1 Cl2 101.1(1), Cl3 Zn2 Cl4 100.9(1). | ||
As the attempts to obtain “Zn2(L)(OR)2” species (where OR is a bulky alkoxide OCtBu2Ph) were unsuccessful, we decided to pursue dinuclear zinc species with a more conventional alkoxide – benzyloxide, Zn2(L)(OBn)2. Zinc benzyloxide complexes are commonly used as precursors in lactide polymerization.7 As the most convenient path to these species generally lies through the protonolysis of preformed zinc alkyl species, the synthesis of Zn2(L)(Et)2 was pursued first. Slow addition of L to the excess diethylzinc solution (6 equivalents) led to the formation of yellow-orange Zn2(L)(Et)2 (5), that was obtained as an analytically pure solid in 95% yield. NMR spectrum of 5 is concentration-dependent. At high concentration (∼60 mM), a single set of peaks, albeit slightly broadened, is observed (for the aromatic region, see Fig. 6A; the full spectrum is given in ESI†). The overall spectrum, that contains five distinct signals in the aromatic region, two distinct signals for the tBu groups, and a single resonance for xanthene methyl groups, is consistent with the fluxional structure of 5. Zn-CH2CH3 methylene appears as a broad signal at 0.64 ppm in 1H NMR spectrum. Intriguingly, low concentration (∼10 mM) spectrum (Fig. 6B) indicates presence of another species, in addition to 5. Comparing the spectrum of 5 with the spectrum of 2·HOR (Fig. 6C) reveals the presence of compound 2 in a low-concentration spectrum. One possible explanation for this observation is the dinuclear – mononuclear equilibrium that is more pronounced at low concentrations due to the volatility of ZnEt2. To probe this hypothesis, we treated the low-concentration spectrum with excess ZnEt2 (4 equivalents), which resulted in the restoration of a single species consistent with Zn2(L)(Et)2 (Fig. 6D).
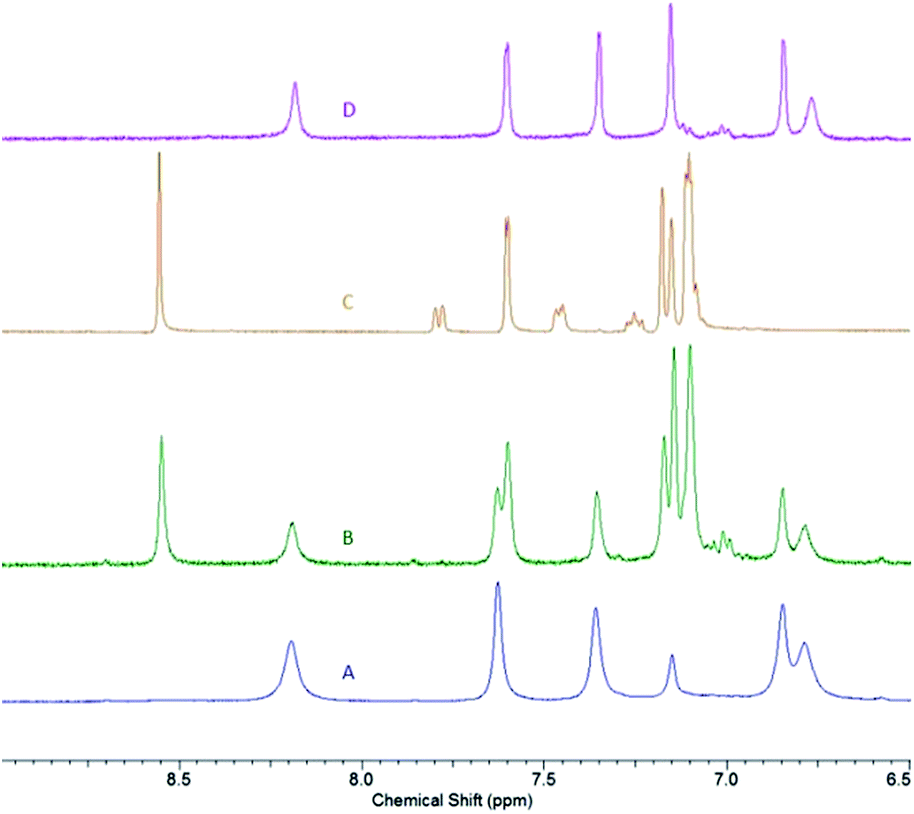 | ||
| Fig. 6 Aromatic region of 1H NMR spectra of: 5 at high concentration (A); 5 at low concentration (B); 2·HOR (C); 5 at low concentration following the addition of excess ZnEt2 (D). | ||
X-ray quality crystals of 5 were obtained by recrystallization from ether. 5 (Fig. 7) is a dinuclear complex exhibiting syn-parallel disposition of the two zinc-ethyl units, with a Zn⋯Zn separation of 4.5 Å. The geometry around zincs is trigonal planar, and the xanthene linker is flat. Zinc-phenoxide, imino, and ethyl distances are unexceptional.25 Overall, 5 constitutes a rare structure of an iminophenolate-bound zinc-ethyl in which the zinc centre is 3-coordinate.25 Based on our unsuccessful attempts to synthesize a mononuclear analogue (see below), we propose that this structure is stabilized by the dinucleating ligand. A notable feature of the structure is the relatively short distance between Zn2 and C1 (methylene bound to Zn2), 3.46 Å, due to the syn-parallel geometry. One could propose that the Zn2–C1 bond formation, which can be easily achieved by a minimal rotation of the N2O2Zn2 chelate towards the N1O1Zn1 chelate, can serve as a first step in the proposed transformation of 5 into 2 and ZnEt2 (Scheme 2).
 | ||
| Fig. 7 X-ray structure (50% ellipsoids) of 5 (top view and side view). H atoms, co-crystallized ether solvent, and an alternative orientation of one of the ethyl groups were omitted for clarity. Selected bond distances (Å) and angles (°): Zn1 O1 1.910(4), Zn1 C1 1.926(6), Zn1 N1 1.991(4), Zn2 O2 1.900(4), Zn2 C2 1.923(6), Zn1 N2 1.992(4), O1 Zn1 C1 126.8(2), O1 Zn1 N1 93.9(2), C1 Zn1 N1 139.0(2), O2 Zn2 C2 127.4(2), O2 Zn2 N2 94.1(2), C2 Zn2 N2 138.4(2). | ||
 | ||
| Scheme 2 Possible mechanism for the formation of complex 2 from complex 5. | ||
Treatment of 5 with two equivalents of BnOH in C6D6 led to the clean formation of di-benzyloxy complex 6 as indicated by 1H and 13C NMR spectroscopy. The NMR spectrum of 6 contains a single and sharp set of dinucleating ligand resonances, consistent with a single isomer in solution. In contrast, benzyloxy resonances appear broadened, which indicates a dynamic process. Such dynamic process, comprising primarily benzyloxy, may involve monomer–dimer equilibrium, in which zincs are bridged by the alkoxide ligand. VT NMR experiments, carried out in the CD2Cl2 solution of 6 down to −75 °C, resolved some of the benzyloxy signals. Our repeated attempts to isolate complex 6 by recrystallization from several different solvents (C6D6, C7D8, CD2Cl2) led to the isolation of yellow crystals of the tetranuclear complex 7 (Fig. 8), that was characterized by X-ray crystallography, 1H NMR spectroscopy, and elemental analysis. Due to the poor solubility of 7, we were not able to obtain its 13C NMR spectrum. In 7, one of the benzyloxy groups (of 6) has been replaced by the hydroxyl group. The “Zn2(L)(OBn)(OH)” fragments then dimerize to form centrosymmetric 7. We note that (i) 7 exhibits syn geometry of the zinc-benzyloxy units in each complex; (ii) the 1H NMR spectrum of 7 differs significantly from that of 6. Whereas hydrolysis of 6 to give 7 can't be ruled out, NMR suggests otherwise as (i) no noticeable amounts of water are detected in BnOH (anhydrous, 99.8%); (ii) NMR monitoring of the decomposition of 6 demonstrated formation of multiple species exhibiting signals in the olefinic (5–6 ppm) region.
 | ||
| Fig. 8 X-ray structure (50% ellipsoids) of 7. H atoms, co-crystallized toluene solvent, and an alternative orientation of two of the tert-butyl groups were omitted for clarity. Selected bond distances (Å) and angles (°): Zn1 O1 1.943(2), Zn2 O1 1.922(2), Zn1 O2 1.931(2), Zn2 O2 1.954(2), Zn2 O3 1.949(2), Zn1 O4 1.937(2), Zn2 O1 Zn1 131.0(1), Zn2 O2 Zn1 120.0(1). | ||
The synthesis of a mononuclear analogue of 5 was also attempted (Fig. 9). An iminophenolate ligand L′ was synthesized, and reacted with excess of diethyl zinc, in order to obtain the Zn(L′)(Et) complex. However, invariably of the amount of diethylzinc used in the reaction (3–6 equivalents), the reaction leads to the formation of two species, none of which possess signals attributable to the ethyl group in the 1H NMR spectrum. Recrystallization leads to the isolation of the major product, bis(homoleptic) Zn(L′)2 complex 8, in 78% yield, that has been characterized by 1H and 13C NMR spectroscopy, elemental analysis, and X-ray crystallography (see ESI†). We note the scarcity of tri-coordinate iminophenolate–zinc–ethyl complexes in the literature. While in some cases such complexes were synthesized, it was shown or hypothesized that they adopt phenoxide-bridged dimeric/polymeric structures in the solid state.25 We also note that in a related study on the synthesis of iminophenolate zinc pre-catalysts for lactide polymerization, bis(homoleptic) complexes were obtained for any ortho-phenolate substituents smaller than cumyl.25c Thus, while the bimetallic cooperativity of L in polymerization could not be directly assessed by a comparison with a mononuclear analogue, L enables bimetallic cooperativity in the formation of the desired diethyl complex, whose mononuclear analogue does not form.
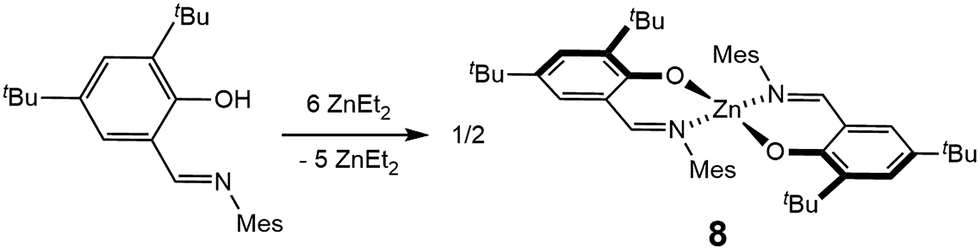 | ||
| Fig. 9 Reaction of L′ with excess ZnEt2 forming the bis(homoleptic) product 8. | ||
Lactide polymerization
We investigated the catalytic activity of Zn2(L)(OBn)2, formed in situ, in the ring-opening polymerization (ROP) of rac-LA. Polymerization attempts were performed in dichloromethane at room temperature by adding benzyl alcohol to 5 followed by rac-LA (see Table 1). Under these conditions, the polymerization of 600 equivalents of rac-lactide reached high conversion of 86% after 24 h, exhibiting comparable activity to other dinuclear zinc catalysts.9b,16 Gel-permeation analysis (GPC) of the PLA samples obtained revealed narrow molecular-weight distribution (PDI ≤ 1.23), signifying a well-controlled polymerization reaction. Increasing the benzyl alcohol ratio to beyond a single equivalent per zinc centre led to “immortal polymerization”,27 yielding PLA samples with narrow MWD and expected MW according to initiator/benzyl alcohol/monomer ratios. Homo-decoupled 1H analysis indicated that 6 led to heterotactically-inclined PLA (Pr = 0.67), as reported with other dinuclear zinc complexes.7g,16| [I]/[BnOH]/[LA] | Temp (°C) | Time (h) | Convb | Mn(calc)c | Mnd | PDI | Pre | |
|---|---|---|---|---|---|---|---|---|
| a Polymerizations performed in CH2Cl2 (entries 1–4, 5 mL) or toluene (entries 5 and 6, 5 mL) employing 5 μmol of catalyst.b Determined by 1H NMR spectroscopy (500 MHz).c Calculated from [LA]/[BnOH] ratio multiplied by monomer conversion.d Determined by GPC analysis calibrated with polystyrene standards and multiplied by a correction factor of 0.58. Values are given in g mol−1.e Determined from the methine region of the HD 1H NMR spectrum.f 6 was formed in situ, by treating 5 with BnOH. | ||||||||
| 1 | 1/2/600 | 25 | 16 | 0.60 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
15420 |
1.18 | 0.66 |
| 2 | 1/2/600 | 25 | 24 | 0.86 | 37200 |
23430 |
1.23 | 0.67 |
| 3 | 1/4/600 | 25 | 24 | 0.80 | 17300 |
15600 |
1.22 | 0.66 |
| 4 | 1/10/1000 | 25 | 24 | 0.69 | 9900 | 5300 | 1.19 | 0.65 |
| 5 | 1/2/600 | 70 | 1 | 0.88 | 38000 |
21700 |
1.43 | 0.58 |
| 6 | 1/2/600 | 70 | 2 | 0.98 | 42300 |
24000 |
1.67 | 0.54 |
Attempting to attain higher activities, polymerization runs were performed at 70 °C in toluene. High conversion of 88% was obtained only after 1 hour yielding PLA with reduced heterotacticity (Pr = 0.58) and higher PDI values. Extending the reaction time to 2 hours led to almost full conversion, however, heterotacticity and dispersity were further decreased. The deterioration of the sample qualities at high conversion might be a result of trans-esterification side reactions, which increase at elevated temperatures.
Conclusions
This study pursued synthesis of new di-zinc complexes, stabilized by a xanthene-bridged bis(iminophenolate) ligand, which could serve as a lactide polymerization catalyst. L demonstrated rich coordination chemistry with various zinc precursors, leading to mononuclear (with zinc alkoxide), syn-dinuclear (with diethyl zinc), or anti-dinuclear (with zinc chloride) complexes. Syn-dinuclear complex Zn2(L)(Et2), obtained via a protonolysis reaction of L with excess diethylzinc, decomposes in solution into a mononuclear complex Zn(L), along with ZnEt2; this reaction can then be reversed by the addition of excess ZnEt2. Interestingly, no mononuclear analogue of Zn2(L)(Et2) could be isolated as the protonolysis reaction of mononucleating iminophenol bearing comparable substituents (L′) with diethylzinc leads instead to the bis(homoleptic) complex Zn(L′)2. Thus, while we could not directly assess cooperative reactivity in catalysis, our dinucleating ligand enables “cooperative synthesis” of two nearby zinc–iminophenolate–ethyl sites, possibly by virtue of sterics. Zn2(L)(Et2) underwent clean reaction with benzyl alcohol to form benzyloxy species Zn2(L)(OBn)2. Zn2(L)(OBn)2 served as an efficient lactide polymerization catalyst, forming heterotactically-inclined PLA. Room-temperature polymerization experiments, carried out in dichloromethane, led to the formation of polymers of relatively narrow MWD, suggesting well-controlled polymerization under these conditions. Polymerization experiments in toluene, carried out at 70 °C, demonstrated higher activity, but led to higher PDI values, and diminished heterotacticity. Our future studies will focus on the reactivity of this and related systems in polymerization of epoxides and copolymerization of epoxides with CO2.Experimental
General
All reactions involving air-sensitive materials were carried out in a nitrogen-filled glovebox. 2,7-di-tert-butyl-9,9-dimethyl-4,5-diaminoxanthene, di-tert-butyl-phenylmethoxide (LiOR) and di-tert-butyl-phenylmethanol (HOR) were synthesized according to previously published procedures.22 Synthesis of L′ has been previously reported as well.26 Di-tert-butylsalicylaldehyde, zinc chloride and diethyl zinc (1.0 M solution in hexanes) were purchased from Sigma and used as received. Rac-lactide was purchased from Sigma, and was purified by crystallization from dry toluene and sublimation. All non-deuterated solvents were purchased from Aldrich and were of HPLC grade. The non-deuterated solvents were purified using an MBraun solvent purification system. Benzene-d6, toluene-d8, dichloromethane-d2, and chloroform-d1 were purchased from Cambridge Isotope Laboratories. All solvents were stored over 3 Å molecular sieves. Compounds were generally characterized by 1H and 13C NMR, X-ray crystallography, and elemental analysis. NMR spectra of the ligands and metal complexes were recorded at the Lumigen Instrument Centre (Wayne State University) on an Agilent 400 MHz Spectrometer in C6D6, C7D8, or CD2Cl2 at room temperature. NMR spectra of PLA samples were recorded on Bruker Advance III 500 MHz spectrometer in CDCl3 at room temperature. Chemical shifts and coupling constants (J) were reported in parts per million (δ) and Hertz respectively. Detailed assignments of the signals in 1H NMR are given in the ESI.† X-ray structures were collected using Bruker Apex2 at the Lumigen Instrument Centre (Wayne State University). Full details on data collection, structure solution and refinement are given in the ESI.† Elemental analysis was performed under ambient air-free conditions by Midwest Microlab LLC. The molecular weights (Mn and Mw) and the molecular mass distributions (Mw/Mn) of the PLA samples were measured by gel permeation chromatography (GPC) at 30 °C, using THF as solvent, a flow rate of eluent of 1 mL min−1, and narrow MW polystyrene standards as reference. The measurements were performed on a Jasco system equipped with an RI 1530 detector. A correction factor of 0.58 was employed for the molecular weight of PLA relative to polystyrene.Synthesis and characterization of L and compounds 1–8
:1 mL), filtered through celite, and the solution was concentrated to approximately 2 mL total volume. Recrystallization at −33 °C yields Li3(L)(μ3-OtBu) (THF) as yellow microcrystalline solid (100 mg, 0.105 mmol, 88% yield). The compound was characterized by 1H and 13C NMR spectroscopy, elemental analysis, and X-ray crystallography. 1H NMR (C6D6, 400 MHz) δ 8.50 (s, 2H), 7.64 (d, 4JHH = 2 Hz, 2H), 7.33 (d, 4JHH = 2 Hz, 2H), 7.15 (d, 4JHH = 2 Hz, 2H), 7.05 (d, 4JHH = 2 Hz, 2H), 3.31 (m, 8H, THF) 1.76 (s, 3H, Me), 1.64 (s, 18H), 1.57 (s, 3H, Me), 1.37 (s, 9H), 1.32 (s, 18H), 1.26 (m, 8H, THF), 1.21 (s, 18H). 13C{1H} NMR (C6D6, 100 MHz) δ 170.02, 168.20, 147.16, 143.32, 141.20, 140.11, 134.17, 131.70, 131.37, 129.43, 128.35, 123.13, 118.12, 68.14, 35.80, 35.52, 35.49, 35.30, 34.99, 34.87, 33.98, 31.80, 31.67, 30.60, 25.75, 25.66, 25.38. Anal. calcd for C57H76Li2N2O4Zn2 × C4H8O: C, 77.27; H, 9.14; N, 2.95. Found: C, 76.71; H, 9.09; N, 2.95.:1 mL) and concentrated in vacuo. Overnight recrystallization at −33 °C afforded X-ray quality crystals of 4 as yellow blocks (67 mg, 0.046 mmol, 43%). 1H NMR (CD2Cl2, 400 MHz) δ 8.86 (br s, 2H), 8.48 (br s, 2H), 7.55–6.90 (m, 6H), 3.45 (q, 3JHH = 8 Hz, 16H), 1.73 (s, 6H), 1.39 (s, 18H), 1.61 (s, 3H), 1.26 (br s, 36H), 1.16 (t, 3JHH = 8 Hz, 24H). Anal. Cald for C69H110Cl4Li2N2O7Zn2 × C6H14: C, 62.03; H, 8.61; N, 1.93. Found: C, 61.76; H, 8.25; N, 1.58.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Science Foundation (NSF) under grant number CHE-1349048. Compounds characterization was carried out at Lumigen Instrument Center at Wayne State University.References
- Biological Inorganic Chemistry: Structure and Reactivity, ed. H. B. Gray, E. I. Stiefel, J. S. Valentine and I. Bertini, University Science Books, Sausalito, CA, 2007 Search PubMed
.
- Homo- and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, Topics in Organometallic Chemistry, ed. P. Kalck, Springer International Publishing, Switzerland, 2016, vol. 59 Search PubMed
- For selected recent reviews/editorials on the cooperative reactivity of bimetallics, see:
(a) J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472–5473 RSC
-
(a) M. H. Chisholm, Pure Appl. Chem., 2010, 82, 1647–1662 CrossRef CAS
-
(a) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS
- For selected reviews on lactide polymerization, see:
(a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2001, 2215–2224 RSC
- For selected references on Zn-containing lactide polymerization catalysts, see:
(a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed
- C. K. Williams, N. R. Brooks, M. A. Hillmeyer and W. B. Tolman, Chem. Commun., 2002, 2132 RSC
-
(a) A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed
- M. Normand, T. Roisnel, J.-F. Carpentier and E. Kirillov, Chem. Commun., 2013, 49, 11692–11694 RSC
- W.-L. Kong and Z.-X. Wang, Dalton Trans., 2014, 43, 9126–9135 RSC
- S. Sun, K. Nie, Y. Tan, B. Zhao, Y. Zhang, Q. Shen and Y. Yao, Dalton Trans., 2013, 42, 2870–2878 RSC
- T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS PubMed
- M. H. Thibault and F.-G. Fontaine, Dalton Trans., 2010, 39, 5688–5697 RSC
- Y. Wang and H. Ma, Chem. Commun., 2012, 48, 6729–6731 RSC
- A. B. Kremer, K. M. Osten, I. Yu, T. Ebrahimi, D. C. Aluthge and P. Mehrkhodavandi, Inorg. Chem., 2016, 55, 5365–5374 CrossRef CAS PubMed
-
(a) I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS PubMed
-
(a) A. Bheemaraju, J. W. Beattie, R. L. Lord, P. D. Martin and S. Groysman, Chem. Commun., 2012, 48, 9595–9597 RSC
-
(a) A. Bheemaraju, J. W. Beattie, Y. Danylyuk, J. Rochford and S. Groysman, Eur. J. Inorg. Chem., 2014, 34, 5865–5873 CrossRef
-
(a) A. Panunzi, F. Giordano, I. Orabona and F. Ruffo, Inorg. Chim. Acta, 2005, 358, 1217–1224 CrossRef CAS
-
(a) J. A. Bellow, M. Yousif and S. Groysman, Comments Inorg. Chem., 2016, 36, 92–122 CrossRef CAS
-
(a) J. A. Bellow, P. D. Martin, R. L. Lord and S. Groysman, Inorg. Chem., 2013, 52, 12335–12337 CrossRef CAS PubMed
- For selected examples of three-coordinate zinc complexes with alkoxide donors, see:
(a) J. S. Lum, P. E. Chen, A. L. Rheingold and L. H. Doerrer, Polyhedron, 2013, 58, 218–228 CrossRef CAS
- J. A. Bellow, M. Yousif, D. Fang, E. G. Kratz, G. A. Cisneros and S. Groysman, Inorg. Chem., 2015, 54, 5624–5633 CrossRef CAS PubMed
-
(a) C. Zhang and Z.-X. Wang, Appl. Organomet. Chem., 2009, 23, 9–18 CrossRef CAS
- L. Clowes, M. Walton, C. Redshaw, Y. Chao, A. Walton, P. Elo, V. Sumerin and D. L. Hughes, Catal. Sci. Technol., 2013, 3, 152–160 CAS
-
(a) T. Aide and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, crystal and refinement data for compounds L, 1–5, 7, and 8. CCDC 1554332–1554340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09207e |
| This journal is © The Royal Society of Chemistry 2017 |