
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First hyperpolarizabilities of Pt(4-ethynylbenzo-15-crown-5)2(bpy) derivatives with the complexation of mono-cations (Li+, Na+, K+) and di-cations (Mg2+, Ca2+): development of a cation detector†
Hai-Ling Yu *a,
Wen-Yong Wangb,
Bo Honga,
Yan-Ling Sia,
Tian-Liang Maa and
Ran Zhenga
*a,
Wen-Yong Wangb,
Bo Honga,
Yan-Ling Sia,
Tian-Liang Maa and
Ran Zhenga
aCollege of Resources and Environmental Science, Jilin Agricultural University, Changchun 130118, People's Republic of China. E-mail: jlauyu@163.com
bJiangSu XinHai Senior High School, Lianyungang City 222000, Jiangsu, People's Republic of China
First published on 30th August 2017
Abstract
Square-planar platinum(II) compounds containing two crown ethers play an important role in the development of nonlinear optical (NLO) materials, liquid crystals, luminescent materials and also in supramolecular chemistry owing to their diverse charge transfer excited states. The coordination of mono-cationic (Li+, Na+, K+) and di-cationic (Mg2+, Ca2+) metals onto the crown seems to be advantageous for the design of NLO-based cation detectors. Pt(4-ethynylbenzo-15-crown-5)2(bpy) (L) and its cation derivatives have been investigated by the density functional theory. The binding of alkali metal cations obviously shrinks the crown ethers due to their strong interaction energies and distorts the crown ethers perpendicular to the bipyridine and Pt plane. Significantly, as compared with L, an obvious decrease appears in the first hyperpolarizability after cation complexing. This can be explained by their variable charge transfer transitions along the y-axis in conjunction with the distribution of holes and electrons. Thus, it can be noted that square-planar platinum(II) compounds merit the widespread use in the fields of optical detectors.
1. Instruction
Over the past 15 years, there has been increasing interest in the properties of square-planar platinum(II) compounds. One key reason is that square-planar platinum(II) complexes exhibit a variety of charge transfer (CT) excited states1–9 such as metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT), and intraligand charge transfer (ILCT). The directional nature of the CT excited states in square-planar platinum(II) complexes is ideal for hole–electron creation and separation, and consequently, these systems merit the considerable use in numerous fields of materials chemistry, including nonlinear optics (NLO), liquid crystals, conducting polymers, luminescent materials, and supramolecular chemistry.10–14 There is a library for platinum(II) compounds with various structures, such as mononuclear, dinuclear, and metallacyclic structures, showing their unique physical, optical, photophysical, conductive, as well as self-assembly properties.15,16 A classical example of the mononuclear square-planar Pt(II) complexes is the platinum(II) alkynyl bipyridine complexes. Because these complexes were found to exhibit a very broad and strong nonlinear absorption in the visible to NIR region, they have potential applications as broadband nonlinear materials.17The photophysical and electrochemical properties of the platinum(II) alkynyl bipyridine complexes are found to be sensitive to the nature of both platinum(II) bipyridine and ancillary ligands. With this regard, the coordination of two 4-ethynylbenzo-15-crown-5 ancillary ligands onto the platinum(II) center18 seems to be advantageous for the development of NLO-based cation detectors.19 Because this type of complexes are agreeable with the NLO-based detecting cations devices that must comprise a signaling moiety (platinum atom and bipyridine as the main part that generates NLO signals) and a cation binding site (e.g. crown ether moieties, usually termed as the receptor). Since Pedersen first reported the synthesis of crown ethers in 1967,20 they occupy a special position in the development of receptors and are widely used in the design of new optical detectors21–23 based on their unique ability to coordinate the cations of alkali and alkaline metals (such as Li+, Na+, K+, Mg2+ and Ca2+),24–27 along with their fairly high selectivity and accessibility.28
Despite the fact that considerable works on the development of platinum(II) complexes as NLO materials have been reported,29–32 works on the design of NLO-based detecting complexes with alkali cations have received less attention. An efficient NLO-based detector should exhibit a substantial “switch-on” or “switch-off” effect upon binding to metal ions. The platinum(II) alkynyl bipyridine complexes and its metal cation derivatives have displayed an obvious contrast in NLO properties. As part of our continuing interest in the design of new NLO-based detectors with crown ether and its corresponding alkali cation derivatives,33 [Pt(4-ethynylbenzo-15-crown-5)2(bpy)] (bpy = 2,2′-bipyridine) (L) was chosen as a parent compound. The platinum(II) alkynyl bipyridine complexes along with its mono-cations (Li+, Na+, and K+) and di-cations alkaline metal (Mg2+ and Ca2+) derivatives (Fig. 1) were investigated using the density functional theory (DFT) with the aim of rationalizing their geometrical and nonlinear optical properties. It is expected that the complexation of L with alkali metals will trigger the modulation of NLO properties such as the first hyperpolarizability. In this case, cation detection is possible by monitoring NLO intensity, which may be of interest for the development of NLO-based probes.
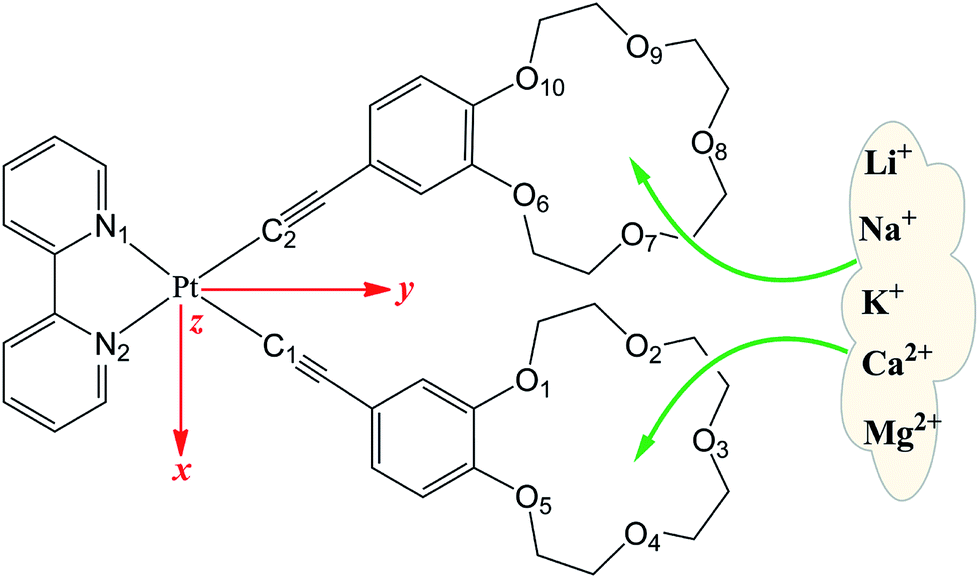 | ||
| Fig. 1 Structures of [Pt(4-ethynylbenzo-15-crown-5)2(bpy)] (bpy = 2,2′-bipyridine) (L) and its metal cation derivatives. | ||
2. Computation details
DFT calculations were performed using the Gaussian 09W program.34 The geometry optimizations of all molecules were fully optimized at the B3LYP functional with the 6-31G** basis set for C, H, and O atoms, 6-311G** for Li, Na, K, Mg, and Ca atoms and the LanL2DZ effective core potential (ECP) basis set for Pt atom. The B3LYP function was used because it successfully reproduced the experimental structures.35,36 All optimized structures reached the true local minimum stationary points. Because there is no imaginary frequency. Interaction energies, enthalpy, Gibbs free energies and natural population analysis (NPA) charge are calculated at the B3LYP/6-31G**/LanL2DZ level.Due to the efficiency and accuracy, the first hyperpolarizability β is calculated by analytical third energy derivatives. The total first hyperpolarizability (βtot) is defined as
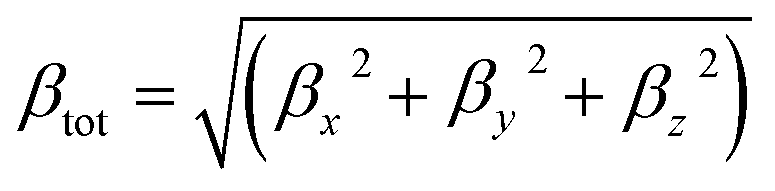 | (1) |
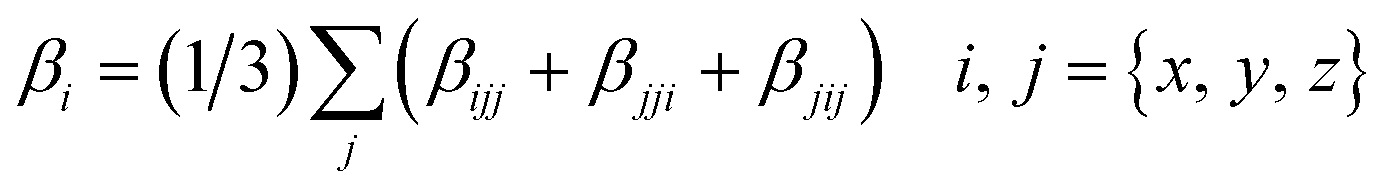 | (2) |
DFT calculations, using conventional functions, sometimes fail to correctly predict the amplitude of hyperpolarizability, which is the result of their wrong asymptotic exchange potentials.37,38 The long-range correct LC-BLYP and CAM-B3LYP functionals are good options for the calculation of hyperpolarizabilities.39 For a contrastive study, the hyperpolarizabilities were also evaluated by the M06-2X functional.40 The basis set used is 6-311+G** except LanL2DZ ECP basis set for Pt atom.
The hyper-Rayleigh scattering (HRS) response is one of the second-order NLO phenomena. Champagne and co-workers41–48 developed an effective method to evaluate the HRS response βHRS(−2ω;ω,ω), which is described as
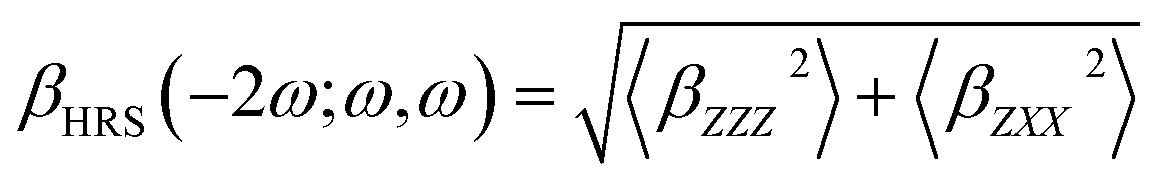 | (3) |
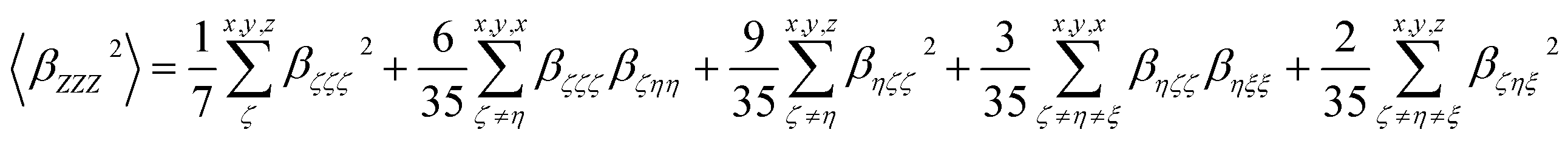 | (4) |
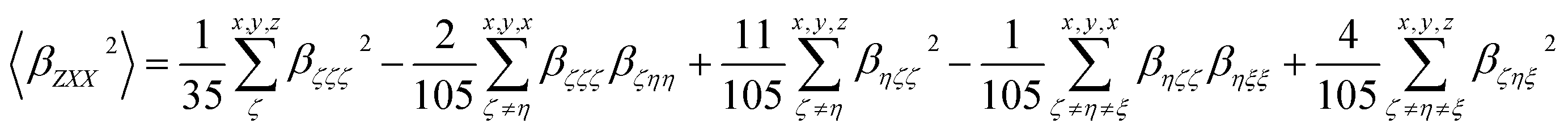 | (5) |
In addition, the depolarization ratio (DR), which gives the molecular geometric information, is responsible for the NLO response and expressed as
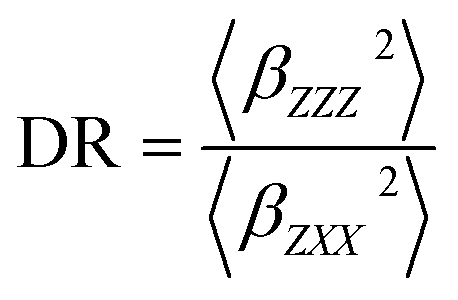 | (6) |
The first hyperpolarizability can also be obtained using the sum-over-states (SOS) methods.49 SOS method is used to separate and discuss the contribution from different excited states. The first hyperpolarizability β of SOS equation can be expressed as follows:50
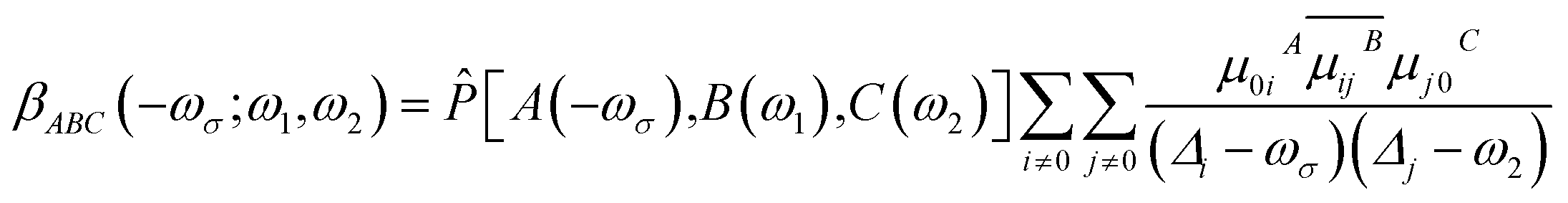 | (7) |
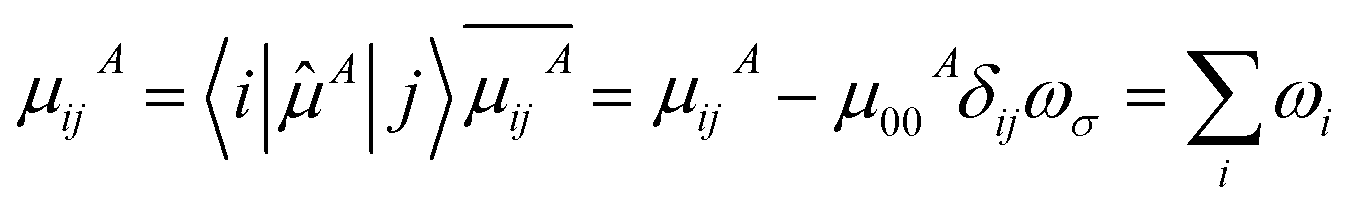 | (8) |
![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif) is permutation operator (for β, there are 3! = 6 permutations). μijA represents the A component of transition dipole moment between state i and j (when i = j, the term simply corresponds to electric dipole moment of state i), and
is permutation operator (for β, there are 3! = 6 permutations). μijA represents the A component of transition dipole moment between state i and j (when i = j, the term simply corresponds to electric dipole moment of state i), and ![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) is dipole moment operator (e.g., x ≡ −x).
is dipole moment operator (e.g., x ≡ −x).
The properties of the excited states were investigated by employing the time-dependent density functional theory (TDDFT), which incorporates electronic screening and relevant correction effect for electronic excitation, to evaluate the excited states.51,52 All electronic excited states are calculated at the CAM-B3LYP/6-311+G**/LanL2DZ level since CAM-B3LYP functional is in satisfactory agreement with the experimental values.53
The NPA charge is evaluated from natural bond orbital (NBO) population analysis using NBO program embedded in Gaussian 09W program, while the charge density difference (CDD) on a surface around a molecule is calculated by the Multiwfn 3.3.8 software.54
3. Results and discussion
3.1 Geometrical structure and thermodynamic analysis
The structures of L, L*(Li+)2, L*(Na+)2, L*(K+)2, L*(Ca2+)2 and L*(Mg2+)2 were optimized at the B3LYP/6-31G** level. The minimum vibrational frequencies of these structures are 1.54, 5.8, 5.48, 3.14, 6.07 and 6.39 cm−1. The small vibration amplitude supports the fact that the optimized structures are thermally stable and at minimum. Selected bond lengths of Pt–C and Pt–N bonds and average O–O bond lengths of crown ethers are listed in Table 1. The Pt–C and Pt–N bond deviations between L and its corresponding experimental values are 0.029 Å and 0.058 Å, respectively, whereas O–O bond deviation is in the range from 0.018 Å to 0.019 Å. As seen from the result, the geometrical parameters of L are very close to the experimental values previously reported by Chi-Ming Che determined from the crystallographic study,55 suggesting that the geometrical calculation of B3LYP method is reliable. For Pt–C and Pt–N bonds, the bond lengths of L are 1.952 Å and 2.120 Å, respectively. In contrast, the Pt–C and Pt–N bonds in metal cation derivatives are slightly longer, reflecting the minimal impact of metal cations on Pt–C and Pt–N bonds. However, as compared with those in L, the average O–O bond lengths of metal cation derivatives became smaller. The deviation of O–O bond lengths between metal cation derivatives and L is in the order of L*(Li+)2 > L*(Na+)2 > L*(K+)2 and L*(Mg2+)2 > L*(Ca2+)2. This indicates that the crown ethers shrink obliviously, especially in L*(Li+)2 and L*(Mg2+)2. This is easy to understand since metal cations have strong interactions with crown ethers. The metal cations also distort the crown ether moieties perpendicular to the bipyridine and Pt plane, which is clearly seen from Fig. S1 in the ESI.†| Compound | Pt–C1 | Pt–C2 | Pt–N1 | Pt–N2 | O–O (1)a | O–O (2)b | Eint |
|---|---|---|---|---|---|---|---|
| a Average O–O bond length of the O1–O2, O2–O3, O3–O4, O4–O5 and O5–O1 bonds.b Average O–O bond length of the O6–O7, O7–O8, O8–O9, O9–O10 and O10–O11 bonds.c Values in the bracket are determined by X-ray analysis from ref. 55. | |||||||
| L | 1.952 (1.981)c | 1.952 (1.981)c | 2.120 (2.062)c | 2.120 (2.062)c | 2.843 (2.861)c | 2.842 (2.861)c | — |
| L*(Li+)2 | 1.954 | 1.954 | 2.125 | 2.125 | 2.599 | 2.599 | −120.2 |
| L*(Na+)2 | 1.954 | 1.954 | 2.124 | 2.124 | 2.727 | 2.728 | −92.9 |
| L*(K+)2 | 1.954 | 1.954 | 2.125 | 2.125 | 2.767 | 2.768 | −63.3 |
| L*(Mg2+)2 | 1.957 | 1.957 | 2.128 | 2.128 | 2.516 | 2.516 | −326.7 |
| L*(Ca2+)2 | 1.957 | 1.957 | 2.128 | 2.128 | 2.706 | 2.706 | −230.6 |
Interaction energies between L and metal cations were considered at the B3LYP/6-31G** level. The results in Table 1 reveal that all complexes have large and negative values of interaction energies, indicating the stability of alkali metal cations derivatives and strong interactions between metal cations and crown ethers. For mono-cation derivatives, the interaction energies are −120.2 kcal mol−1 (L*(Li+)2) < −92.9 kcal mol−1 (L*(Na+)2) < −63.3 kcal mol−1 (L*(K+)2), which are two times higher compared to the values reported by Gao.56 This is reasonable since L and its metal cation derivatives have two crown ether fragments, and the interaction energy is the sum of two interaction energies between the crown ether and metal cation.
The interaction energies present the same sequence of the deviation of O–O bond lengths. The smaller interaction energy of complex L*(K+)2 can be attributed to its structure of K atoms out of the plane of the five O atoms of the crown ethers. Moreover, for di-cations derivatives, the interaction energies are −326.7 kcal mol−1 (L*(Mg2+)2) < −230.6 kcal mol−1 (L*(Ca2+)2), which holds the same truth with the deviation of O–O bond lengths. These values support the oblivious influence of alkali metal cations and the degree of the shrinkage on the crown ethers.
Table 2 summarizes the main thermodynamic data for the complexation reaction of the alkali ion by the L form. The Gibbs free energies (T = 298.15 K and P = 1 atm) of these reaction are exothermic, and the most exothermic reaction is the complexation of the Mg2+ and Ca2+ cations. Note that the Gibbs free energies (in absolute value) decrease monotonically with the cation size for mono-cations (Li+, Na+, K+) and di-cations (Mg2+, Ca2+) complexes, which is similar with the trend of interaction energy. Indeed, the enthalpy also behaves similarly as the cation size.
| Complex | ΔHθ | ΔGθ |
|---|---|---|
| L*(Li+)2 | −207.5 | −198.3 |
| L*(Na+)2 | −156.9 | −142.0 |
| L*(K+)2 | −94.6 | −80.9 |
| L*(Mg2+)2 | −616.2 | −604.3 |
| L*(Ca2+)2 | −428.7 | −413.3 |
3.2 Charge distribution
Atomic charge is one of the simplest and most intuitive description of charge distribution and has great significance in theory and practical applications.57 The global charge of the different fragments (Fi) of compounds L and its metal cation derivatives was computed at the B3LYP/6-31G**/LanL2DZ level using the NPA method (Table S1, ESI†). For all compounds, the negative charge is mainly located at the crown ether (F1, F2) and alkynyl (F5, F6) moieties, which is respectively attributed to the large electronegativity of oxygen and the rich electron-density of π conjunction. The negative charge and cavity of crown ether of L lead to its ability to coordinate the cations of alkali metals. In contrast, the benzene (F3, F4), bipyridine (F8) and Pt (F7) moieties are positively charged. Upon complexing with alkali metal cation, all fragments (F1 to F8) become positive and the electron density decreases (Fig. 2) because the complexation leads mainly to electron transfer from all fragments to an alkali metal cation. The most modification of the charge distribution is the crown ether (F1, F2), benzene (F3, F4), and bipyridine (F8) moieties. For the crown ether (F1, F2) moieties, the modification decreases monotonically with the cation size of mono-cations (Li+, Na+, K+) and di-cations (Mg2+, Ca2+), respectively. In cases of Pt (F7) atoms and alkynyl (F5, F6) moieties, they play the role of spectator and are almost unaffected by the complexation. Thus, the coordination of alkali metal cation onto L is also characterized by charge reorganizations.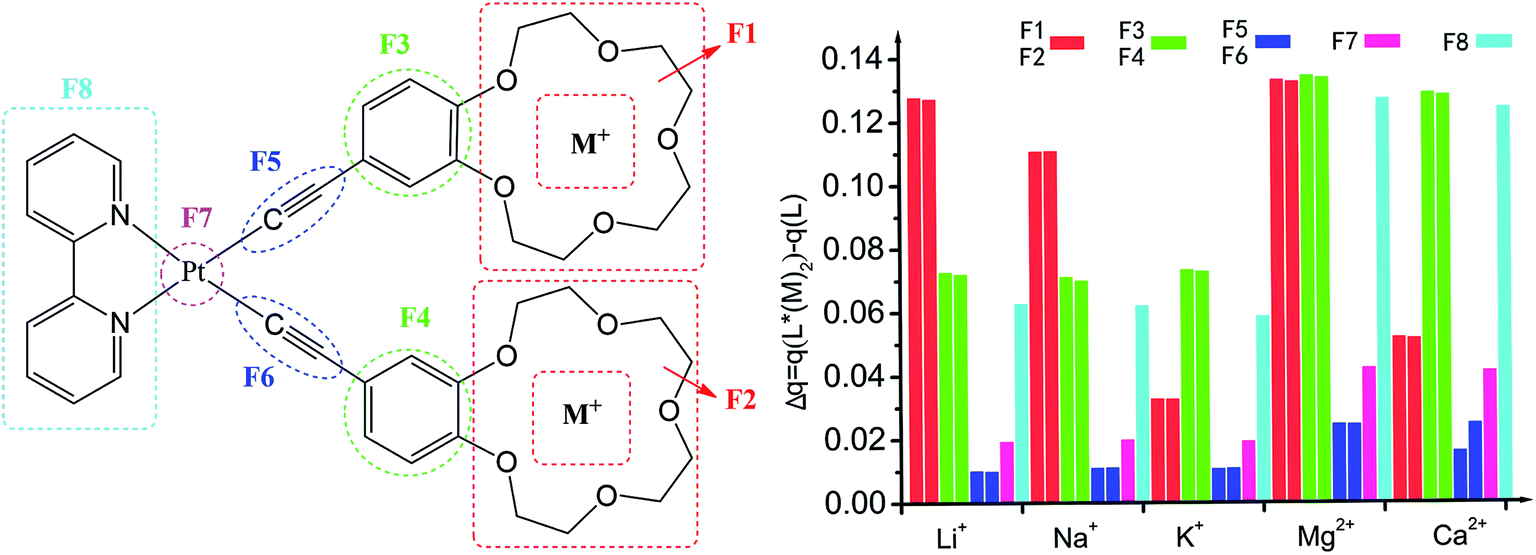 | ||
| Fig. 2 The NPA charge for the fragments (S1–S8) of L and L*(M+/2+)2 determined at the B3LYP/6-31G*/LanL2DZ level, Δq = q(L*(M+/2+)2) − q(L). | ||
3.3 First hyperpolarizability
At the molecular level, the second-order NLO response comes from the second-order NLO coefficient, i.e. the first hyperpolarizability (β). Previous theoretical investigations have revealed that the second-order NLO properties of crown ether derivatives can be switched by cation complexing.33 By combining the switches of nonlinear optical responses, macrocyclic crown ethers present a more practical and reliable chance to be a cation detector. In order to rationalize the different NLO behaviors of L and its metal cation derivatives, the first hyperpolarizabilities of six molecules have been calculated theoretically at the DFT/6-311++G** levels using the CAM-B3LYP, M06-2X and LC-BLYP functionals. The results are reported in Fig. 3 and Tables S2–S5 (ESI).† Comparison to CAM-B3LYP functional, the M06-2X and LC-BLYP functionals seem to underestimate the first hyperpolarizabilities with the exception of L*(Mg2+)2 and L*(Ca2+)2 (L*(Mg2+)2 ≈ L*(Ca2+)2). However, the first hyperpolarizabilities obtained with three functionals show the similar decrease trends of L > L*(Mg2+)2 ≈ L*(Ca2+)2 > L*(Li+)2 ≈ L*(Na+)2 > L*(K+)2, demonstrating the relative reliability of three DFT methods. Therefore, in the next discussion, only the CAM-B3LYP functional is taken as an example to shed light on the change of the first hyperpolarizability. All first hyperpolarizabilities are calculated with the ultrafine integration grid (99, 590). The first hyperpolarizabilities are also calculated at the CAM-B3LYP level with the superfine grid (the superfine grid specification is (150, 974) for the first two rows of the periodic table and (225, 974) for later elements) (Table S3, ESI†) to check the convergence of the size of the integration grid. The first hyperpolarizabilities of the ultrafine grid are almost consistent with the values of the superfine grid, indicating that the ultrafine integration grid is acceptable and convergent.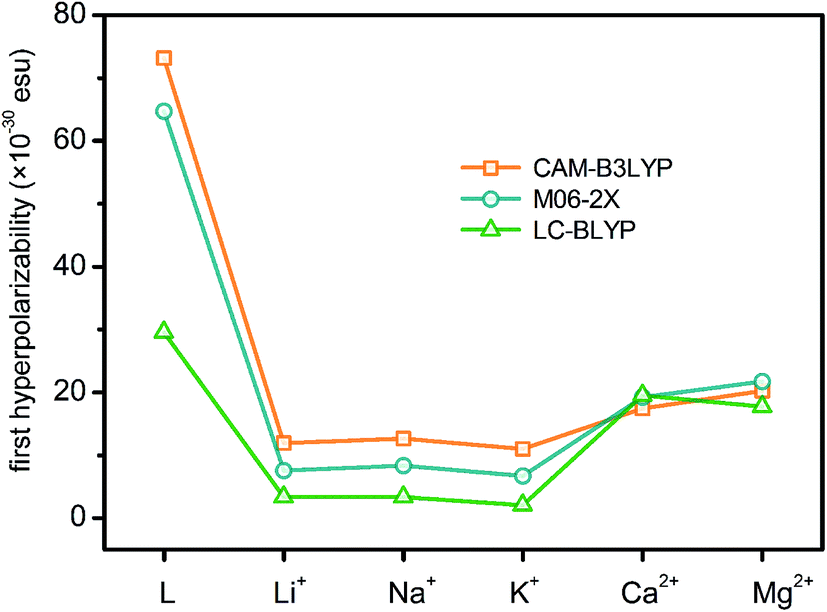 | ||
| Fig. 3 First hyperpolarizability of L and its cation derivatives. | ||
First hyperpolarizability is a third rank tensor that can be described by a 3 × 3 × 3 matrix. For the static first hyperpolarizability, the 3 × 3 × 3 = 27 components can be reduced to 10 components by Kleinman's symmetry. With these 10 components, the contribution of each one to the first hyperpolarizability is obtained. The β values in three Cartesian axes, which directly reflect the direction of the charge transfer, can also be calculated. The important components of the first hyperpolarizability are listed as ESI (Tables S2–S5).† As seen from the results, the βxxy and βyyy tensorial components with some contribution of βyyz are the dominant ones that describe the βtot values with respect to the rest tensorial components, which inevitably leads to the largest βy tensorial component. For L, L*(Li+)2, L*(Na+)2 and L*(K+)2, the βy values are positive, whereas for L*(Mg2+)2 and L*(Ca2+)2, the βy values are negative. It suggests that during a polarization process, the direction of electronic charge transition among L, L*(Li+)2, L*(Na+)2, L*(K+)2 and L*(Mg2+)2, L*(Ca2+)2 is opposite. It is expected that the electronic charge transition is migrated from 2,2′-bipyridine and Pt moieties (15-crown-5 moiety) to the 15-crown-5 moiety (2,2′-bipyridine moiety) since the βy values are positive (negative). For the metal cation derivatives, the first hyperpolarizabilities shown in Fig. 3 are insensitive to the alkali metals in the same group. However, an obviously decrease in β after cation complexing is observed with respect to compound L. It reveals that the first hyperpolarizability is sensitive to the cation complexing.
Table 3 reports the static and dynamic (λ = 1064 nm) HRS first hyperpolarizabilities (βHRS) and the depolarization ratios and the βHRS contrast ratios for L and its cation derivatives. The variations of the static and dynamic βHRS values are similar to those of βtot values. It can be stated that the cation complexing leads to the small static and dynamic βHRS values, in particular the mono-cations (Li+, Na+, K+) with βHRS(L)/βHRS(L*(M+/2+)2) contrast ratio decreases by about a factor of 4.4. It is interesting to note that the effects of alkali/complexation on the βHRS values are much smaller and mainly in different direction of Champagne's investigations.19,58 In any case, the obvious variations of the first hyperpolarizability contrasts gives complex L a chance to access NLO-based detection of cations. When considering the complexation process, the alkali metal cation induces a remarkable decrease in the values of static DR. In the case of di-cations (Mg2+, Ca2+) derivatives, the DR values are even smaller, demonstrating that they depart more from ideal one-dimensional structures of which DR = 5. These variations of DR can be rationalized in terms of a large change of geometrical and electronic structure. Since the measurements are performed at 1064 nm, the dynamic βHRS and DR are calculated at λ = 1064 nm. As compared with the static βHRS and DR, a strong enhancement is clearly seen at λ = 1064 nm, in particular in the case of compound L. Compound L is impacted by electronic resonance since it presents a maximum absorption at 482 and 436 nm, whereas the 532 nm second-harmonic generation wavelength is within the absorption band.
| ∞ | 1064 | ∞ | |||
|---|---|---|---|---|---|
| βHRS | DR | βHRS | DR | βHRS(L)/βHRS(L*(M+/2+)2) | |
| L | 28.0 | 6.8 | 124.3 | 8.7 | — |
| L*(Li+)2 | 6.4 | 2.8 | 14.1 | 3.8 | 4.4 |
| L*(Na+)2 | 6.6 | 3.0 | 15.1 | 4.1 | 4.2 |
| L*(K+)2 | 6.3 | 2.6 | 15.0 | 4.0 | 4.4 |
| L*(Mg2+)2 | 11.6 | 2.2 | 18.6 | 2.1 | 2.4 |
| L*(Ca2+)2 | 13.0 | 2.3 | 20.9 | 2.2 | 2.2 |
3.4 Analysis of excited state
The SOS method is generally not recommended for evaluation of first hyperpolarizability due to its less accuracy.59 However, the advantage of SOS is that the contribution from different states can be separated. The combination of the SOS approach (Fig. S2, ESI†) with the low-lying excited states, which contributes obviously to first hyperpolarizability, has been listed in Table 4.| Complex | S | ΔE | fos | MO transition |
|---|---|---|---|---|
| L | 2 | 2.8436 | 0.1760 | HOMO−1 → LUMO (83%), HOMO−3 → LUMO (10%) |
| L*(Li+)2 | 7 | 4.4291 | 0.8069 | HOMO → LUMO+10 (34%), HOMO−1 → LUMO+9 (15%), HOMO → LUMO+6 (12%) |
| L*(Na+)2 | 7 | 4.4184 | 0.7504 | HOMO → LUMO+10 (18%), HOMO → LUMO+5 (13%), HOMO−1 → LUMO+9 (10%), HOMO → LUMO+8 (10%) |
| L*(K+)2 | 7 | 4.4173 | 0.7712 | HOMO → LUMO+9 (24%), HOMO → LUMO+10 (23%) |
| L*(Mg2+)2 | 9 | 4.2446 | 0.6993 | HOMO → LUMO+6 (44%), HOMO−3 → LUMO+5 (22%), HOMO−2 → LUMO+6 (10%) |
| L*(Ca2+)2 | 9 | 4.2199 | 0.7716 | HOMO → LUMO+6 (23%), HOMO → LUMO+8 (13%), HOMO−3 → LUMO+5 (12%), HOMO → LUMO+4 (11%) |
For compound L, the S2 excited state has a large contribution to first hyperpolarizability. It is mainly made up of HOMO−1 → LUMO. These molecular orbitals (MO) involved in the main charge transfer transition are illustrated in Fig. 4. It is obvious that the transition of L can be assigned to the charge transfer from ethynylbenzo crown moiety to Pt and 2,2′-bipyridine moiety. The direction of charge transfer is opposite to the y-axis, which inevitably reflects the positive βy values. However, for metal cation derivatives, the transition of molecular orbital (MO) pairs is multiple and more complex. The most straightforward way to deal with this problem is to introduce the charge density difference (CDD) between the excited state and ground state (Fig. 5), which can be exactly evaluated as Δρ(r) = ρele(r) − ρhole(r). In single-electron excitation process, an electron leaves A and goes to B, they are named as “hole” and “electron”, respectively. The density distribution of holes and electrons are
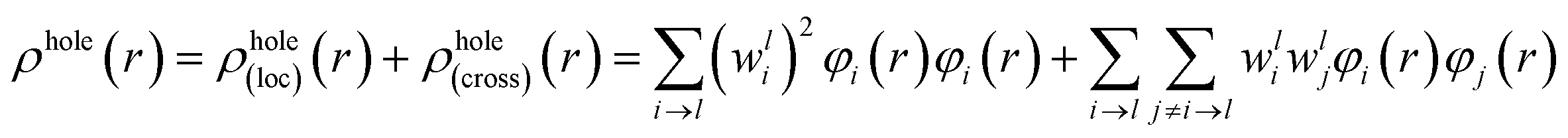 | (9) |
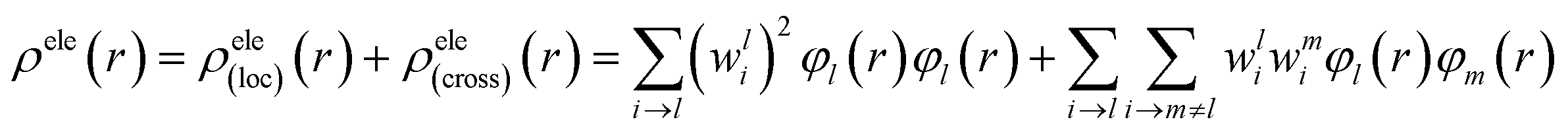 | (10) |
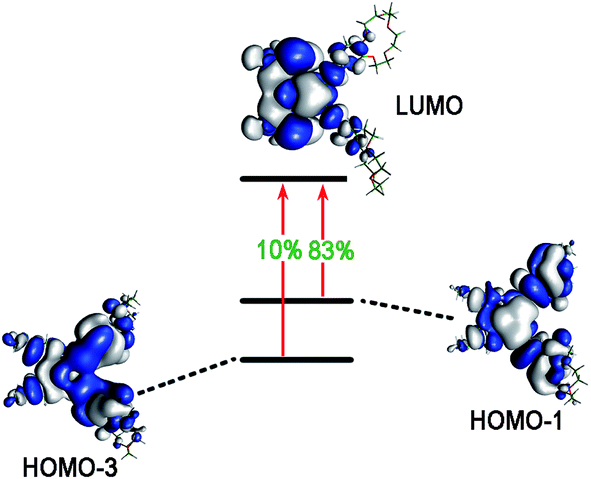 | ||
| Fig. 4 The molecular orbitals of L involved in the crucial excited states. | ||
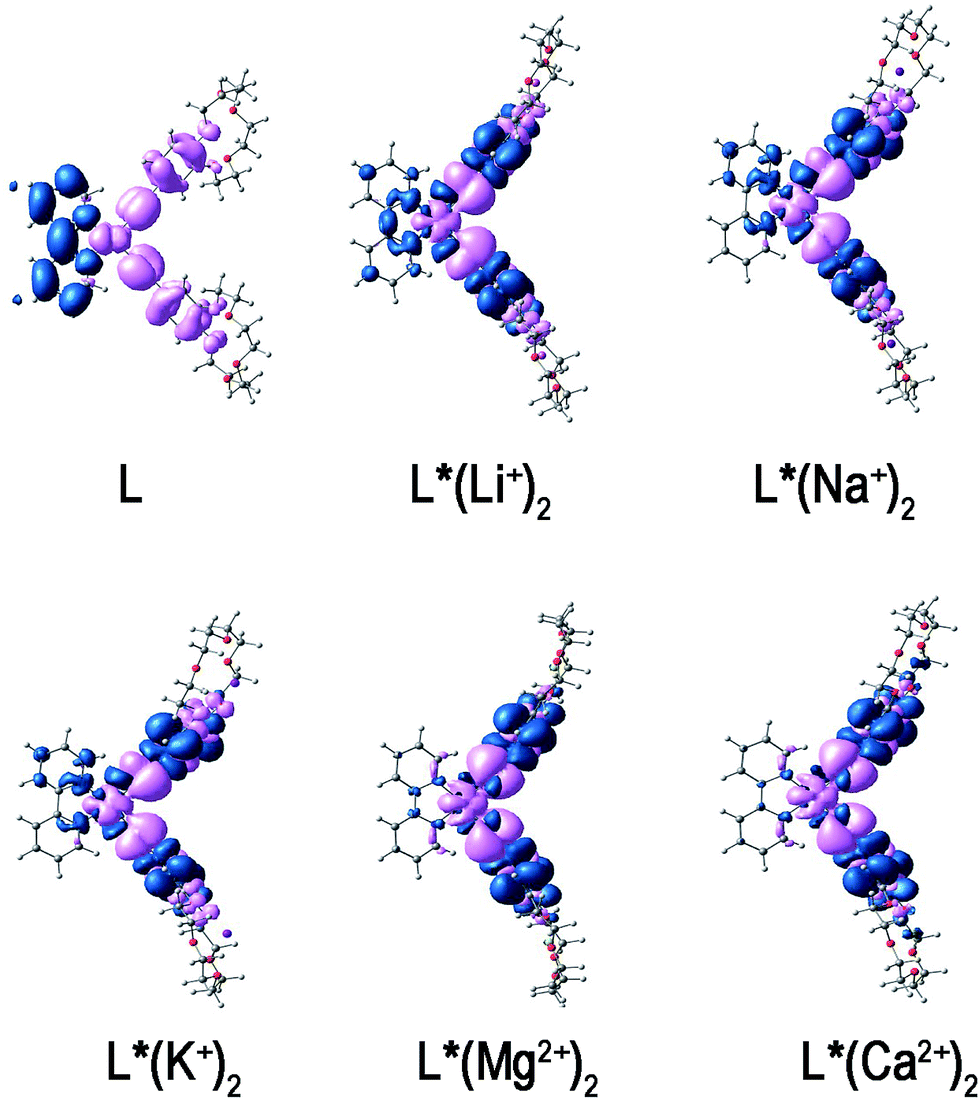 | ||
| Fig. 5 The CDD between the excited and ground states. Pink and blue colors indicate the depletion and the accumulation of electron density, respectively. | ||
From the CDD plots, it is quite obvious that for compound L, there is a charge transfer from ethynylbenzo crown moiety to Pt and 2,2′-bipyridine moiety. This result is in accordance with the MO analysis. It suggests that the regions of the density depletion and increment and the electronic transition involved in excited states can be easily identified according to the CDD analysis. For mono-cation (Li+, Na+, K+) derivatives, the distribution of holes is located on the Pt atom and ethynyl moiety, whereas the distribution of electrons is located on the bipyridine moiety with some on the phenyl moiety. Due to the extra and opposite charge transfer from Pt atom and ethynyl moiety to phenyl moiety, the strength of charge transfer to bipyridine moiety is weakened. It then reflects the smaller positive βy values of mono-cation (Li+, Na+, K+) derivatives. In contrast, the distribution of electrons of di-cations (Mg2+, Ca2+) derivatives is located on the phenyl moiety, while the distribution of holes is located on the Pt atom and ethynyl moiety. Obviously, there is a little confusion between the distribution of holes and electrons. A contrary direction of the charge transfer of di-cations (Mg2+, Ca2+) derivatives is shown as compared to L and its mono-cation (Li+, Na+, K+) derivatives. Thus, the βy values of di-cations (Mg2+, Ca2+) derivatives become smaller and negative.
From the complex SOS expression, Oudar has established the two-level model that linked between β and a low-lying charge transfer transition.60,61 In this two-level model, the first hyperpolarizability is proportional to the optical intensity and inversely proportional to the cube of the transition energy. Hence, the low excitation energy is the decisive factor in the β value. The transition energies at the low-lying and crucial states, which are contributed the most to the hyperpolarizability, are listed in Table 4. The transition energy increases in the order of L < L*(Mg2+)2 ≈ L*(Ca2+)2 < L*(Li+)2 ≈ L*(Na+)2 ≈ L*(K+)2. As seen from the results, when the metal cation coordinates to L, it brings larger transition energy and reduces the possibility of electron transition, thus generates a smaller β value.
4. Conclusion
Systematic DFT calculations have been conducted Pt(4-ethynylbenzo-15-crown-5)2(bpy) (bpy = 2,2′-bipyridine) complex and its derivatives with the complexation of mono-cations (Li+, Na+, K+) and di-cations (Mg2+, Ca2+). The binding of alkali/alkaline metal cations shrinks the crown ether obviously and distort them perpendicular to the bipyridine and Pt plane. Clearer charge reorganizations are characterized by the coordination of alkali metal cation onto L. The complexation of mono-cations and di-cations can induce a significant decrease of the first hyperpolarizability. This first hyperpolarizability contrast can be explained by different charge transfer transition from the standpoint of the distribution of holes and electrons. Therefore, the second-order NLO behaviors can be switched by different groups of alkali metal cations. These results will broaden the scope of the optical molecular detectors and inspired us to investigate further.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Science and Technology Research Project of the “13th Five-Year” Education Department of Jilin Province ([2016] 171), the financial support from the National Natural Science Foundation of China (No. 31502109), and the National Natural Science Foundation of China (No. 21503091). Additionally, we gratefully acknowledge the technical support from the group of Professor Yong-Qing Qiu and the usage of the computer and software from the Institute of Functional Material Chemistry of Northeast Normal University.References
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529–1533 CrossRef CAS.
- J. V. Slageren, A. Klein and S. Záliš, Coord. Chem. Rev., 2002, 230, 193–211 CrossRef.
- H. Y. Wang, C. Zheng and P. Q. Dai, RSC Adv., 2016, 6, 114078–114085 RSC.
- A. Klein, J. V. Slageren and S. Záliš, Eur. J. Inorg. Chem., 2003, 10, 1917–1938 CrossRef.
- S. W. Lai, M. C. W. Chan, K. K. Cheung and C. M. Che, Organometallics, 1999, 18, 3327–3336 CrossRef CAS.
- F. Q. Guo, W. F. Sun, Y. Liu and K. Schanze, Inorg. Chem., 2005, 44, 4055–4065 CrossRef CAS PubMed.
- D. K. Crites Tears and D. R. McMillin, Coord. Chem. Rev., 2001, 211, 195–205 CrossRef CAS.
- R. Büchner, J. S. Field and R. J. Haines, Inorg. Chem., 1997, 36, 3952–3956 CrossRef.
- J. Manna, C. J. Kuehl, J. A. Whiteford, P. J. Stang, D. C. Muddiman, S. A. Hofstadler and R. D. Smith, J. Am. Chem. Soc., 1997, 119, 11611–11619 CrossRef CAS.
- V. W. W. Yam, K. K. W. Lo and K. M. C. Wong, J. Organomet. Chem., 1999, 578, 3–30 CrossRef CAS.
- K. Onitsuka, M. Fujimoto, N. Ohshiro and S. Takahashi, Angew. Chem., Int. Ed., 1999, 38, 689–692 CrossRef CAS.
- P. Nguyen, P. Gómez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS PubMed.
- U. Belluco, R. Bertani, R. A. Michelin and M. Mozzon, J. Organomet. Chem., 2000, 600, 37–55 CrossRef CAS.
- K. M. C. Wong and V. W. W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed.
- V. W. W. Yam and K. M. C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- V. W. W. Yam, V. K. M. Au and S. Y. L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- P. K. M. Siu, S. W. Lai, W. Lu, N. Y. Zhu and C. M. Che, Eur. J. Inorg. Chem., 2003, 3, 2749–2752 CrossRef.
- B. Champagne, A. Plaquet, J. L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101–8103 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- H. G. Löhr and F. Vögtle, Acc. Chem. Res., 1985, 18, 65–72 CrossRef.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rice and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- R. Boulatov, B. Du, E. A. Meyers and S. G. Shore, Inorg. Chem., 1999, 38, 4554–4558 CrossRef CAS PubMed.
- S. T. Liddle and K. Izod, Organometallics, 2004, 23, 5550–5559 CrossRef CAS.
- Y. Lp, J. Yamauchi and N. Azuma, J. Coord. Chem., 1997, 42, 291–301 CrossRef.
- C. Lichtenberg, P. Jochmann, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2011, 50, 5753–5756 CrossRef CAS PubMed.
- C. I. C. Esteves, R. M. F. Batista, M. M. M. Raposo and S. P. G. Costa, Dyes Pigm., 2016, 135, 134–142 CrossRef CAS.
- M. Y. Zhang, C. H. Wang, W. Y. Wang, N. N. Ma, S. L. Sun and Y. Q. Qiu, J. Phys. Chem. A, 2013, 117, 12497–12510 CrossRef CAS PubMed.
- A. A. Melekhova, D. V. Krupenya, V. V. Gurzhiy, A. S. Melnikov, P. Y. Serdobintsev, S. I. Selivanov and S. P. Tunik, J. Organomet. Chem., 2014, 763–764, 1–5 CrossRef CAS.
- Y. Fan and D. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 6162–6171 CAS.
- C. Makedonas and C. A. Mitsopoulou, Inorg. Chim. Acta, 2007, 360, 3997–4009 CrossRef CAS.
- H. L. Yu, W. Y. Wang, B. Hong, Y. Zong and Y. L. Si, Phys. Chem. Chem. Phys., 2016, 18, 26487–26494 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- D. Herebian, K. E. Wieghardt and F. Neese, J. Am. Chem. Soc., 2003, 125, 10997–11005 CrossRef CAS PubMed.
- D. Espa, L. Pilia, L. Marchiò, F. Artizzu, A. Serpe, M. L. Mercuri, D. Simão, M. Almeida, M. Pizzotti, F. Tessore and P. Deplano, Dalton Trans., 2012, 41, 3485–3493 RSC.
- B. Champagne, E. A. Perpète, S. J. A. van Gisbergen, G. J. Snijders, C. Soubra-Ghaoui, K. A. Robins and B. Kirtman, J. Chem. Phys., 1998, 109, 10489–10495 CrossRef CAS.
- B. Champagne, E. A. Perpète and D. Jacquemin, J. Phys. Chem. A, 2000, 104, 4755–4763 CrossRef CAS.
- M. Torrent-Sucarrat, J. M. Anglada and J. M. Luis, J. Chem. Theory Comput., 2011, 7, 3935–3943 CrossRef CAS PubMed.
- L. E. Johnson, L. R. Dalton and B. H. Robinson, Acc. Chem. Res., 2014, 47, 3258–3265 CrossRef CAS PubMed.
- M. Guillaume, B. Champagne, N. Markova, V. Enchev and F. Castet, J. Phys. Chem. A, 2007, 111, 9914–9923 CrossRef CAS PubMed.
- F. Mancois, L. Sanguinet, J. L. Pozzo, M. Guillaume, B. Champagne, V. Rodriguez, F. Adamietz, L. Ducasse and F. Castet, J. Phys. Chem. B, 2007, 111, 9795–9802 CrossRef CAS PubMed.
- A. Plaquet, M. Guillaume, B. Champagne, L. Rougier, F. Mancois, V. Rodriguez, J. L. Pozzo, L. Ducasse and F. Castet, J. Phys. Chem. C, 2008, 112, 5638–5645 CAS.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J. L. Pozzo and V. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- F. Mancois, J.-L. Pozzo, J. Pan, F. Adamietz, V. Rodriguez, L. Ducasse, F. Castet, A. Plaquet and B. Champagne, Chem.–Eur. J., 2009, 15, 2560–2571 CrossRef CAS PubMed.
- E. Bogdan, L. Rougier, L. Ducasse, B. Champagne and F. Castet, J. Phys. Chem. A, 2010, 114, 8474–8479 CrossRef CAS PubMed.
- E. Bogdan, A. Plaquet, L. Antonov, V. Rodriguez, L. Ducasse, B. Champagne and F. Castet, J. Phys. Chem. C, 2010, 114, 12760–12768 CAS.
- F. Castet, E. Bogdan, A. Plaquet, L. Ducasse, B. Champagne and V. Rodriguez, J. Chem. Phys., 2012, 136, 024506 CrossRef PubMed.
- B. Champagne and B. Kirtman, J. Chem. Phys., 2006, 125, 024101 CrossRef PubMed.
- K. Sasagane, F. Aiga and R. Itoh, J. Chem. Phys., 1993, 99, 3738–3778 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 302, 375–382 CrossRef CAS.
- R. Liu, A. Azenkeng, D. Zhou, Y. H. Li, K. D. Glusac and W. F. Sun, J. Phys. Chem. A, 2013, 117, 1907–1917 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. K. M. Siu, S. W. Lai, W. Lu, N. Zhu and C. A. Che, Eur. J. Inorg. Chem., 2003, 2749–2752 CrossRef CAS.
- Y. Gao, S. L. Sun, H. L. Xu, L. Zhao and Z. M. Su, RSC Adv., 2014, 4, 24433–24438 RSC.
- T. Lu and F. W. Chen, Acta Phys.-Chim. Sin., 2012, 28, 1–18 CAS.
- A. Plaquet, B. Champagne and F. Castet, Molecules, 2014, 19, 10574–10586 CrossRef CAS PubMed.
- K. Sasagane, F. Aiga and R. Itoh, J. Chem. Phys., 1993, 99, 3738–3778 CrossRef CAS.
- J. L. Oudar, J. Chem. Phys., 1977, 67, 446–457 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04919f |
| This journal is © The Royal Society of Chemistry 2017 |