DOI:
10.1039/C7RA09124A
(Paper)
RSC Adv., 2017,
7, 46812-46822
Preparation and evaluation of surface-grafted block copolymers and random copolymers via surface-initiated atom transfer radical polymerization for hydrophilic/ion-exchange stationary phases
Received
17th August 2017
, Accepted 18th September 2017
First published on 4th October 2017
Abstract
Mixed-mode chromatographic (MMC) stationary phases must be functionalized with at least two functional groups to yield multiple interactions. The present study proposed a novel approach of grafting copolymers with monomers with different properties via surface initiated-atom transfer radical polymerization (SI-ATRP) for the development of MMC stationary phases. In the synthesis, block copolymers and random copolymers containing sodium 4-styrenesulfonate (NASS) and dimethylaminoethyl methacrylate (DMAEMA) units were controllably grafted onto a silica surface via SI-ATRP for the preparation of hydrophilic/ion-exchange stationary phases. Upon investigation of their retention behaviors under different chromatographic conditions (i.e. water content, salt concentration and pH in mobile phase, column temperature) compared to a typical diol column, both the block and random copolymer stationary phases presented mixed-mode retention mechanisms involving hydrophilic and ion-exchange interactions. Furthermore, various solutes, such as basic β-agonists, strong polar nucleosides, organic acids, and a real sample of safflower injection, were employed to evaluate the separation selectivities of the stationary phases; similar selectivities and good separation efficiencies were achieved on the two copolymer columns. In conclusion, the method of surface-grafted copolymers via SI-ATRP possesses potential for further applications in the development of various MMC stationary phases.
1. Introduction
Chromatographic technology plays an important role in the analysis of complex samples, and the stationary phase is a core factor of high-performance liquid chromatography (HPLC).1 However, traditional single-mode stationary phases, such as those used in reverse-phase liquid chromatography (RPLC), ion-exchange chromatography (IEC), and hydrophilic interaction chromatography (HILIC), only provide a single interaction mechanism, leading to limited capacity for the analysis of very complex mixtures. Mixed-mode chromatography (MMC), which involves multiple interactions between the stationary phase and analytes, possesses good flexibility, high selectivity and broader analyte coverage for the simultaneous separation of diverse compounds; thus, it has the ability to meet the higher separation requirements of complex samples.2
To date, various MMC stationary phases have been reported, such as RPLC/IEC,3–7 RPLC/HILIC,8–10 HILIC/IEC,11,12 and RPLC/HILIC/IEC.13 Theoretically, in MMC, the stationary phase must be functionalized with at least two functional groups to yield multiple interactions with analytes. In terms of synthesis methods, the reported strategies for the preparation of MMC stationary phases are as follows: (i) the universal method is immobilization of a small molecule bearing at least two different groups,14–16 e.g. benzimidazole, which contains hydrophobic and ionic groups, to functionalize silica for an RPLC/IEC stationary phase;14 (ii) two different organic molecules are mixed to react with the substrate. For example, a RPLC/weak cation-exchange (WCX) stationary phase was prepared by mixing two silyl ligands with n-octadecyl and carboxyl groups;17 moreover, a zwitterionic stationary phase with a controllable ratio of positively charged groups to negatively charged groups was prepared by adjusting the proportion of two silylations, which respectively installed tertiary amino groups and carboxyl groups;18 (iii) a polymer-based substrate is synthesized by traditional in situ radical polymerization, such as poly(allylimidazole)-grafted silica, for an RPLC/anion-exchange stationary phase.19 However, monolayer-functional MMC stationary phases prepared by the first two approaches all provide limited interaction sites to analytes due to lower bonding capacity, resulting in poor selectivity.20,21 Moreover, conventional polymer stationary phases show lower column efficiency due to their heterogeneous structures and higher mass-transfer resistance.22 Recent progress has been made in polymerization synthesis; in particular, “living/controllable” surface-initiated atom transfer radical polymerization (SI-ATRP), which can offer well-controlled polymer thickness, desired compositions and molecular architectures to ensure good separation efficiency and selectivity of stationary phases, is an effective technique to develop novel separation materials.23 However, currently reported studies all use one monomer to form homopolymers on a substrate via SI-ATRP for the synthesis of single-mode stationary phases, such as grafting of poly(3-sulfopropyl methacrylate) for IEC24 and poly(octadecyl acrylate) for RPLC.25
In addition to homopolymers, SI-ATRP has been successfully applied in the preparation of advanced multifunctional materials by copolymerization of different types of monomers.26 In MMC, the stationary phase should possess at least two different functional groups to achieve multiple mode separation. Herein, we explored an effective strategy to develop MMC stationary phases by grafting copolymers with monomers with different properties via SI-ATRP, installing different functional groups on the surface of the substrate to provide multiple interactions with analytes. In addition to providing relatively accurate polymer thickness, SI-ATRP enables control and variation of the polymer structure (e.g. random copolymers, block copolymers, and gradient copolymers) by different polymerization processes. In fact, the surface structure of the stationary phase affects the separation efficiency. However, to date, no report in the literature has investigated the effects of copolymer structures on the chromatographic performance of a stationary phase prepared by grafting of copolymers via SI-ATRP.
In the present work, using different reported literature approaches for the preparation of MMC stationary phases, copolymers containing negatively charged sodium 4-styrenesulfonate (NASS) and positively charged dimethylaminoethyl methacrylate (DMAEMA) were grafted via SI-ATRP onto the surface of silica for the preparation of HILIC/IEC stationary phases. By utilizing the intrinsic property of SI-ATRP, where the synthesized polymer chain ends contain terminal radically transferable atoms that can be used for re-initiation of the next monomer to produce block copolymers,27 we adopted sequential grafting of NASS and DMAEMA units onto a silica surface via two-step SI-ATRP to obtain block copolymer-functionalized silica (poly(NASS-b-DMAEMA)@silica). In an alternative approach, we prepared random copolymer-functionalized silica (poly(NASS-co-DMAEMA)@silica) with a mixture of NASS and DMAEMA to undergo one-step SI-ATRP. During synthesis, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of NASS to DMAEMA units was controlled using block copolymerization and random copolymerization. The prepared silica materials were packed as HPLC columns to test the chromatographic performance. The influences of water content, salt concentration, pH of the mobile phase and column temperature on the retention behaviors of the two copolymer columns were investigated in detail. Furthermore, diverse solutes (basic β-agonists, strong polar nucleosides, organic acids, and real sample safflower injection) were used to compare the separation selectivities of the two copolymer columns, with the objective of observing the influences of the copolymer structures on the surface properties of the MMC stationary phases and studying their separation efficiencies.
:1 ratio of NASS to DMAEMA units was controlled using block copolymerization and random copolymerization. The prepared silica materials were packed as HPLC columns to test the chromatographic performance. The influences of water content, salt concentration, pH of the mobile phase and column temperature on the retention behaviors of the two copolymer columns were investigated in detail. Furthermore, diverse solutes (basic β-agonists, strong polar nucleosides, organic acids, and real sample safflower injection) were used to compare the separation selectivities of the two copolymer columns, with the objective of observing the influences of the copolymer structures on the surface properties of the MMC stationary phases and studying their separation efficiencies.
2. Experimental
2.1 Instruments and materials
All chromatographic experiments were carried out on a Shimadzu HPLC System (Kyoto, Japan), which consisted of two LC-20ATvp pumps, an SCL-10A system controller, a UV detector, and a Class VP 5.03 chromatographic workstation. An elemental analyzer (Varios EL III, Elementar Co., Ltd, Germany) was used to measure the chemical compositions of the stationary phases.
Spherical silica (5 μm diameter, 12 nm pore size and 310 m2 g−1 surface area) was used as the stationary phase substrate and was purchased from Fuji Silysia Chemical (Kasugai, Japan). Furthermore, 4-(chloromethyl)phenyltrimethoxysilane and γ-(2,3-epoxypropoxy)propytrimethoxysilane were purchased from J&K Chemical Reagent Co. Ltd (Beijing, China); copper(I) bromide (CuBr, >98%) and 2,2-bipyridyl (dpy, >99.5%) were purchased from Sinopharm Chemical Reagent Factory (Shanghai, China). Sodium 4-styrenesulfonate (NASS, >95.0%) and 2-dimethylaminoethyl methacrylate (DMAEMA, 98%) were purchased from Aladdin (Shanghai, China) and distilled under vacuum. Test solutes: nucleosides, benzoic acid and its analogs were obtained from Aladdin (Shanghai, China); β-agonists were kindly offered by the Ningxia Center for Disease Control; safflower injection was obtained from Lonch Group Wanrong Pharmaceutical Co., Ltd (Shanxi, China). HPLC-grade acetonitrile (ACN) from Kermel (Tianjin, China) and water purified by a Millipore purification system (Molsheim, France) were used as HPLC mobile phases.
2.2 Synthesis of the two copolymer stationary phases
2.2.1 Immobilization of initiator onto silica. Silica (6.0 g) was suspended in 90 mL of dried toluene; then, 4-(chloromethyl)phenyltrimethoxysilane (2.4 mL, 10.89 mmol) was added. Under nitrogen atmosphere, the reaction mixture was mechanically stirred and refluxed at 110 °C for 24 h. The initiated silica (silica–Cl) was filtered and washed with methanol and acetone in sequence and finally dried under vacuum at 50 °C before polymerization.
2.2.2 Preparation of the block copolymer stationary phase. As shown in Fig. 1a, the reaction was carried out by mixing silica–Cl (1.0 g) with dpy (0.156 g, 1.02 mmol) and the first monomer NASS (0.5 g, 2.4 mmol) in 20 mL methanol/water (v/v, 1:1) solution. The reaction mixture was ultrasonicated and then purged with nitrogen; CuBr (0.0717 g, 0.43 mmol) was added, the mixture was deoxygenated by a freeze–vacuum–thaw process, and then the reaction was performed at 35 °C under constant stirring for 3 h. After that, the product was filtered and repeatedly washed with methanol to remove excess reagents. Subsequently, the resulting product as the next initiator, dpy (0.156 g, 1.02 mmol), and DMAEMA (0.4 mL, 2.4 mmol) were mixed in methanol/water (v/v, 1:1) solution and bubbled with nitrogen; then, CuBr (0.0717 g, 0.43 mmol) was added, followed by stirring for 3 h at 35 °C. The product was washed with EDTA Na2 solution, to remove copper, repeatedly washed with water and methanol, and dried under vacuum at 50 °C overnight. Finally, the block copolymer poly(NASS-b-DMAEMA)-functionalized silica was obtained.
 |
| | Fig. 1 Synthesis of poly(NASS-b-DMAEMA)@silica (a) and poly(NASS-co-DMAEMA)@silica (b). | |
2.2.3 Preparation of the random copolymer stationary phase. Random copolymer poly(NASS-co-DMAEMA)-modified silica was prepared by a simple procedure (Fig. 1b). Silica–Cl (1.0 g), dpy (0.156 g, 1.02 mmol), and a mixture of NASS (0.5 g, 2.4 mmol) and DMAEMA (0.4 mL, 2.4 mmol) were dissolved in 20 mL methanol/water (1:1, v/v). The mixture was purged with nitrogen and CuBr (0.0717 g, 0.43 mmol) was added. After deoxygenation by freezing–vacuum–thaw, the reaction was performed for 3 h at 35 °C under stirring. The product was filtered, sequentially washed with EDTA Na2 solution, water and methanol, and finally dried under vacuum at 50 °C overnight.
2.3 Preparation of the diol-modified silica
Diol-modified silica as a control was synthesized according to ref. 28. Briefly, a mixture of silica (1.0 g), γ-(2,3-epoxypropoxy)propytrimethoxysilane (1 mL, 4 mmol) and 20 mL dry toluene was stirred and refluxed at 110 °C for 6 h. Then, the diol-modified silica was obtained by filtration, hydrolyzed in 50 mL of 0.1 mol L−1 sulfuric acid at 60 °C for 2 h, and finally washed with water until neutral and dried under vacuum.
2.4 Elemental analysis of silica materials
The elemental compositions of the silica materials with different polymerization methods were determined by elemental analysis. According to the S and N contents, the grafting amounts of NASS and DMAEMA were calculated by the following equations, respectively:29| | |
Grafted NASS = S% × 106/Sp(cal)% × (1 − S%/Sp(cal)%) × M1 × A
| (1) |
| | |
Grafted DMAEMA = N% × 106/Np(cal)% × (1 − N%/Np(cal)%) × M2 × A
| (2) |
where S% and N% represent the S and N percentages determined by elemental analysis, respectively; Sp(cal)% and Np(cal)% are the calculated weight percentages of S in NASS and N in DMAEMA, respectively; M1 and M2 are the formula weights of the NASS and DMAEMA monomers, respectively; and A is the specific surface area of the prepared silica in units of m2 g−1.
2.5 Column packing
The block copolymer-, random copolymer-, and diol-modified silica materials were respectively slurry-packed into a stainless steel column (100 mm × 4.6 mm i.d.) using isopropanol as the slurry solvent and methanol as the propulsion solvent under 40 MPa for 40 min.
2.6 Chromatographic conditions
For chromatographic evaluations, the mobile phase was prepared by mixing acetonitrile (ACN) and 20 mM ammonium formate solution (NH4COOH) at different volume ratios. The flow rate of the mobile phase was set at 1.0 mL min−1, and the column temperature was maintained at 30 °C unless otherwise specified. The detection wavelength was set at 240 nm. All test samples were first dissolved in the initial mobile phase at known concentrations and directly injected into the HPLC system for further analysis.
3. Results and discussion
3.1 Characterization of prepared stationary phases
In SI-ATRP, the polymer chains grow from the initiator, and an organic halide is always used as the initiator.30,31 As shown in Fig. 1, the initiator-functionalized silica (silica–Cl) was first prepared via a silyation reaction; then, the polymer chains grew from the initiators according to the ATRP mechanism. Normally, the polymer thickness is manipulated by the concentration of monomers and polymerization time.32 Herein, because both NASS and DMAEAM are ion-type monomers and possess similar polymerization rates to transform into copolymer chains, the grafting densities of the NASS and DMAEMA units in poly(NASS-b-DMAEMA) or poly(NASS-co-DMAEMA) were similar when the ratio of NASS to DMAEAM was controlled at 1:1 and the polymerization time was maintained at 3 h during the different polymerization processes. The grafting amounts of NASS and DMAEMA were calculated according to the element contents of S and N determined by elemental analysis (Table 1). Clearly, the grafting amounts of NASS were almost the same, while the amounts of DMAEMA on the block copolymer column were slightly lower than that on the random copolymer column. This demonstrates that the Cl on the initiator is slightly lost during block copolymerization, resulting in a decrease of bonding density for the next monomer.33
Table 1 Elemental contents of copolymer-modified silica materials
| Samples |
Measured elemental contents (%) |
Calculated grafting amounts (μmol m−2) |
| C |
H |
S |
N |
NASS |
DMAEMA |
| Silica |
0.16 |
0.306 |
— |
— |
— |
— |
| Silica–Cl |
3.88 |
0.41 |
— |
— |
— |
— |
| Block copolymer column |
20.70 |
2.36 |
1.60 |
0.61 |
1.78 |
1.50 |
| Random copolymer column |
20.95 |
2.46 |
1.54 |
0.73 |
1.72 |
1.83 |
3.2 Chromatographic behaviors on the two copolymer columns
NASS and DMAEMA are negatively and positively charged monomers, respectively; thus, the obtained stationary phases are essentially zwitterionic. The chromatographic properties of the new stationary phases were first evaluated in HILIC mode using six β-agonists as the test solutes; the results were compared with those from a diol column, which is a typical HILIC column. Theoretically, in HILIC, the retention time of the analyte exhibits a decreasing trend as the water content in the mobile phase increases. As shown in Fig. 2, the retention of the tested solutes on the three columns clearly displayed decreases as the water content increased from 11% to 15%, demonstrating typical HILIC retention mechanisms.
 |
| | Fig. 2 Effects of water content on the retention of β-agonists. Mobile phase, ACN-H2O; flow rate, 1.0 mL min−1; column temperature, 30 °C; detector wavelength, 254 nm. | |
Furthermore, to obtain a deeper understanding of the chromatographic properties of the two copolymer columns, it was necessary to clarify the retention mechanisms of polar solutes on the columns. At present, three models, partitioning mechanism,34 adsorption mechanism,35 and mixed-mechanism,36 are often used to elucidate the retention behavior in HILIC. Three empirical equations have been used to describe the partitioning retention mechanism for the retention of solutes based on partitioning between the mobile phase and a water-rich layer of the polar stationary phase (eqn (3)), the surface adsorption mechanism in normal phase chromatography systems (eqn (4)), and the mixed mechanism, which combines the partitioning mechanism and adsorption mechanism (eqn (5)).
| | |
logk = logkw − Sφ
| (3) |
| | |
logk = a + m1φ − m2logφ
| (5) |
where
φ refers to the concentration of water in the mobile phase in HILIC mode;
k is the retention factor of test solutes at different water contents;
kw and
kB are the retention factors using the pure weaker eluting solvent; and
a is an empirical constant.
The linear regression analysis for the retention factors of six β-agonists vs. water content on the three columns, based on eqn (3), (4) and (5), respectively, and the corresponding correlation coefficients are listed in Table 2. It was found that the linear regression of solutes on the diol column fitted to eqn (3), confirming that the retention behaviors of β-agonists followed the hydrophilic partitioning mechanism, as expected. However, for all solutes, the fitting of eqn (5) was better than those of eqn (3) and (4) on the two copolymer columns; this result demonstrated that a mixed-mechanism process rather than a simple partitioning or adsorption process was responsible for the retention on these columns. Due the structures of the functionalities on the surfaces of the stationary phases, NASS and DMAEMA can provide H-bonding and ion-exchange sites; therefore, multiple interactions between the developed stationary phases with basic β-agonists would result in multiple mechanisms.
Table 2 Correlation coefficients of β-agonists obtained by eqn (3)–(5)
| Solute |
Correlation coefficients |
| Block copolymer column |
Random copolymer column |
Diol column |
| Eqn (3) |
Eqn (4) |
Eqn (5) |
Eqn (3) |
Eqn (4) |
Eqn (5) |
Eqn (3) |
Eqn (4) |
Eqn (5) |
| Rectopamine |
0.829 |
0.800 |
0.988 |
0.927 |
0.960 |
0.98 |
0.997 |
0.987 |
0.953 |
| Phenylethylamine A |
0.980 |
0.945 |
0.995 |
0.963 |
0.952 |
0.991 |
0.951 |
0.929 |
0.893 |
| Clenbuterol |
0.969 |
0.949 |
0.989 |
0.970 |
0.959 |
0.992 |
0.990 |
0.986 |
0.944 |
| Formoterol |
0.984 |
0.974 |
0.992 |
0.982 |
0.977 |
0.990 |
0.983 |
0.960 |
0.891 |
| Salbutamol |
0.989 |
0.990 |
0.997 |
0.986 |
0.981 |
0.990 |
0.995 |
0.989 |
0.967 |
| Metaproterenol |
0.988 |
0.990 |
0.998 |
0.952 |
0.969 |
0.990 |
0.992 |
0.990 |
0.966 |
The separation chromatograms are shown in Fig. 3; under the same chromatographic conditions, it can be seen that the six β-agonists were separated better and retained more strongly on the two copolymer columns than on the diol column. Moreover, the elution orders of the test solutes exhibited the same trend on the two copolymer columns, while those of clenbuterol and formoterol, salbutamol and metaproterenol were reversed on the diol column. Theoretically, the retention of solutes in HILIC depends on their polarities; therefore, the elution order of the test solutes should be formoterol, clenbuterol, metaproterenol and salbutamol according to logP, which is a parameter to measure the polarity of a solute.37 Conventional diol columns retain solutes mainly based on hydrophilic interactions; as a result, the retention orders of the test solutes on this column corresponded to their polarities. However, for the two copolymer columns with sulfonic and tertiary amine group-based zwitterionic functionalities, the retention was due to multiple retention mechanisms composed of hydrophilic and electrostatic interactions, which led to deviation from the retention orders derived by the logP values of the β-agonists in HILIC mode and stronger retention as well as better resolution than on the diol column. In addition, the solutes on the two copolymer columns presented similar elution times (about 20 min) and selectivities; for example, the selectivity factors (α) of clenbuterol/formoterol and salbutamol/metaproterenol were 1.48 and 1.55 on the block copolymer column and 1.53 and 1.59 on the random copolymer column, respectively. This may be because the prepared silica materials containing block copolymers and random copolymers possessed the same compositions, resulting in similar interaction strengths.
 |
| | Fig. 3 Separation of β-agonists on the block copolymer column (A), random copolymer column (B) and diol column (C) at a temperature of 30 °C. Mobile phase, ACN-20 mM NH4COOH (88/12, v/v), pH 6.8; flow rate, 1.0 mL min−1; detection wavelength, 240 nm. Solutes: (1) rectopamine; (2) phenylethylamine A; (3) clenbuterol; (4) formoterol; (5) salbutamol; (6) metaproterenol. | |
3.3 Effect of buffer salt concentration in the mobile phase on the retention
In this study, the functionalized monomers, NASS with sulfonic groups and DMAEMA with tertiary amine groups, were expected to provide ion-exchange interactions with charged solutes. Normally, the salt concentration can affect the ionic strength and change the retention behavior of analytes. Among different salts in HILIC, ammonium formate is widely used due to its compatibility with mass spectrometry and its relatively good solubility in organic solvents. Herein, the effects of ammonium formate concentration on the retention of β-agonists were examined; the separation chromatograms on the three columns are displayed in Fig. 4. When the ammonium formate concentration was increased from 20 to 60 mM, as shown in Fig. 4C and F, the retention of six β-agonists on the diol column remained constant due to the absence of ionic exchange sites; however, for the two copolymer columns, the retention obviously decreased as higher ammonium formate concentrations were used in the mobile phase (Fig. 4D and E). Under the given conditions, the β-agonists (all pKa values above 9.2) were partly ionized and positively charged; therefore, they could interact with negatively charged sulfonic groups through electrostatic attraction and with positively charged tertiary amine groups through electrostatic repulsion. With increasing salt concentration, increasingly large amounts of buffer salt gathered around the charged groups, leading to suppression of the electrostatic interactions (attraction and repulsion),38 resulting in weaker retention at higher salt concentrations on the two copolymer columns.
 |
| | Fig. 4 Chromatograms of β-agonists using 20 and 60 mM NH4COOH in the mobile phase and plots of logk vs. logc on the block copolymer column (A, D and G), random copolymer column (B, E and H) and diol column (C, F and I). Mobile phase, ACN-20-80 mM NH4COOH (86/14, v/v); the other chromatographic conditions were the same as in Fig. 3. Solutes: (1) rectopamine; (2) phenylethylamine A; (3) clenbuterol; (4) formoterol; (5) salbutamol; (6) metaproterenol. | |
To further investigate the ionic strength between the two copolymer columns, the simple empirical stoichiometric displacement model was used to test the linear relationship between logk and logc, and the linear regression is described by eqn (6).39
| | |
logk = −slogc + constant
| (6) |
where
k and
c represent the retention factor of the solute and salt concentration in the mobile phase, respectively. The slope (
s) of log
k vs. log
c is always used to measure the intensity of hydrophilic and ion-exchange interactions between the stationary phase and the solute. In general, the absolute value of
s would be close to 0 when hydrophilic interaction plays a primary role and close to 1 when the ion-exchange interaction dominates. Herein, the effects of ammonium formate at various concentrations (from 20 to 80 mM) on the retention of β-agonists were studied in detail.
Fig. 4G–I display the linear relationships between log
k and log
c on the three columns, and the relevant parameters are listed in
Table 3. The six β-agonists showed
s values ranging from 0.122 to 0.705 on the block copolymer column, 0.122 to 0.658 on the random copolymer column and 0.109 to −0.002 on the diol column. Clearly, the two copolymer columns possessed multiple mechanisms of hydrophilic interaction and ion-exchange, while the diol column provided only hydrophilic interactions with the β-agonists. Moreover, the
s values of the test solutes on the block copolymer column were close to those on the random copolymer column, which demonstrated that the prepared columns by block copolymerization or random copolymerization could not only enable multiple interactions toward charged polar solutes, but could also provide similar interaction strengths. This may result from the similar surface properties of the block copolymer- and random copolymer-functionalized silica materials. SI-ATRP is a controllable polymerization technique; although two polymerization methods were used to form different copolymer structures on the prepared silica materials, the bonding amounts of negatively charged sulfonic groups and positively charged tertiary amine groups on the block copolymer column were very consistent with that on the random copolymer column due to control of the 1
:
1 ratio of NASS to DMAEMA in the different polymerization reactions. Hence, the similar surface properties,
i.e. polarity and charge, resulted in similar interaction strengths with charged solutes and presented similar sensitivities to salt concentration.
Table 3 Slopes (s) and correlation coefficients (R2) for plots of logk vs. logc of the three columns
| Column solute |
Block copolymer column |
Random copolymer column |
Diol column |
| s |
R2 |
s |
R2 |
s |
R2 |
| Rectopamine |
0.122 |
0.999 |
0.122 |
0.998 |
0.009 |
0.996 |
| Phenylethylamine A |
0.705 |
0.997 |
0.658 |
0.995 |
−0.018 |
0.995 |
| Clenbuterol |
0.702 |
0.997 |
0.684 |
0.992 |
−0.013 |
0.993 |
| Formoterol |
0.720 |
0.995 |
0.569 |
0.991 |
−0.008 |
0.992 |
| Salbutamol |
0.583 |
0.994 |
0.552 |
0.992 |
−0.003 |
0.994 |
| Metaproterenol |
0.608 |
0.980 |
0.519 |
0.980 |
−0.002 |
0.994 |
3.4 Effect of mobile phase pH
The effect of the pH of the mobile phase on the two copolymer columns was investigated; as shown in Fig. 5, the retentions of all basic β-agonists displayed decreasing trends on the developed columns, whereas the retention remained almost constant on the diol column as the pH increased from 4.0 to 8.0. This behavior on the two copolymer columns can be explained by two main causes: (i) for the solutes, basic β-agonists carried less positive charge as the pH increased, resulting in weaker electrostatic attraction and decreased retention of β-agonists; (ii) for the stationary phases, the pH-responsive poly(DMAEMA) in random copolymers and block copolymers could alter the surface properties of the stationary phase between hydrophilicity at low pH for protonation and hydrophobicity at high pH for deprotonation.40 Therefore, with increasing mobile phase pH, the retention of β-agonists decreased with the increasing hydrophobicity of the copolymers. This indicated that the introduction of DMAEMA units into block copolymers and random copolymers can confer additional pH-sensitive characteristics for regulating retention behavior during separation.
 |
| | Fig. 5 Effects of pH on the retention factors (k) of β-agonists. Columns: block copolymer column (A), random copolymer column (B), and diol column (C). Mobile phase, ACN-20 mM NH4COOH (86/14, v/v), other chromatographic conditions were the same as in Fig. 3. | |
3.5 Temperature-sensitive characteristics of the developed copolymer columns
It is well known that column temperature can affect the retention behaviors of solutes. Theoretically, in HILIC mode, the retention of solute decreases as the column temperature increases. To investigate the separation of β-agonists on three columns in HILIC mode at temperatures 20 °C and 60 °C, as shown in Fig. 6 (from A to F), only the diol column followed the rule where the retention of β-agonists decreased as the column temperature was changed from 20 °C to 60 °C; on the two copolymer columns, the retentions of the test solutes obviously increased at 60 °C, deviating from the normal rule of the effect of temperature on retention in HILIC mode. In addition, from the separation chromatograms of the two copolymer columns, higher resolution was clearly observed at a temperature of 60 °C than at 20 °C, suggesting that the separation efficiency could be improved simply by adjusting the column temperature. The relationships of the retention factors (k) of β-agonists with different column temperatures were investigated, as shown in Fig. 6(G–I). When the temperature was increased from 20 °C to 60 °C, the k value of each β-agonist gradually increased on the two copolymer columns, whereas the diol column still demonstrated the expected behavior of decreasing k value with increasing column temperature. This abnormal phenomenon was mainly ascribed to the fact that temperature-sensitive DMAEMA units are present in the block copolymers and random copolymers. At present, it has been documented that the extended poly(DMAEMA) chains gradually fold as the environmental temperature increases, inducing a positive charge on the DMAEMA hidden inside the copolymer chains by compacting it.41 Thus, the obtained results for the two copolymer columns can be explained by the fact that the electrostatic attraction between the basic β-agonist and negatively charged NASS units was strengthened because the folded poly(DMAEMA) chains induced more NASS units to be exposed to the surrounding water, resulting in enhancement of retention of the β-agonist with increasing column temperature. Overall, the introduction of temperature-responsive monomers into block copolymers or random copolymers can endow them with thermo-sensitive features to improve the selectivity of the stationary phase during analysis.
 |
| | Fig. 6 Chromatograms of β-agonists at temperatures of 20 °C and 60 °C, and effects of column temperatures on the retention factors (k) of β-agonists on the block copolymer column (A, D and G), random copolymer column (B, E and H), and diol column (C, F and I). Mobile phase, ACN-20 mM NH4COOH (86/14, v/v), pH 6.8; flow rate, 1.0 mL min−1; detection wavelength, 240 nm. Solutes: (1) rectopamine; (2) phenylethylamine A; (3) clenbuterol; (4) formoterol; (5) salbutamol; (6) metaproterenol. | |
3.6 Separation selectivity of the prepared copolymer columns
The above experiments mainly investigated the retention mechanisms based on basic β-agonists as the test solutes. Herein, to better explore the chromatographic properties of the prepared copolymer columns, highly polar nucleosides, organic acids, and a real sample of safflower injection were used to further evaluate the separation efficiencies of the developed columns.
As strong polar compounds, nucleosides are often used to evaluate the chromatographic properties of HILIC stationary phases.42 Usually, the classical HILIC column with diol functional groups is particularly suitable for the analysis of nucleosides. In this study, the developed mixed-mode columns were employed to separate six nucleosides in HILIC mode. As shown in Fig. 7, the two copolymer columns also provided effective separation of these nucleosides and exhibited the same elution order as the diol column, which retained highly polar analytes according to their hydrophilicity; this demonstrated that the retention of highly polar solutes on both copolymer columns followed the hydrophilic retention mechanism. Furthermore, in the analysis of nucleosides, not only the separation resolutions but also the retention times were very similar on our developed columns; this result further confirmed that the two copolymer materials with the same zwitterionic functionality possessed favorable hydrophilicity in the analysis of highly polar compounds.
 |
| | Fig. 7 Separation of nucleosides on the block copolymer column (A), random copolymer column (B) and diol column (C) at a temperature of 30 °C. Mobile phase, ACN-20 mM NH4COOH (95/5, v/v), pH 6.8; flow rate, 1.0 mL min−1; detection wavelength, 240 nm. Solutes: (1) uridine; (2) deoxythymide; (3) deoxyadenosine; (4) adenosine; (5) deoxyguanosine; (6) guanosine. | |
The chromatographic efficiencies of the two copolymer columns were determined by the separation of benzoic acid and its analogs. As shown in Fig. 8, the three columns exhibited significantly different selectivities for these organic acids under the same elution conditions. Clearly, the retention strength followed the order of random copolymer column > diol column > block copolymer column. The elution orders of the five organic acids were all in accord with their hydrophilicities on the three columns; brombenzeneacetic acid and benzoic acid were co-eluted on the diol column and block copolymer column owing to their similar hydrophilicities, whereas better separation of these solutes was observed on the random copolymer column. As mentioned in Section 3.1, the grafting amounts of NASS were almost the same on the two copolymer columns; however, the amounts of positively charged DMAEMA on the block copolymer column were slightly lower than that on the random copolymer column due to the gradual loss of initiator. The random copolymer column has a higher grafted capacity of DMAEMA and possesses more tertiary amino groups, leading to increased electrostatic attraction to acidic analytes; as a result, this column was more suitable for the separation of acidic solutes and, reasonably, achieved better selectivity. Overall, the grafting amounts of functional monomers on the stationary phase affect the separation efficiency. Therefore, when block polymerization of SI-ATRP is adopted to a develop mixed-mode stationary phase, the concentration or polymerization time of the post-monomer should be appropriately increased to control the grafting amounts, which can overcome the factors that adversely affect separation efficiency.
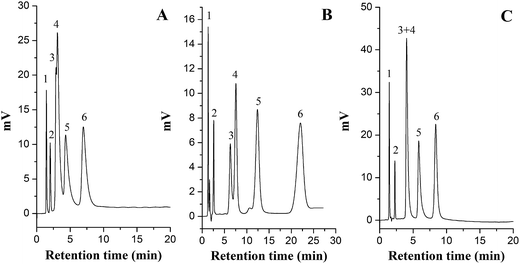 |
| | Fig. 8 Separation of organic acids on the block copolymer column (A), random copolymer column (B) and diol column (C) at a temperature of 30 °C. Mobile phase, ACN-20 mM NH4COOH (95/5, v/v), pH 6.8; flow rate, 1.0 mL min−1; detection wavelength, 240 nm. Solutes: (1) solvent peak; (2) aminobenozic acid; (3) brombenzeneacetic acid; (4) benzoic acid; (5) parabromobenzoic acid; (6) bromobenzoic acid. | |
Safflower injection, which is a popular Chinese traditional medicine and contains a large number of water-soluble compounds, is appropriately retained on HILIC columns. Herein, our prepared HILIC/IEC columns were applied in the separation of safflower injection to evaluate the capacity of the columns to analyze real samples. As shown in Fig. 9, almost fourteen peaks were detected by the home-made diol column, which is slightly higher than the dozen peaks in reported studies using commercial amino columns and the developed HILIC column.43 Compared with the diol column and the reported columns under the same chromatographic conditions, our prepared HILC/IEC copolymer columns showed better resolution, and the detected peak numbers were more than twice those of the diol column. In addition, it can be seen that the two copolymer columns afforded almost the same peaks and exhibited similar separation efficiencies. This further proved that the prepared stationary phases containing block copolymers and random copolymers provided near-multiple interaction strengths, obtaining similar selectivities in the separation of safflower injection. It can be seen that the two copolymer columns both have latent capacities for the analysis of complex samples.
 |
| | Fig. 9 Separation of safflower injection on the block copolymer column (A), random copolymer column (B) and diol column (C). Mobile phase, (A) ACN, (B) 5 mM NH4COOH, pH 6.8; gradient elution: 0 to 50 min: 5% to 30% B; 50 to 60 min: 30% B; flow rate, 1.0 mL min−1; detection wavelength, 288 nm. | |
4. Conclusion
Two novel stationary phases were respectively prepared by grafting block copolymers and random copolymers on the surface of silica via SI-ATRP using two functional monomers, NASS and DMAEMA. To investigate the chromatographic properties and applicabilities of the stationary phases, several conclusions were obtained as follows. (1) The grafting amounts of negatively charged sulfonic groups and positively charged tertiary amino groups on the surfaces of the stationary phases were controlled by the ratio of NASS to DMAEMA during block copolymerization and random copolymerization. This indicates that these controlled preparation methods enable adjustment of the selectivity of the stationary phase in the analysis of real samples. (2) Both the block copolymer and random copolymer columns were found to demonstrate hydrophilic and ion-exchange retention mechanisms by investigating the influences of water and salt content in the mobile phase on the retention behaviors, indicating that SI-ATRP is an effective method for the preparation of mixed-mode stationary phases. (3) The introduction of DMAEMA units in the copolymers additionally endowed the block copolymer and random copolymer columns with pH and temperature-sensitive characteristics, which enabled further adjustment of the separation selectivity at the optimal mobile phase pH and column temperature. (4) Similar selectivity was observed on the two copolymer columns in the separation of basic β-agonists, strong polar nucleosides, and a real sample of safflower injection. However, for acidic solutes, poor separation was obtained on the block copolymer column; this may be inhibited by increasing the concentration of the post monomer through block copolymerization. Generally, it can be seen that the methods of surface-grafting block copolymers or random copolymers via SI-ATRP have outstanding abilities for the development of novel mixed-mode stationary phases.
Conflicts of interest
The authors have declared no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21475104 and 21775121), the Industry Development Project by the Science and Technology Department of Shaanxi Province (No. 2016GY-214), and the Major Innovation Projects for Building First-class Universities in China's Western Region (ZKZD2017003).
References
- S. Sun, X. Zhang, Q. Han, W. Wan and M. Ding, Talanta, 2016, 149, 187–193 CrossRef CAS PubMed.
- M. Gilar, Y. Q. Yu, J. Ahn, J. Fournier and J. C. Gebler, J. Chromatogr. A, 2008, 1191, 162–170 CrossRef CAS PubMed.
- X. L. Li, Z. M. Guo, Q. Y. Sheng, X. Y. Xue and X. M. Liang, Analyst, 2012, 137, 2774–2776 RSC.
- Z. Zhang, Y. H. Hao, J. Ding, S. N. Xu, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2015, 1416, 64–73 CrossRef CAS PubMed.
- K. Peng, Q. Wang, W. Chen, D. Xia, Z. Zhou, Y. Wang, Z. Jiang and F. Wu, RSC Adv., 2016, 6, 100891–100898 RSC.
- Z. M. Guo, P. J. Zhang, F. F. Zhang, B. C. Yang and X. M. Liang, J. Chromatogr. A, 2012, 1246, 129–136 CrossRef PubMed.
- S. Tang, L. Wang, H. Han, H. Qiu, X. Liu and S. Jiang, RSC Adv., 2013, 3, 7894–7901 RSC.
- Q. Wang, M. Ye, L. Xu and Z. G. Shi, Anal. Chim. Acta, 2015, 888, 182–190 CrossRef CAS PubMed.
- X. Q. Qiao, L. Zhang, N. Zhang, X. Wang, X. Y. Qin, H. Y. Yan and H. Y. Liu, J. Chromatogr. A, 2015, 1400, 107–116 CrossRef CAS PubMed.
- K. Ohyama, Y. Inoue, N. Kishikawa and N. Kuroda, J. Chromatogr. A, 2014, 1371, 257–260 CrossRef CAS PubMed.
- T. Liang, Q. Fu, A. J. Shen, H. Wang, Y. Jin, H. Xin, Z. M. Guo and X. M. Liang, J. Chromatogr. A, 2015, 1388, 110–118 CrossRef CAS PubMed.
- L. Z. Qiao, S. G. Wang, H. Li, Y. H. Shan, A. B. Dou, X. Z. Shi and G. W. Xu, J. Chromatogr. A, 2014, 1360, 240–247 CrossRef CAS PubMed.
- Y. Li, J. Yang, J. Jin, X. Sun, L. Wang and J. Chen, J. Chromatogr. A, 2014, 1337, 133–139 CrossRef CAS PubMed.
- M. Sun, J. J. Feng, C. N. Luo, X. Liu and S. X. Jiang, Talanta, 2013, 105, 135–141 CrossRef CAS PubMed.
- M. G. Kiseleva and P. N. Nesterenko, J. Chromatogr. A, 2001, 920, 87–93 CrossRef CAS PubMed.
- S. Wongyai, Chromatographia, 1994, 38, 485–490 CAS.
- X. M. Cai, Z. M. Guo, X. Y. Xue, J. Y. Xu, X. L. Zhang and X. M. Liang, J. Chromatogr. A, 2012, 1228, 242–249 CrossRef CAS PubMed.
- X. D. Cheng, Y. H. Hao, X. T. Peng, B. F. Yuan, Z. G. Shi and Y. Q. Feng, Talanta, 2015, 141, 8–14 CrossRef CAS PubMed.
- M. Sun, H. Qiu, L. Wang, X. Liu, S. Jiang, M. Sun, H. Qiu, L. Wang, X. Liu and S. Jiang, J. Chromatogr. A, 2009, 1216, 3904–3909 CrossRef CAS PubMed.
- L. J. Wang, W. L. Wei, Z. N. Xia, X. Jie and Z. Z. Xia, Trends Anal. Chem., 2016, 80, 495–506 CrossRef CAS.
- L. Zhang, Q. Dai, X. Q. Qiao, C. Y. Yu, X. Y. Qin and H. Y. Yan, Trends Anal. Chem., 2016, 82, 143–163 CrossRef CAS.
- P. Hemstrom, M. Szumski and K. Irgum, Anal. Chem., 2006, 78, 7098–7103 CrossRef PubMed.
- K. Matyjaszewksi and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef.
- E. Unsal, B. Elmass, B. Caglayan, M. Tuncel, S. Patir and A. Tuncel, Anal. Chem., 2006, 78, 5868–5875 CrossRef CAS PubMed.
- A. Mallik, M. Rahman, M. Czaun, M. Takafuji and H. Ihara, J. Chromatogr. A, 2008, 1187, 119–127 CrossRef CAS PubMed.
- H. Wang, M. Song and T. Hang, ACS Appl. Mater. Interfaces, 2016, 8, 2881–2898 CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- L. A. Jurado, J. Mosely and H. W. Jarrett, J. Chromatogr. A, 2002, 971, 95–104 CrossRef CAS PubMed.
- A. Mizutani, K. Nagase, A. Kikuchi, H. Kanazawa, Y. Akiyama, J. Kobayashi, M. Annaka and T. Okano, J. Chromatogr. A, 2010, 1217, 522–529 CrossRef CAS PubMed.
- K. Matyjaszewski, P. J. Miller, N. Shukla, B. Immaraporn, A. Gelman, B. B. Luokala, T. M. Siclovan, G. Kickelbick, T. Vallant, H. Hoffmann and T. Pakula, Macromolecules, 1999, 32, 8716–8724 CrossRef CAS.
- Q. Q. Gai, F. Qu, Z. J. Liu, R. J. Dai and Y. K. Zhang, J. Chromatogr. A, 2010, 1217, 5035–5042 CrossRef CAS PubMed.
- D. Xu, X. C. Dong, H. Y. Zhang, H. S. Wang, P. Jiang and M. Zhang, J. Sep. Sci., 2012, 35, 1573–1581 CrossRef CAS PubMed.
- L. Ran, L. Wu and Z. Zhang, Prog. Polym. Sci., 2014, 39, 124–144 CrossRef.
- A. J. Alpert, J. Chromatogr., 1990, 499, 177–196 CrossRef CAS PubMed.
- P. Hemstrom and K. Irgum, J. Sep. Sci., 2006, 29, 1784–1821 CrossRef PubMed.
- P. Jandera, T. Hajek, V. Skerikova and J. Soukup, J. Sep. Sci., 2010, 33, 841–852 CrossRef CAS PubMed.
- L. Sun, L. Zhang, Y. L. Zhu, S. H. Wang and X. Wang, Chin. J. Chromatogr., 2008, 20, 709–713 Search PubMed.
- A. J. Alpert, Anal. Chem., 2008, 80, 62–76 CrossRef CAS PubMed.
- J. Ståhlberg, J. Chromatogr. A, 1999, 855, 3–55 CrossRef.
- Y. Shen, L. Qi, X. Wei, R. Zhang and L. Mao, Polymer, 2001, 52, 3725–3731 CrossRef.
- C. Sakamoto, Y. Okada, H. Kanazawa, E. Ayano, T. Nishimura, M. Ando, A. Kikuchi and T. Okano, J. Chromatogr. A, 2004, 1030, 247–253 CrossRef CAS PubMed.
- X. T. Peng, B. F. Yuan and Y. Q. Feng, J. Sep. Sci., 2011, 34, 3123–3130 CrossRef CAS PubMed.
- Y. Y. Li, L. Xu, T. Chen, X. Y. Liu, Z. G. Xu and H. X. Zhang, Anal. Chim. Acta, 2012, 726, 102–108 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab and
Yinmao Wei
ab and
Yinmao Wei







