
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atmospheric chemistry of CH3O: its unimolecular reaction and reactions with H2O, NH3, and HF†
Mei-Ling Wei‡
a,
Xing-Feng Tan‡b,
Zheng-Wen Long*a and
Bo Long *ab
*ab
aDepartment of Physics, Guizhou University, Guiyang, 550025, China. E-mail: zwlong@gzu.edu.cn
bCollege of Material Science and Engineering, GuizhouMinzu University, Guiyang, 550025, China. E-mail: longbo@gzmu.edu.cn; wwwltcommon@sina.com
First published on 13th December 2017
Abstract
We have investigated the hydrogen atom transfer processes of CH3O to CH2OH without catalyst and with water, ammonia, and hydrofluoric acid as catalysts using ab initio methods, density functional theory (DFT) methods, and canonical variational transition state theory with small curvature tunneling (CVT/SCT). Herein, we have performed the benchmark barrier heights of the title reactions using W3X-L//CCSD(T)-F12a/VDZ-F12 methods. We have also performed the calculations of the combination of MPW-type, PBE-type exchange, M05-type, M06-type functional, and composite theoretical model chemistry methods such as CBS-QB3 and G4. We found that the M05-2X/aug-cc-pVTZ, mPW2PLYP/MG3S, M05-2X/aug-cc-pVTZ, and M06-2X/MG3S methods are performed better in different functionals with the unsigned errors (UEs) of 0.34, 0.02, 0.05, and 0.75 kcal mol−1 for its unimolecular reaction and reactions with H2O, NH3, and HF, respectively. The calculated results show that NH3 exerts the strongest catalytic role in the isomerization reaction of CH3O to CH2OH, compared with H2O and HF. In addition, the calculated rate constants show that the effect of tunneling increases the rate constant of the unimolecular reaction of CH3O by 102–1012 times in the temperature range of 210–350 K. Moreover, the variational effects of the transition state are obvious in CH3O + NH3. The calculated results also show that the direct unimolecular reaction of CH3O to CH2OH is dominant in the sink of CH3O, compared with the CH3O + H2SO4, CH3O + HCOOH, CH3O + H2O, CH3O + NH3, and CH3O + HF reactions in the atmosphere. The present results provide a new insight into catalysts that not only affect energy barriers, but have influences on tunneling and variational effects of transition states. The present findings should have broad implications in computational chemistry and atmospheric chemistry.
1. Introduction
Alkoxy radicals have received a great amount of attention because they play a key role in both combustion and atmospheric chemistry.1 The methoxy radical (CH3O) is one of the simplest alkoxy radicals.2 CH3O is produced from OH-initiated oxidation of CH4.2 In the atmosphere, CH3O undergoes unimolecular isomerization and decomposition and bimolecular reaction.1 While CH3O dominantly reacts with O2, responsible for the formation of HCHO and HO2, the CH2OH + O2 reaction is 104 times faster than the CH3O + O2 reaction, where CH2OH is formed through the hydrogen atom transfer of CH3O.3–5 Therefore, Radford stated that the isomerization reaction of CH3O could be an important process for the loss of CH3O.6 Exploring the unimolecular isomerization of CH3O is required to estimate the fate of CH3O in the atmosphere.The reaction kinetics and dynamics of methoxy radicals (CH3O) has been extensively investigated for both experimental and theoretical methods in the literature.1,7–19 However, the kinetics of the unimolecular isomerization reaction of CH3O remains unclear. With regard to the unimolecular reaction of CH3O, the energy barrier is very high, in the range of 26–36 kcal mol−1, depending on different theoretical methods.14,16,20–22 For example, Batt et al.16 estimated an energy barrier of 26.1 kcal mol−1, Tachikawa et al.22 reported an energy barrier of 32.88 kcal mol−1 calculated by CCSDST4/D95V**, Saebo et al.14 reported an energy barrier of 36 kcal mol−1 calculated by MP3/6-31G**. This uncertainty of energy barrier of CH3O unimolecular isomerization leads to the difficulty in quantitatively estimating the rate constant of CH3O unimolecular isomerization reaction. In addition, CH3O unimolecular isomerization is a hydrogen atom transfer (HAT) reaction. In particular, tunneling effects play a critical role in reaction kinesics for hydrogen transfer reactions,23,24 such as the unimolecular reactions of Criegee intermediates,25–28 CH3OH + OH,29 OH + H2SO4⋯NH3,30 unimolecular rearrangement of Rh(PH3)2ClCH4,31 H/D + CO,32,33 H/D + CH3OH,34 and Al + 3H2O.35 Therefore, it is necessary to reevaluate kinetics of the unimolecular isomerization reaction of CH3O.
The other issue is that there are some reports that water and atmospheric acids can remarkably decrease the energy barrier of hydrogen atom transfer reaction.36 More over water, sulfuric acid, and formic acid have been reported as catalysts to reduce the isomerization of methoxy to 25.7, 2.3, and 4.2 kcal mol−1, respectively.36 In particular, the calculated results are 25.7 kcal mol−1 by Buszek et al.36 and 22.9 kcal mol−1 by Kumar et al.5 at the CCSD(T)/aug-cc-pVTZ//QCISD/6-31G(d) and CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ, respectively. It is noted that the reported water catalytic CH3O isomerization of the energy barrier has difference of 2–3 kcal mol−1. This results lead to the inaccuracy of evaluating the kinetics of the methoxy unimolecular isomerization reaction. In addition, the catalytic effect of ammonia is better than water in the literature.37 Hydrofluoric acid is an important inorganic acid in the atmosphere. So, we calculated the H2O, NH3, and HF as catalysts in the unimolecular isomerization reaction of CH3O.
In this work, we investigated the hydrogen atom transfer processes of CH3O to CH2OH catalyzed by water, ammonia, and hydrofluoric acid using ab initio methods and density functional theory (DFT) methods, and canonical variational transition state theory with small curvature tunneling (CVT/SCT). We studied following reactions:
| CH3O → CH2OH | (1) |
| CH3O + M → CH2OH + M | (2) |
2. Computational methods
2.1. Benchmark calculation
It is of great necessity for studying the atmospheric reactions with high-accurate electronic structure methods to obtain quantitative results. We used the CCSD(T)-F12a/VDZ-F12 (ref. 38–40) and QCISD/VTZ41 methods for optimizing the reactants, pre-reactive complexes, transition states, post-reactive complexes, and products and calculating their corresponding frequencies. Single point energy calculations were carried out using the W2X42 and W3X-L42 methods at the CCSD(T)-F12a/VDZ-F12 and QCISD/VTZ optimized geometries, respectively. We have obtained the benchmark barrier heights of hydrogen atom transfer reactions for CH3O to CH2OH by different catalysts at the W3X-L//CCSD(T)-F12a/VDZ-F12 level as our best estimate. It is worth noting that W3X-L composite methods have been used in the reactions of Criegee intermediates with water,25 SO2 with OH,43 and HO2 with XCHO44 to obtain rate constants with experimental accuracy.2.2. Composite method calculation
Quantum chemical composite methods have developed because they approaches CCSD(T)/CBS.45 We used G4,45 unrestricted coupled cluster spin contamination corrected [UCCSD(T)], and unrestricted Brueckner doubles [UBD(T)] variations of the Weizmann-1 theory (W1), named as W1U and W1BD,46 and the CBS-QB3 (ref. 47) method.2.3. Density functional theory (DFT) calculation
We studied different functionals: (1) the generalized gradient approximation (GGA) such as B98 ref. 48) and BP86 (ref. 49–51; (2) depending on the density of Laplace or kinetic energy density of meta-GGA such as M11-L;52 (3) hybrid GGA with the addition of Hartree–Fock (HF) exchange to non-local information of occupied orbital such as BMK,53 HSEh1PBE,54–57 MPW1K,58–60 and MPW3LYP;61 (4) global-hybrid meta-GGA: B3LYP,62 M05-2X,63 M06-HF,64 and M06-2X;65 (5) range-separated hybrid meta GGA such as M11 (ref. 66) and range-separated meta-NGA such as MN12-SX;67 (6) double hybrid density functional using from both occupied and virtual orbital such as B2PLYP,68 B2PLYPD,69 and mPW2PLYP.70 These computations were finished using aug-cc-pVTZ,71–73 maug-cc-pVTZ,74 MG3S,75 and ma-TZVP76 basis sets.2.4. Reaction kinetics
The rate constants were calculated using canonical variational transition-state theory with small curvature tunneling (CVT/SCT).77–81 We selected the best functional for every specific reactions to do direct dynamics calculations by comparing with our best estimate. The unimolecular rate constants of CH3O to CH2OH was calculated by M05-2X/aug-cc-pVTZ, while the bimolecular rate constants of the CH3O + H2O, CH3O + NH3, and CH3O + HF reactions were calculated using mPW2PLYP/MG3S, M05-2X/maug-cc-pVTZ, and M06-2X/MG3S, respectively. Scale factors82 were used to scale all directly calculated harmonic vibrational frequencies, which are 0.964, 0.972, 0.964, and 0.970 for M05-2X/aug-cc-pVTZ, mPW2PLYP/MG3S, M05-2X/maug-cc-pVTZ, and M06-2X/MG3S, respectively.The optimization and frequency calculations of all geometries including reactants, transition states, and products calculated were carried out with the Gaussian 09 (ref. 83) suites of programs. The high level geometry optimization calculations are performed using the Molpro 2012 (ref. 84) suites of programs. Rate constants were calculated using the Polyrate 2010A85 and Gaussrate 2009 (ref. 86) dynamics codes.
3. Results and discussion
We have obtained the benchmark barrier heights of CH3O to CH2OH without catalyst and with water, ammonia, and hydrofluoric acid as catalysts using W3X-L//CCSD(T)-F12a/VDZ-F12 methods. We defined the unsigned error (UE) to determine which is the best functional, and UE is the absolute value of the difference between the computed barrier heights by different methods and the benchmark barrier heights calculated by W3X-L//CCSD(T)-F12a/VDZ-F12.3.1. The unimolecular isomerization of CH3O
The unimolecular isomerization of CH3O into CH2OH occurs via the transfer of the hydrogen atom of CH3 group to the oxygen atom in CH3O responsible for the formation of CH2OH as shown in Fig. 1. The unimolecular isomerization of CH3O into CH2OH has been extensively studied using different theoretical methods; the previous calculated results indicated that the barrier heights of the unimolecular isomerization of CH3O into CH2OH are varies between 26.1 and 36.0 kcal mol−1.14,16,20–22 Therefore, higher-level theoretical methods are required to obtain quantitative results. Herein, we use the benchmark calculation of beyond-CCSD(T) to obtain reliable results. The main results are summarized in Table 1 and Fig. 1, while all the results are provided in Table S1 (ESI).† The calculated results by W3X-L//CCSD(T)-F12a/VDZ-F12 indicate that the barrier height of the reaction is 29.56 kcal mol−1 in Table 1. Fig. 2 shows that the results are calculated by various density functional methods and ab initio methods, where the UEs are 0.13, 0.18, 0.32, and 0.34 kcal mol−1 using W1U, CBS-QB3, W1BD, and M05-2X/aug-cc-pVTZ, respectively; this results reflect slight changes for different theoretical methods. Therefore, the barrier height of the unimolecular isomerization of CH3O into CH2OH is computed to be 29.56 kcal mol−1 (W3X-L//CCSD(T)-F12a/VDZ-F12), which should be reliable. The W2X//CCSD(T)-F12a/VDZ-F12 result is 29.64 kcal mol−1, which agrees well with the value of 29.56 kcal mol−1; this shows that the electronic structures in the unimolecular reaction of CH3O to CH2OH do not represent a multireference character. Additionally, Table 1 shows that the barrier height of CH3O to CH2OH by W3X-L//QCISD/cc-pVTZ is calculated to be 28.99 kcal mol−1, which is 0.57 kcal mol−1 different from the value (29.56 kcal mol−1) calculated by W3X-L//CCSD(T)-F12/VDZ-F12; this result shows that the optimized geometries and calculated frequencies at the QCISD/cc-pVTZ are in adequate agreement with CCSD(T)-F12a/VDZ-F12 results, which has been observed in the SO2 + OH reaction,43 while the QCISD/cc-pVTZ-optimized geometries and calculated frequencies are reliable in the reactions of Criegee intermediates with H2O.25 Since the unimolecular reaction of CH3O to CH2OH and the SO2 + OH reaction are open-shell systems, the reliability of QCISD-optimized geometries and calculated frequencies for open-shell systems should be particular concerned to obtain quantitative results in competition with experimental accuracy with an error bar of 0.1–0.2 kcal mol−1.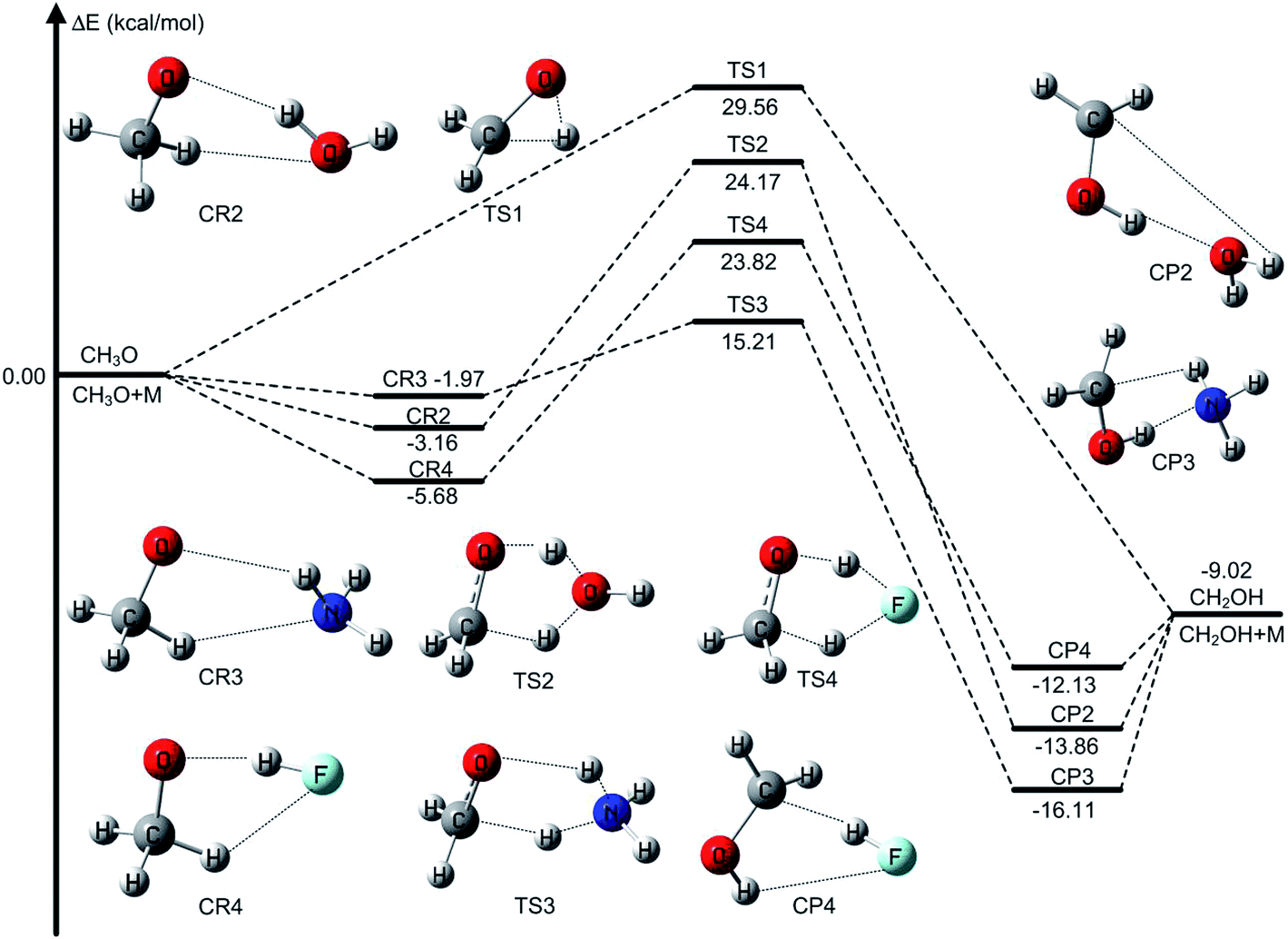 | ||
| Fig. 1 Variation in potential energy surface for the reactants, intermediates, transition states, and products of the CH3O isomerization into CH2OH in the without catalysis and catalyzed by water, ammonia, and hydrofluoric acid reactions at the W3X-L//CCSD(T)-F12a/VDZ-F12 level. | ||
| Methods | TS1 | UE | TS2 | UE | TS3 | UE | TS4 | UE |
|---|---|---|---|---|---|---|---|---|
| a Unsigned error (UE) obtained via the absolute value of the difference between the computed barrier heights and the benchmark barrier heights. | ||||||||
| W3X-L//CCSD(T)-F12a/VDZ-F12 | 29.56 | 0.00 | 24.17 | 0.00 | 15.21 | 0.00 | 23.82 | 0.00 |
| W2X//CCSD(T)-F12a/VDZ-F12 | 29.64 | 0.08 | 24.26 | 0.09 | 15.26 | 0.05 | 24.30 | 0.48 |
| W2X//QCISD/cc-pVTZ | 29.07 | 0.49 | 23.82 | 0.35 | 14.65 | 0.56 | 23.86 | 0.04 |
| W3X-L//QCISD/cc-pVTZ | 28.99 | 0.57 | 23.74 | 0.43 | 14.62 | 0.59 | 23.32 | 0.50 |
| W1U | 29.69 | 0.13 | 24.72 | 0.55 | 15.32 | 0.11 | 25.39 | 1.57 |
| W1BD | 29.88 | 0.32 | 24.88 | 0.71 | 15.47 | 0.26 | 25.11 | 1.29 |
| mPW2PLYP/MG3S | 31.33 | 1.77 | 24.15 | 0.02 | 15.48 | 0.27 | 22.92 | 0.90 |
| M06-2X/MG3S | 30.39 | 0.83 | 23.19 | 0.98 | 15.71 | 0.50 | 23.07 | 0.75 |
| G4 | 30.09 | 0.53 | 25.87 | 1.70 | 15.97 | 0.76 | 25.55 | 1.73 |
| CBS-QB3 | 29.74 | 0.18 | 25.24 | 1.07 | 15.06 | 0.15 | 27.62 | 3.80 |
| M05-2X/aug-cc-pVTZ | 29.90 | 0.34 | 21.35 | 2.82 | 15.16 | 0.05 | 20.41 | 3.41 |
| M05-2X/maug-cc-pVTZ | 30.04 | 0.48 | 21.65 | 2.52 | 15.40 | 0.19 | 20.80 | 3.02 |
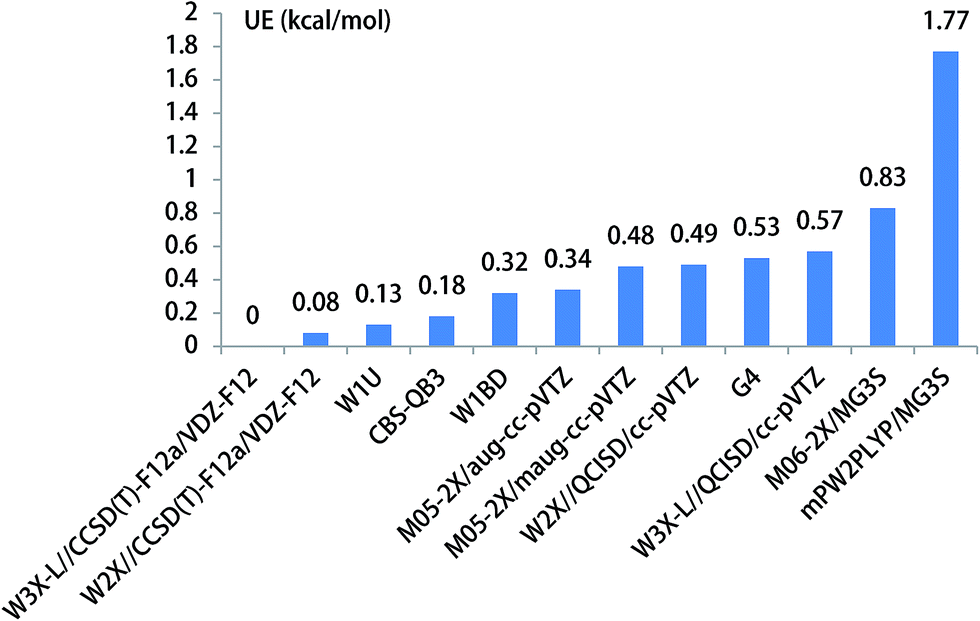 | ||
| Fig. 2 The unsigned error for the energy barrier of unimolecular isomerization reaction of CH3O to CH2OH. | ||
3.2. The bimolecular reactions of CH3O with H2O, NH3, and HF
The unimolecular isomerization of CH3O into CH2OH catalyzed by H2O, NH3, and HF occurs via the prereactive complex before the corresponding transition state and subsequently undergo the postreactive complex responsible for the formation of CH2OH as shown in Fig. 1. For example, when water acts as a catalyst, the hydrogen atom of CH3O is transferred to the oxygen atom in H2O and simultaneously the hydrogen atom of H2O is transferred to the terminal oxygen atom in CH3O responsible for the formation of CH2OH. The recent investigations have indicated that the energy barrier with water catalysis is 25.7 (ref. 36) and 22.9 (ref. 22) kcal mol−1 at the CCSD(T)/aug-cc-pVTZ//QCISD/6-31G(d) and CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ, respectively, which is 2.8 kcal mol−1 difference between the CCSD(T)/aug-cc-pVTZ//QCISD/6-31G(d) and CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculated results; this reflects that higher-level theoretical methods are necessary to obtain reliable results. Thus, we calculate the energy barrier of the unimolecular isomerization of CH3O into CH2OH with water as a catalyst usingW3X-L//CCSD(T)-F12a/VDZ-F12 theoretical method. The computed energy barrier is 24.17 kcal mol−1 in Table 1. Additionally, Fig. 3 shows that the UEs are 0.02, 0.55, and 0.71 kcal mol−1 for the mPW2PLYP/MG3S, W1U, and W1BD theoretical methods, respectively, comparing with the barrier height of TS2 calculated by W3X-L//CCSD(T)-F12a/VDZ-F12.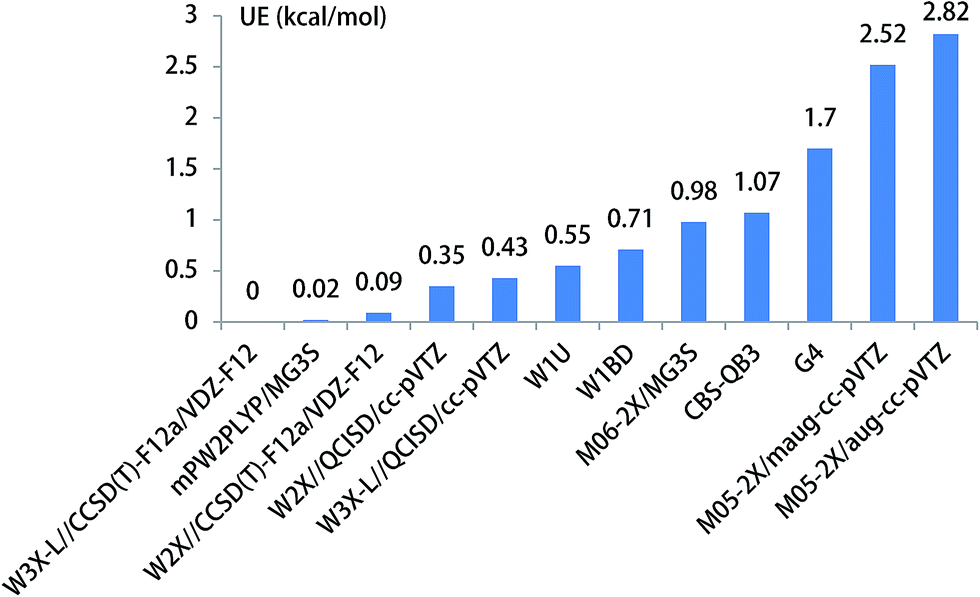 | ||
| Fig. 3 The unsigned error for the energy barrier of CH3O + H2O. | ||
Previous investigations have shown that ammonia has remarkably catalytic role in hydrogen transfer processes in the H2SO4⋯NH3 + OH,30 CF3OH + NH3,37 SO3 + H2O + NH3 (ref. 87) reactions. Herein, we investigate the CH3O + NH3 reaction, resulting in the formation CH2OH and NH3, where NH3 is acted as a catalyst. The optimized geometries of the transition state TS3 are provided in Fig. 1. The energy barrier of the CH3O + NH3 reaction is 15.21,15.26 kcal mol−1 using the best W3X-L//CCSD(T)-F12a/VDZ-F12 method, W2X//CCSD(T)-F12a/VDZ-F12 method in Table 1, which shows that the beyond-CCSD(T) calculations are not necessary for obtain quantitative results; this shows that there are not multireference features in the CH3O + NH3 reaction. In addition, the QCISD-optimized geometries and frequency calculations are still not adequate accurate to obtain quantitative results because the UE of W3X-L//QCISD/VTZ is 0.59 kcal mol−1, comparing with the results calculated by W3X-L//CCSD(T)-F12a/VDZ-F12 in Table 1. The calculated results also shows that NH3 has much stronger catalytic ability in the isomerization reaction of CH3O to CH2OH than H2O because the energy of the CH3O + NH3 reaction is about 9 kcal mol−1 lower than that of the CH3O + H2O reaction, which also agree with the previous investigation in the CF3OH + NH3 reaction.37 It is noted the UE of M05-2X/aug-cc-pVTZ is only 0.05 kcal mol−1 in Table 1 and Fig. 4. Thus, the M05-2X/aug-cc-pVTZ theoretical method is chosen to do direct dynamics calculations in the CH3O + NH3 reaction.
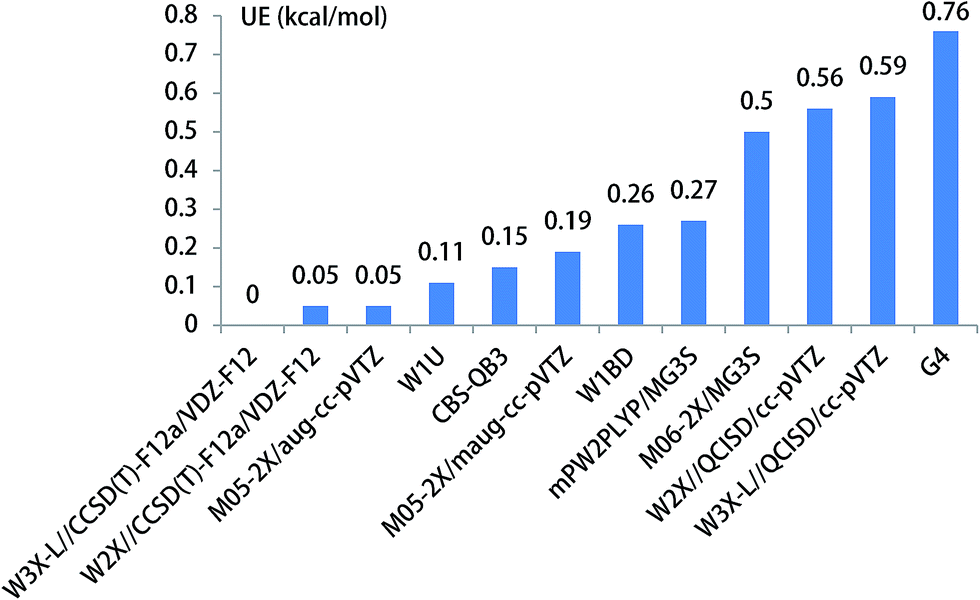 | ||
| Fig. 4 The unsigned error for the energy barrier of CH3O + NH3. | ||
When HF is acted as a catalyst in the CH3O + HF reaction responsible for the formation of CH2OH, the energy barrier is decreased to 23.82 kcal mol−1 in the CH3O + HF reaction from 29.56 kcal mol−1 in the unimolecular reaction of CH3O to CH2OH at the W3X-L//CCSD(T)-F12a/VDZ-F12 level in Table 1. In addition, it is particularly noted that the difference in the energy of the CH3O + HF reaction between W3X-L/CCSD(T)-F12 and W2X//CCSD(T)-F12a/VDZ-F12 is about 0.5 kcal mol−1, which shows that there are certain multireference features for the transition state TS4; this reveals that different catalyst may lead to the variation of nature of electronic structures in the transition states. Also, the W3X-L//QCISD/VTZ energy barrier is estimated to be 23.32 kcal mol−1, which is about 0.5 kcal mol−1 different from the W3X-L//CCSD(T)-F12a/VDZ-F12 in TS4; this shows that the QCISD/VTZ-optimized geometries and calculated frequencies still present unreliable results in estimating rate constants quantitatively for hydrogen transfer systems. It is noted that the CBS-QB3 result is 27.62 kcal mol−1 and the M05-2X/aug-cc-pVTZ result is 20.41 kcal mol−1 as listed in Table 1. The difference between CBS-QB3 and M05-2X/aug-cc-pVTZ is about 7.21 kcal mol−1. However, compared with the benchmark result of 23.82 kcal mol−1, the CBS-QB3 method overestimates the barrier height, while the M05-2X/aug-cc-pVTZ method underestimates the barrier in TS4. The UE of M06-2X/MG3S is about 0.75 kcal mol−1, which is the best functional for the CH3O + HF reaction as shown in Table 1 and Fig. 5.
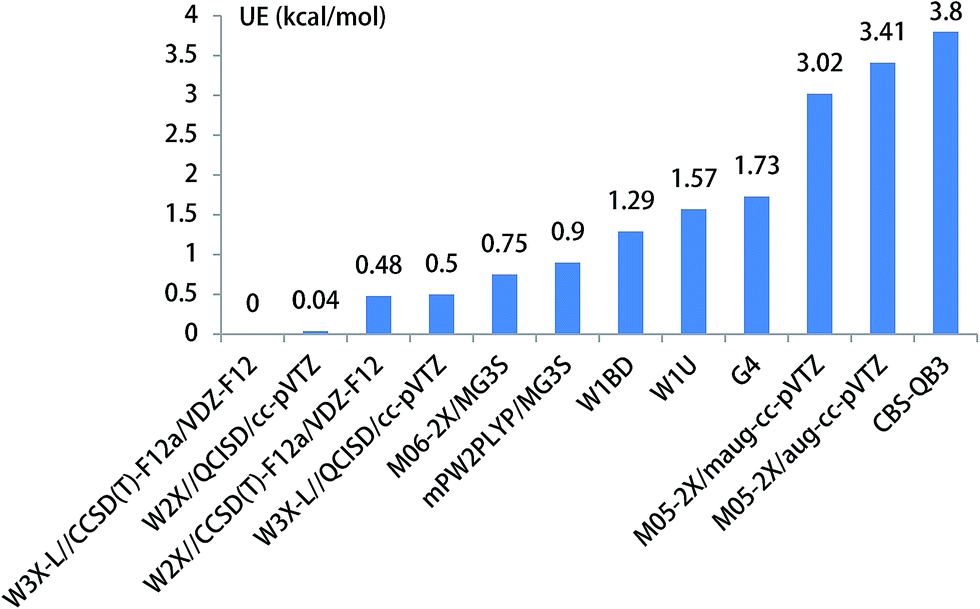 | ||
| Fig. 5 The unsigned error for the energy barrier of CH3O + HF. | ||
3.3. Rate constants
The calculated rate constants are presented in Table 2, where lists that the rate constants of the four reactions investigated herein are calculated using canonical variational transition-state theory with small curvature tunneling (CVT/SCT) in the temperature range of 210–350 K. Tunneling transmission coefficients are listed in Table 2, which shows that the tunneling transmission coefficients are very large for the hydrogen atom transfer process at 210 K. Furthermore, the tunneling transmission coefficient in the unimolecular isomerization of CH3O to CH2OH is even larger than the other reaction; in particular it is 3.29 × 1012 at 210 K (Table 2). It is also noted that tunneling effects are very remarkable in the CH3O unimolecular isomerization into CH2OH, CH3O + H2O, and CH3O + HF reactions, while the CH3O + NH3 reaction is not remarkable. For example, the tunneling coefficients are 4.71 × 104, 6.46 × 102, and 9.18 × 103 remarkable in the CH3O unimolecular isomerization into CH2OH, CH3O + H2O, and CH3O + HF reactions, while the tunneling coefficient in the CH3O + NH3 reaction is only 5.85 at 298 K (Table 2). It is particular noted that the energy barrier in the CH3O + NH3 reaction is the lowest of the four reactions; this shows that although NH3 exerts the strongest catalytic role in the CH3O unimolecular isomerization into CH2OH for three different catalysts, NH3 also reduces tunneling and consequently that the rate constants of the CH3O + NH3 reaction is still slow.| T/K | ΓTS1 | κTS1SCT | k1 | ΓTS2 | κTS2SCT | k2 | ΓTS3 | κTS3SCT | k3 | ΓTS4 | κTS4SCT | k4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a ΓTS1, ΓTS2, ΓTS3, and ΓTS4 are the rate constant ratios of canonical variational transition state to transition state theory in the CH3O unimolecular isomerization, CH3O + H2O, CH3O + NH3, and CH3O + HF reactions. κTS1SCT, κTS2SCT, κTS3SCT, and κTS4SCT are tunneling coefficients in the CH3O unimolecular isomerization, CH3O + H2O, CH3O + NH3, and CH3O + HF reactions. | ||||||||||||
| 210 | 0.96 | 3.29 × 1012 | 2.90 × 10−6 | 0.95 | 5.34 × 107 | 1.36 × 10−30 | 0.53 | 1.73 × 102 | 3.11 × 10−27 | 0.92 | 6.87 × 109 | 4.53 × 10−27 |
| 230 | 0.96 | 1.07 × 1010 | 5.26 × 10−6 | 0.95 | 1.18 × 106 | 4.20 × 10−30 | 0.56 | 4.51 × 101 | 2.02 × 10−26 | 0.92 | 9.48 × 107 | 6.89 × 10−27 |
| 250 | 0.96 | 1.06 × 108 | 1.06 × 10−5 | 0.96 | 6.26 × 104 | 1.42 × 10−29 | 0.59 | 1.85 × 101 | 1.25 × 10−25 | 0.93 | 3.01 × 106 | 1.13 × 10−26 |
| 270 | 0.96 | 2.57 × 106 | 2.41 × 10−5 | 0.96 | 6.54 × 103 | 5.12 × 10−29 | 0.61 | 1.02 × 101 | 6.93 × 10−25 | 0.93 | 1.86 × 105 | 2.02 × 10−26 |
| 290 | 0.97 | 1.30 × 105 | 6.12 × 10−5 | 0.96 | 1.15 × 103 | 1.92 × 10−28 | 0.63 | 6.68 × 100 | 3.38 × 10−24 | 0.94 | 1.98 × 104 | 3.89 × 10−26 |
| 298 | 0.97 | 4.71 × 104 | 9.15 × 10−5 | 0.96 | 6.46 × 102 | 3.27 × 10−28 | 0.63 | 5.85 × 100 | 6.14 × 10−24 | 0.94 | 9.18 × 103 | 5.17 × 10−26 |
| 310 | 0.97 | 1.20 × 104 | 1.72 × 10−4 | 0.96 | 3.02 × 102 | 7.27 × 10−28 | 0.64 | 4.92 × 100 | 1.44 × 10−23 | 0.94 | 3.29 × 103 | 8.07 × 10−26 |
| 330 | 0.97 | 1.83 × 103 | 5.28 × 10−4 | 0.96 | 1.08 × 102 | 2.72 × 10−27 | 0.66 | 3.91 × 100 | 5.37 × 10−23 | 0.94 | 7.87 × 102 | 1.78 × 10−25 |
| 350 | 0.97 | 4.20 × 102 | 1.73 × 10−3 | 0.96 | 4.88 × 101 | 9.91 × 10−27 | 0.67 | 3.26 × 100 | 1.78 × 10−22 | 0.94 | 2.54 × 102 | 4.09 × 10−25 |
The variational effects are also different from each other in Table 2. Of particular interest is the obvious variational effects in the CH3O + NH3, leading in further decreasing the rate constants of the CH3O + NH3 reaction. Thus, the catalyst not only has influences on the energy barriers, but affects on tunneling and variational effects of transition states.
It is worth noting that the rate constants of these reactions are increased with the increase of temperature. At 298 K, the rate constants of the CH3O isomerization into CH2OH, CH3O + H2O, CH3O + NH3, and CH3O + HF reactions are 9.15 × 10−5 s−1, 3.27 × 10−28 cm3 per molecule per s, 6.14 × 10−24 cm3 per molecule per s, and 5.17 × 10−26 cm3 per molecule per s, respectively. In addition, note that k4 is estimated to be 6.89 × 10−27–4.09 × 10−25 cm3 per molecule per s between 230 and 350 K, while k3 is computed 2.02 × 10−26–1.78 × 10−22 cm3 per molecule per s between 230 and 350 K; this shows k3 is larger than k4. However, in 210 K k4 is calculated to be 4.53 × 10−27 cm3 per molecule per s, which is slightly larger than that of k3 (3.11 × 10−27 cm3 per molecule per s) because the tunneling of TS4 is 6.87 × 109, which is much larger than that of TS3 (1.73 × 102).
3.4. Atmospheric implications
The calculated atmospheric lifetimes are provided in Table 3. With regard to the unimolecular reaction, τTS1 is calculated by , where k1 is the unimolecular rate constant of the reaction TS1, while for bimolecular reactions, τTS2, τTS3, τTS4 are calculated by
, where k1 is the unimolecular rate constant of the reaction TS1, while for bimolecular reactions, τTS2, τTS3, τTS4 are calculated by 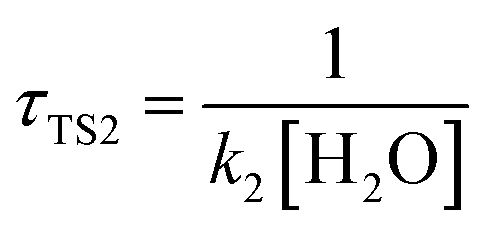 ,
, 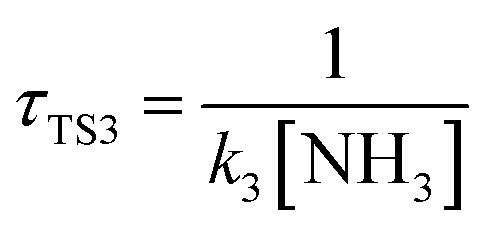 ,
, 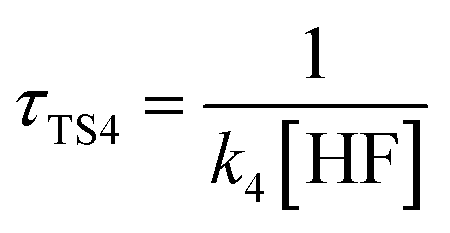 , where k2, k3, and k4 are the bimolecular rate constants of the reactions TS2, TS3, and TS4, respectively, and [H2O] is the concentration of H2O is 4.4 × 1017 molecule per cm3,88 [NH3] is the concentration of NH3 is 1.32 × 1012 molecule per cm3,89,90 and [HF] is the concentration of HF is 1.8 × 107 molecule per cm3,88 respectively in Table 3. With regard to the bimolecular reactions of CH3O + H2O, CH3O + NH3, and CH3O + HF, the atmospheric lifetimes are determined by both the rate constant and the corresponding concentrations of these catalysts in the atmosphere.
, where k2, k3, and k4 are the bimolecular rate constants of the reactions TS2, TS3, and TS4, respectively, and [H2O] is the concentration of H2O is 4.4 × 1017 molecule per cm3,88 [NH3] is the concentration of NH3 is 1.32 × 1012 molecule per cm3,89,90 and [HF] is the concentration of HF is 1.8 × 107 molecule per cm3,88 respectively in Table 3. With regard to the bimolecular reactions of CH3O + H2O, CH3O + NH3, and CH3O + HF, the atmospheric lifetimes are determined by both the rate constant and the corresponding concentrations of these catalysts in the atmosphere.
| T/K | τTS1a | τTS2b | τTS3b | τTS4b |
|---|---|---|---|---|
a For the unimolecular reaction,  , where k1 is the unimolecular rate constant of the reaction TS1.b For bimolecular reactions, , where k1 is the unimolecular rate constant of the reaction TS1.b For bimolecular reactions,  , , 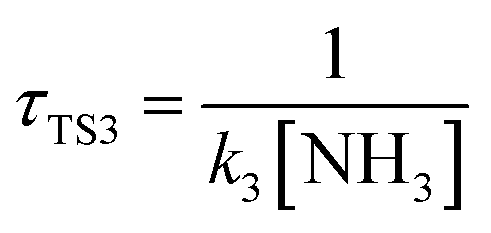 , ,  , where k2, k3, and k4 are the bimolecular rate constants of the reactions TS2, TS3, and TS4, respectively, and [H2O] is the concentration of H2O is 4.4 × 1017 molecule per cm3,88 [NH3] is the concentration of NH3 is 1.32 × 1012 molecule per cm3,94,95 and [HF] is the concentration of HF is 1.8 × 107 molecule per cm3,88 respectively. , where k2, k3, and k4 are the bimolecular rate constants of the reactions TS2, TS3, and TS4, respectively, and [H2O] is the concentration of H2O is 4.4 × 1017 molecule per cm3,88 [NH3] is the concentration of NH3 is 1.32 × 1012 molecule per cm3,94,95 and [HF] is the concentration of HF is 1.8 × 107 molecule per cm3,88 respectively. |
||||
| 210 | 3.4 × 105 | 1.7 × 1012 | 2.4 × 1014 | 1.2 × 1019 |
| 230 | 1.9 × 105 | 5.4 × 1011 | 3.8 × 1013 | 8.1 × 1018 |
| 250 | 9.4 × 104 | 1.6 × 1011 | 6.1 × 1012 | 4.9 × 1018 |
| 270 | 4.1 × 104 | 4.4 × 1010 | 1.1 × 1012 | 2.8 × 1018 |
| 290 | 1.6 × 104 | 1.2 × 1010 | 2.2 × 1011 | 1.4 × 1018 |
| 298 | 1.1 × 104 | 7.0 × 109 | 1.2 × 1011 | 1.1 × 1018 |
| 310 | 5.8 × 103 | 3.1 × 109 | 5.3 × 1010 | 6.9 × 1017 |
| 330 | 1.9 × 103 | 8.4 × 108 | 1.4 × 1010 | 3.1 × 1017 |
| 350 | 5.8 × 102 | 2.3 × 108 | 4.3 × 109 | 1.4 × 1017 |
The calculated results show that the direct unimolecular reaction of CH3O to CH2OH dominates the sink of CH3O. In particular, the atmospheric lifetime of the direct unimolecular reaction of CH3O to CH2OH is 5.8 × 102 s at 350 K (Table 3). The rate constants of the CH3O + H2SO4 and CH3O + HCOOH reactions are 9.12 × 10−14, 4.19 × 10−16 cm3 per molecule per s, respectively at 298 K.36 In the atmosphere, the concentration of H2SO4 is in the range of 104–4 × 108 molecules·per cm3.91–93 When the upper limit concentration of sulfuric acid is considered, the atmospheric lifetime of CH3O in the CH3O + H2SO4 reaction is 2.7 × 104 s at 298 K. The gas-phase concentration of formic acid is 1.1 × 1011 molecules·per cm3.93 The corresponding atmospheric lifetime is 2.2 × 104 s at 298 K. However, the atmospheric lifetime is 1.1 × 104 s for the direct unimolecular isomerization reaction of CH3O to CH2OH at 298 K, which shows that the direct unimolecular reaction of CH3O to CH2OH can compete well with the corresponding bimolecular reaction of CH3O + H2SO4 and CH3O + HCOOH.
4. Concluding remarks
The unimolecular reaction of CH3O to CH2OH catalyzed by different catalysts has been investigated by combining with W3X-L//CCSD(T)-F12a/VDZ-F12 benchmark calculations, the validated density functional, and canonical variational transition-state theory with small curvature tunneling. The main conclusions are extracted from the results as follows.(1) We considered significant pathways for the isomerization of CH3O to CH2OH via the reactions with water, ammonia, and hydrofluoric acid. The results show that different catalysts can decrease the energy barrier of the unimolecular isomerization of CH3O to CH2OH. The reductions of energy barriers for the isomerization of CH3O to CH2OH catalyzed by water, ammonia, and hydrofluoric acid are 5.39, 14.35, and 5.74 kcal mol−1, respectively, comparing with the energy barrier of the isomerization of CH3O to CH2OH without catalyst. Thus, the result shows that ammonia has the best catalytic ability among the three catalysts.
(2) We tabulate the unsigned error (UE) of the tested methods as listed in Table 1. The calculated results also show that the different functionals with basis sets have different accuracy. Among the functionals, the best method for the unimolecular isomerization of methoxy to hydroxymethyl and the bimolecular reaction of CH3O with NH3 are M05-2X/aug-cc-pVTZ. And, the best method for the bimolecular reactions of CH3O with H2O and HF are mPW2PLYP/MG3S and M06-2X/MG3S, respectively.
(3) The calculated rate constants show that catalysts can affect variational effects of transition states and tunneling. In addition, we show that the atmospheric lifetime of CH3O is mainly determined by the direct unimolecular reaction of CH3O to CH2OH due to tunneling, which has not been previously considered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by National Natural Science Foundation of China (41775125 and 41165007), Science and Technology Foundation of Guizhou Province & Guizhou Minzu University, China([2015]7211), Science and Technology Foundation of Guizhou Provincial Department of Education, China ([2015]350).References
- J. J. Orlando, G. S. Tyndall and T. J. Wallington, Chem. Rev., 2003, 103, 4657–4690 CrossRef CAS PubMed.
- A. R. Ravishankara, Annu. Rev. Phys. Chem., 1988, 39, 367–394 CrossRef CAS.
- W. B. DeMore, D. M. Golden, R. F. Hampson, C. J. Howard, M. J. Kurylo, M. J. Molina, A. R. Ravishankara and S. P. Sander, Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling (Evaluation Number 11), JPL Publication 94-26, Jet Propulsion Laboratory, Pasadena, CA, 1994 Search PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, J. Phys. Chem., 1997, 26, 521 CAS.
- P. Kumar, P. Biswas and B. Bandyopadhyay, Phys. Chem. Chem. Phys., 2016, 18, 27728–27732 RSC.
- H. E. Radford, Chem. Phys. Lett., 1980, 71, 195 CrossRef CAS.
- J. Chai, H. Hu, T. S. Dibble, G. S. Tyndall and J. J. Orlando, J. Phys. Chem. A, 2014, 118, 3552–3563 CrossRef CAS PubMed.
- H. Hu and T. S. Dibble, J. Phys. Chem. A, 2013, 117, 14230–14242 CrossRef CAS PubMed.
- O. S. And and M. Sato, J. Phys. Chem. A, 2002, 106, 8124–8132 CrossRef.
- J. M. Bofill, S. Olivella, A. Solé and J. M. Anglada, J. Am. Chem. Soc., 1999, 121, 1337–1347 CrossRef CAS.
- S. Dertinger, A. Geers, J. Kappert, J. Wiebrecht and F. Temps, Faraday Discuss., 1995, 102, 31–52 RSC.
- S. C. Foster, P. Misra, T. Y. D. Lin, C. P. Damo, C. C. Carter and T. A. Miller, J. Phys. Chem., 1988, 92, 5914–5921 CrossRef CAS.
- P. J. Wantuck, R. C. Oldenborg, S. L. Baughcum and K. R. Winn, J. Phys. Chem., 1987, 91, 18–23 Search PubMed.
- S. Saebo, L. Radom and H. F. S. Iii, J. Phys. Chem., 1983, 78, 845–853 CrossRef CAS.
- D. Gutman, N. Sanders and J. E. Butler, J. Phys. Chem., 1982, 86, 66–70 CrossRef CAS.
- L. Batt, J. P. Burrows and G. N. Robinson, Chem. Phys. Lett., 1981, 78, 467–470 CrossRef CAS.
- L. Batt, Int. J. Chem. Kinet., 1979, 11, 977–993 CrossRef CAS.
- J. L. Heicklen, Environ. Sci. Technol., 1976, 10, 310 CrossRef.
- J. A. Kerr, J. G. Calvert and K. L. Demerjian, Chem. Br., 1972, 8, 252–257 CAS.
- G. F. Adams, R. J. Bartlett and G. D. Purvis, Chem. Phys. Lett., 1982, 87, 311 CrossRef CAS.
- H. Tachikawa, Chem. Phys. Lett., 1993, 212, 27–31 CrossRef CAS.
- H. Tachikawa, S. Lunell, C. Tornkvist and A. Lund, J. Mol. Struct.: THEOCHEM, 1994, 304, 25–33 CrossRef.
- M. C. Smith, W. Chao, K. Takahashi, K. A. Boering and J. J.-M. Lin, J. Phys. Chem. A, 2016, 120, 4789–4798 CrossRef CAS PubMed.
- A. C. Davis and J. S. Francisco, J. Phys. Chem. A, 2010, 114, 11492–11505 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- Y. Fang, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, J. Chem. Phys., 2017, 146, 134307 CrossRef PubMed.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, J. Chem. Phys., 2016, 145, 234308 CrossRef PubMed.
- G. T. Drozd, T. Kurtén, N. M. Donahue and M. I. Lester, J. Phys. Chem. A, 2017, 121, 6036–6045 CrossRef CAS PubMed.
- R. J. Shannon, M. A. Blitz, A. Goddard and D. E. Heard, Nat. Chem., 2013, 5, 745–749 CrossRef CAS PubMed.
- B. Long, X. F. Tan, Y. B. Wang, J. Li, D. S. Ren and W. J. Zhang, ChemistrySelect, 2016, 16, 1421–1430 CrossRef.
- J. Espinosa-Garcia, C. J. Corchado and D. G. Truhlar, J. Am. Chem. Soc., 1997, 119, 9891–9896 CrossRef CAS.
- K. Hiraoka, T. Sato, S. Sato, N. Sogoshi, T. Yokoyama, H. Takashima and S. Kitagawa, Astrophys. J., 2002, 577, 265–270 CrossRef CAS.
- H. Hidaka, M. Watanabe, A. Kouchi and N. Watanabe, Astrophys. J., 2009, 702, 291–300 CrossRef CAS.
- T. P. M. Goumans and J. Kastner, J. Phys. Chem. A, 2011, 115, 10767–10774 CrossRef CAS PubMed.
- S. Alvarez-Barcia, J. R. Flores and J. Kastner, J. Phys. Chem. A, 2014, 118, 78–82 CrossRef CAS PubMed.
- R. J. Buszek, A. Sinha and J. S. Francisco, J. Am. Chem. Soc., 2011, 133, 2013–2015 CrossRef CAS PubMed.
- B. Long, X. F. Tan, Z. W. Long, D. S. Ren and W. J. Zhang, Chin. J. Chem. Phys., 2011, 24, 16–21 CrossRef CAS.
- T. B. Adler, G. Knizia and H. J. Werner, J. Chem. Phys., 2007, 127, 221106–221110 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H. J. Werner, J. Chem. Phys., 2008, 128, 084102–084113 CrossRef PubMed.
- G. Knizia, T. B. Adler and H. J. Werner, J. Chem. Phys., 2009, 130, 054104–054123 CrossRef PubMed.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS.
- B. Chan and L. Radom, J. Chem. Theory Comput., 2015, 11, 2019–2119 CrossRef PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2017, 19, 8091–8100 RSC.
- B. Long, X. F. Tan, J. L. Bao, D. M. Wang and Z. W. Long, Int. J. Chem. Kinet., 2016, 49, 130–139 CrossRef.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108–084119 CrossRef PubMed.
- E. C. Barnes, G. A. Petersson, M. J. Frisch Jr and J. M. Martin, J. Chem. Theory Comput., 2009, 5, 2687–2693 CrossRef CAS PubMed.
- H. E. Daniel and K. N. Houk, J. Phys. Chem. A, 2005, 109, 9542–9553 CrossRef PubMed.
- H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2016, 3, 117–124 CrossRef.
- A. D. Boese and J. M. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed.
- A. F. Izmaylov, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2006, 125, 8207–8357 Search PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 1133–1357 CrossRef PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 5121–5129 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810–2817 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 16187–16191 RSC.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108–034123 CrossRef PubMed.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397–3406 RSC.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 4398–4401 RSC.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- J. M. Anglada, J. Gonzalez and M. Torrent-Sucarrat, Phys. Chem. Chem. Phys., 2011, 13, 13034–13045 RSC.
- B. J. Lynch, Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2003, 107, 1384–1388 CrossRef CAS.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 CrossRef CAS.
- Y. P. Liu, G. C. Lynch, T. N. Truong, D. H. Lu, D. G. Truhlar and B. C. Garrett, J. Am. Chem. Soc., 1993, 115, 2408–2415 CrossRef CAS.
- T. Yu, J. Zheng and D. G. Truhlar, J. Phys. Chem. A, 2012, 116, 297–308 CrossRef CAS PubMed.
- J. Zheng and D. G. Truhlar, Faraday Discuss., 2012, 157, 59–88 RSC.
- J. L. Bao, R. Meana-Paneda and D. G. Truhlar, J. Phys. Chem., 2015, 6, 5866–5881 CAS.
- J. L. Bao, P. Sripa and D. G. Truhlar, Phys. Chem. Chem. Phys., 2016, 18, 1032–1041 RSC.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs Search PubMed.
- J. Zheng, S. Zhang, B. J. Lynch, J. C. Corchado, Y. Y. Chuang, P. L. Fast, W. P. Hu, Y. P. Liu, G. C. Lynch, K. A. Nguyen, C. F. Jackels, A. Fernandez-Ramos, B. A. Ellingson, V. S. Melissas, J. Villa, I. Rossi, L. Coitino, J. Pu, T. V. Albu, R. Steckler, B. C. Garrett, A. D. Issacson and D. G. Truhlar, POLYRATE – version, 2010-A, University of Minnesota, Minneapolis, 2013 Search PubMed.
- J. Zheng, S. Zhang, J. C. Corchado, Y. Y. Chuang, E. L. Coitiño, B. A. Ellingson and D. G. Truhlar, GAUSSRATE – version 2009-A, University of Minnesota, Minneapolis, 2009 Search PubMed.
- B. Bandyopadhyay, P. Kumar and P. Biswas, J. Phys. Chem. A, 2017, 121, 3101–3108 CrossRef CAS PubMed.
- G. Brasseur and S. Solomon, Aeronomy of the Middle Atmosphere: Chemistry and Physics of the Stratosphere and Mesosphere, Springer, New York, 3rd edn, 2005 Search PubMed.
- E. C. Tuazon, A. M. Winer and J. N. Pitts, Environ. Sci. Technol., 1981, 15, 1232–1237 CrossRef CAS PubMed.
- W. P. Robarge, J. T. Walker and R. B. McCulloch, Atmos. Environ., 2002, 36, 1661–1674 CrossRef CAS.
- F. L. Eisele and D. J. Tanner, J. Geophys. Res., 1993, 98, 9001–9010 CrossRef CAS.
- S. Mikkonen, S. Romakkaniemi, J. N. Smith, H. Korhonen, T. Petäjä, C. Plass-Duelmer, M. Boy, P. H. McMurry, K. E. J. Lehtinen, J. Joutsensaari, A. Hamed, R. L. Mauldin Iii, W. Birmili, G. Spindler, F. Arnold, M. Kulmala and A. Laaksonen, Atmos. Chem. Phys., 2011, 11, 11319–11334 CrossRef CAS.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- E. C. Tuazon, A. M. Winer and J. N. Pitts, Environ. Sci. Technol., 1981, 15, 1232–1237 CrossRef CAS PubMed.
- W. P. Robarge, J. T. Walker and R. B. McCulloch, Atmos. Environ., 2002, 36, 1661–1674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1 listing barrier heights at different theoretical methods and Table S2 providing Cartesian coordinates. See DOI: 10.1039/c7ra09167b |
| ‡ Mei-Ling Wei and Xing-Feng Tan contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |