
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of rhodium content on the behavior of Rh/SiO2–Al2O3 catalysts for selective ring opening of decalin
Silvana A. D'Ippolitoa,
Laurence Pirault-Royb,
Catherine Especelb,
Florence Epron *b and
Carlos L. Piecka
*b and
Carlos L. Piecka
aInstituto de Investigaciones en Catálisis y Petroquímica (INCAPE) (FIQ-UNL, CONICET), Colectora Ruta Nac. No. 168 – Paraje El Pozo, CP 3000, Santa Fe, Argentina
bInstitut de chimie des milieux et des matériaux de Poitiers (IC2MP), Université de Poitiers, UMR 7285 CNRS, 4, rue Michel Brunet, 86073 Poitiers cedex 9, France. E-mail: florence.epron@univ-poitiers.fr
First published on 4th October 2017
Abstract
The influence of the Rh content of catalysts supported on SiO2–Al2O3 (SIRAL 70 and 80, with 70 and 80 wt% of SiO2) on the activity and selectivity for selective ring opening of decalin was studied at 325 and 350 °C. An optimum metal content was found at 1.5 wt% of rhodium. Both supports present moderate and strong acid sites. For the selective opening of decalin, the best metal/support combination was found with Rh(1.5)/SIRAL 70 balancing the support acidity together with the hydrogenolytic activity of the metal. A linear correlation was established between the amount of coke deposited and the formation of dehydrogenated compounds.
Introduction
Light cycle oil (LCO) is an unwanted liquid residue of low commercial value produced on Fluid Catalytic Cracking (FCC) units. LCO is composed by hydrocarbons with the same number of carbon atoms than diesel. However, the amount of aromatic and polyaromatic components turns the oil unusable as diesel due to environmental specifications. Moreover, LCO fractions have low cetane index. Conversion into diesel by selective ring opening has been proposed. The removal of sulphur and nitrogen compounds as well as the transformation of aromatics into naphthenics could be achieved by conventional technologies.1 Nevertheless, it is necessary to open the ring of the naphthenic compounds in order to achieve an adequate cetane index.2–4 LCO mainly consists of aromatic compounds with two rings, therefore, decalin can be considered as a model molecule.5 Calemma et al.6 reported that the results obtained in the catalytic hydroconversion of desulfurized and dearomatized LCOs are in agreement with those obtained when using decalin as a model molecule.Bifunctional catalysts are generally used to achieve the selective ring opening (SRO) of six-membered naphthenic molecules.7–16 Acid sites catalyze cracking, isomerization and dealkylation, while the metal sites promote de/hydrogenation and hydrogenolysis.17 Hydrogenolysis of cycloalkanes, especially four and five-membered rings cycloalkanes, was first studied in depth on Pt/Al2O3 by François Gault and his collaborators,18–20 who laid the foundation of the reaction mechanisms that are still used by the heterogeneous catalysis community. Studies using supported metallic catalysts including Ir, Pt, Ru and Rh showed that the hydrogenolytic activity displays the following order: Pt < Rh < Ir < Ru,21,22 while the ring opening capacity depends on a large number of factors, such as particle size,23 the nature and acidity of the support24–26 and of the naphthenic compound.2,27 In general, the use of large pore zeolites favors the ring-opening reaction.28 Galadima and Muraza pointed out that zeolites promoted by metals are more active, and can achieve up to 100% conversion of a model feed with a selectivity greater than 70% to hydrocarbons suitable to improve the cetane number.29 Nevertheless, with zeolites-based materials, limitation occurs by deactivation due to coke formation.30–32 Amorphous silica-alumina can advantageously substitute zeolites as metal support, reducing excessive cracking activity.33–36 However, some key issues such as the role of the acid and textural properties of support, the effect of the feed composition on catalyst stability and the influence of reaction parameters such as temperature and hydrogen pressure to improve performance on diesel hydrocarbons need to be addressed.
By screening different Rh catalytic systems supported on SiO2–Al2O3 with two different Si/Al ratios, this work aims to determine the most suitable metal–support combination to direct the hydrogenolytic capacity of the metal towards the endocyclic C–C bond rupture of decalin and to inhibit the exocyclic rupture.
Experimental
Catalysts preparation
Commercial SiO2–Al2O3 provided by SASOL (SIRAL 70 and 80) were used as support. The main characteristics of these supports are the following: SIRAL 70, 28.5 wt% Al2O3 and 71.5 wt% SiO2, 377 m2 g−1; SIRAL 80, 21.0 wt% Al2O3 and 79.0 wt% SiO2, 337 m2 g−1. The support was firstly calcined at 450 °C for 4 h in air flow (60 cm3 min−1), and then impregnated with HCl (0.2 mol L−1). After one hour at rest, the required amount of RhCl3(aq.) precursor solution was added in order to obtain a metallic charge of 1.0, 1.5 and 2.0 wt% of Rh. The suspension was stirred gently for 1 h, then dried at 70 °C in a thermostatic bath until a dry powder was obtained. This powder was left overnight in an oven at 120 °C. Finally, the catalysts were calcined (air, 60 cm3 min−1, 300 °C, 4 h) and reduced (H2, 60 cm3 min−1, 500 °C, 4 h). The catalysts were labelled as Rh(X)/Sy, where “x” is the theoretical wt% content of Rh and “y” the SIRAL support number (SIRAL 70 or SIRAL 80).Temperature-programmed desorption (TPD) of pyridine
This test was used to measure the amount and strength of the acid sites. Samples of 200 mg were impregnated with a pyridine excess. The samples were then rinsed and the excess of physisorbed pyridine was eliminated by heating the sample in a nitrogen stream at 110 °C for 1 h. Then the temperature was raised at a rate of 10 °C min−1 to a final value of 750 °C. To measure the amount of desorbed pyridine, the reactor exhaust was connected to a flame ionization detector (FID). The error associated to the peak position and areas has been determined to be of about 7%.37Fourier transform infrared (FTIR) spectroscopy of adsorbed pyridine
Self-supported wafers with a diameter of 16 mm and a weight of 15 ± 4 mg were used. Each wafer was placed in the analysis quartz cell equipped with KBr windows and pretreated in air flow at 450 °C (heating ramp of 2 °C min−1) for 12 h. Then the temperature was decreased at 200 °C and the cell containing the wafer was treated under ultra-high vacuum for 1 h for removing all physisorbed species. The cell was then cooled to 150 °C, a reference spectrum was measured and pyridine was injected into the system (250 Pa, 10 min). The non-adsorbed excess was eliminated by application of a vacuum (2 × 10−3 Pa, 60 s). The IR absorption spectra were measured in a Nicolet 750 Magna instrument (2 cm−1 resolution). The amount of Brønsted and Lewis acid sites was determined from the area of the bands at 1455 cm−1 (Lewis sites) and 1546 cm−1 (Brønsted sites), using the molar extinction coefficient values previously determined (εL = 1.28 cm μmol−1 for the pyridine Lewis band38 and εB = 1.13 cm μmol−1 for Brønsted acid sites39).Isomerization of 3,3-dimethyl-but-1-ene (33DM1B)
The reaction was performed in a U shape microreactor. The feed was generated by passing a nitrogen stream through a saturator containing the liquid reagent and immersed in an ice bath at 0 °C. The catalyst (100 mg) was pretreated in situ by reduction with H2 (60 cm3 min−1, 450 °C, 1 h). The sample was then cooled in N2 (30 cm3 min−1) to the reaction temperature (100 or 125 °C). Then the feed from the saturator was injected. The reagent partial pressure and flow rate were set at 20.9 kPa and 15.27 mmol h−1, respectively. The products were analyzed with an on-line gas chromatograph with a FID detector (AlphaMOS PR2100).Temperature-programmed reduction (TPR)
TPR experiments were carried out at atmospheric pressure using a reductive mixture of H2 in N2 (5% H2 v/v, 10 cm3 STP min−1) after in situ calcination at 400 °C under flowing air for 1 h. Samples (200 mg) were heated at 10 °C min−1 from 25 up to 700 °C. The reactor outlet was connected to a TCD (thermal conductivity detector) in order to obtain the TPR signal.Transmission electron microscopy (TEM)
Catalysts were dispersed in ethanol and deposited on a copper grid before characterization by Transmission Electron Microscopy (TEM) analysis (TEM/STEM JEOL 2100 UHR) with a resolution of 0.19 nm. Images were taken with a Gatan CCD camera. The diameter of each particle was manually measured from the acquired TEM images using ImageJ software, by assuming a circular perimeter. The measuring error was estimated to be of 10% of the particle diameters due to calibration of the TEM and to the manual image analysis. At minimum 500 particles were measured in order to have a statistically significant result. Then, the mean surface diameter d (Å) was determined by d = Σ(nidi3)/Σ(nidi2), where ni is the number of particles and di the individual particle size. Considering spherical particles, the dispersion D was calculated from the mean surface diameter from D (%) = 100 × 6 (VRh/aRh)/d, where VRh and aRh are the volume occupied by a Rh atom in the bulk and the surface area occupied by a Rh atom, respectively, with VRh = 13.78 Å3 and aRh = 7.58 Å2.40CO chemisorption
Metal dispersion was determined by dynamic CO chemisorption. Calibrated adsorbate pulses were injected in a stream of nitrogen that flowed over the sample. These pulses were sent to the reactor until the sample was saturated. Before the experiment, the sample (150 mg) was reduced under H2 at 500 °C (10 °C min−1) for 1 h. Then nitrogen was made to flow over the sample for 1 h at 500 °C in order to eliminate adsorbed hydrogen. Furthermore, the sample was cooled down to room temperature in nitrogen and pulses of 0.6 μmole of CO were sent to the reactor. An estimation of Rh dispersion was obtained assuming a stoichiometry of one CO per surface Rh atom.41–43Cyclopentane (CP) hydrogenolysis
The reaction was performed at atmospheric pressure. Before the reaction, samples were reduced with H2 at 500 °C for 1 h. 80 mg of catalyst were put in contact with hydrogen at a 36 cm3 min−1 flow rate and a 0.36 cm3 h−1 CP current at 250 °C for 2 h. Products were analyzed online by a Shimadzu 2014 gas chromatograph equipped with a Phenomenex ZB-1 capillary column.Decalin ring opening
The reaction was conducted in a stainless steel autoclave reactor at either 325 or 350 °C. The hydrogen pressure was set at 3 MPa. The vessel was stirred at 1360 rpm, and 1 g of catalyst was put in contact with 25 cm3 of decalin. The decalin composition was 37.5% of cis isomer, with a trans/cis ratio of 1.63. The equipment was described in detail previously, and the lack of diffusional limitations due to mass transfer was checked.44 Samples collected after 6 h reaction time were analyzed using a Shimadzu 2014 gas chromatograph (with a Phenomenex ZB-5 capillary column) connected to a FID. The reaction of decalin led to a complex mixture of products containing more than 200 components. The identification of all the reaction products was performed using a Saturn 2000 mass spectrometer coupled to a Varian 3800 gas chromatograph using the same Phenomenex column. The reaction products analyzed at the end of the reaction (6 h) were categorized as: cracking products (C1–C9); ring opening (RO) products (C10); ring contraction (RC) products; and dehydrogenated heavy products, including naphthalene.45The experimental reproducibility (catalytic test and analysis) was checked by (i) performing the same experiment twice and (ii) for each experiment by injecting three times the same collected sample. The conversion and yields in the various products were calculated by averaging the three values. Thus, it was estimated that the conversion and yield values were obtained with a precision of ± 1.5%.
Differential scanning calorimetry (DSC)
This procedure was carried out on a SDT-Q600 manufactured by TA instruments. Approximately 10 mg of the coked catalyst were tested in a 50 cm3 min−1 air flow, and heated from 30 °C until 600 °C with a ramp of 10 °C min−1.Temperature-programmed oxidation (TPO)
Around 40 to 60 mg of the coked catalyst were first charged into a quartz micro reactor. Then the carbon was burned in an oxidizing stream (40 cm3 min−1 of diluted oxygen, 5% O2 in N2). The temperature of the cell was increased from 30 °C to 700 °C with a heating rate of 10 °C min−1. The outlet gases were fed to a methanation reactor where CO2 and CO were quantitatively transformed into CH4 in the presence of H2, and the stream was analysed using a FID detector. The catalysts carbon concentration was calculated from the area of the TPO trace by comparison to calibration experiments performed on catalysts with a known carbon concentration.46Results and discussion
Table 1 shows that the weight percentages of the deposited Rh are lower than the expected theoretical values, with lower Rh contents generally found on SIRAL 80. The metallic dispersion determined by CO chemisorption is virtually the same for all catalysts, although it slightly decreases as the Rh content increases in both series. TEM pictures displayed in Fig. 1 show that the particle distribution of Rh(1.5)/S70 is in agreement with the chemisorption result, with a mean particle size of 3.3 nm. On the contrary, Rh(1.5)/S80 presents a larger mean particle size, 4.1 nm, which corresponds to a dispersion of roughly 27%. This value is slightly lower than the one determined by CO chemisorption (33%), due to the presence of large particles, which contributes significantly to the calculation of the mean surface diameter but a little to the chemisorption.40 Furthermore, the hypothesis of a stoichiometry CO/Rh = 1 could also explain this divergence. It is well known that CO may not only adsorb linearly on Rh atom, the predominant form, but also in the form of bridged species between two neighboring atoms on large particles, as well as gem-dicarbonyl species, clearly identified by Fourier Transform Infrared spectroscopy (FTIR).47,48| Rh contenta (wt%) | Dispersion (%) | TOF (s−1) | μmol CP g−1 s−1 | |||||
|---|---|---|---|---|---|---|---|---|
| S70 | S80 | S70 | S80 | S70 | S80 | S70 | S80 | |
| a Determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). | ||||||||
| Rh(1.0) | 0.91 | 0.86 | 38 | 35 | 0.286 | 0.297 | 9.62 | 8.69 |
| Rh(1.5) | 1.10 | 1.20 | 34 | 33 | 0.297 | 0.244 | 10.79 | 9.39 |
| Rh(2.0) | 1.64 | 1.44 | 31 | 32 | 0.243 | 0.258 | 11.99 | 11.56 |
 | ||
| Fig. 1 TEM pictures and particle size distributions of (A) Rh(1.5)/S70 (874 particles analyzed); (B) Rh(1.5)/S80 (570 particles analyzed). | ||
The presence of larger particles on Rh(1.5)/S80 than on Rh(1.5)/S70, as shown by the histograms presented in Fig. 1, should increase the proportion of bridged CO species to the detriment of linear CO species on Rh atoms, which would have led to an underestimation of the dispersion value by CO chemisorption. The formation of gem-dicarbonyl species may better explain the discrepancy between the dispersion values from TEM and CO chemisorption analysis. Gem-dicarbonyl species (RhI–(CO)2) may also result from the oxidation of Rh in the presence of CO involving hydroxyl groups of support due to the Rh–Rh bond disruption in the presence of CO.49 This has been observed on alumina and MgO.49,50 The RhI–(CO)2/Rh0–CO ratio fluctuates with rhodium particle size, it is favored on very small particles,49 and also with contact time with CO, long contact time favoring the disruption phenomenon.50 Thus, in the present study, the lower dispersion from TEM pictures compared with the results of CO chemisorption could be due to the presence of very small metal particles, able to adsorb two molecules of CO per metal atom and not visible on the TEM picture. Both phenomenon will contribute to the discrepancy between the dispersion calculated by TEM and the one calculated by CO chemisorption considering a stoichiometry of 1: small particles are not taken into account for the calculation of the mean particle size, leading on the one hand to an overestimation of the mean particle size, and underestimation of the derived-dispersion (27%) and, on the other hand, to the formation of gem-dicarbonyl, which causes an overestimation of the dispersion by CO chemisorption (33%). Consequently, the actual dispersion of Rh in the Rh(1.5)/S80 sample is probably between 27 and 33% and can be roughly estimated to be around 30%. Then it can be inferred that the particle distribution is heterogeneous in the Rh(1.5)/S80 sample, with very small metallic and non-visible particles as well as very large ones. In conclusion, CO chemisorption gives a rough estimation of the dispersion of Rh at the support surface as does the analysis of TEM pictures, especially for catalysts with wide size distribution such as the Rh(1.5)/S80 sample. The heterogeneity of the sample supported on S80, illustrated by the size distribution range (1–8 nm), observed by TEM and the large standard deviation (1.8 nm), may be explained by the presence of a higher SiO2 content in S80 than in S70, since it is well known that metal dispersion is better on alumina than on silica.51
Cyclopentane (CP) hydrogenolysis is a useful model reaction to estimate the metallic activity of a catalyst and requires big metallic particle ensembles.52,53 The CP reaction rate (μmoles of CP converted per second and per gram of catalyst) increases with Rh contents, as expected. TOF values are similar for both series given the small differences in metallic dispersion values. Since the reaction is only catalyzed by the metallic function, it is practically not influenced by the support.
The S70 support has a slightly greater surface area than the S80, being 377 and 337 m2 g−1 respectively. As shown in a previous work, both supports present X-ray diffraction spectra characteristic of an amorphous silica-alumina structure.54 Moreover, from pyridine TPD experiments, it was shown that both supports exhibit moderate acidity sites, corresponding to pyridine desorption peak between 300 and 500 °C, and strong acid sites (desorption at T > 500 °C) in comparable concentration. Weak acid sites (desorption at T < 300 °C) are also present in lesser quantity. Although both supports display a quite comparable total acidity, S70 possesses a slightly higher ratio of strong acid sites.54 In order to complete these previous results, the concentration and strength of Lewis and Brønsted acid sites were evaluated by FTIR of adsorbed pyridine (Py) by using different desorption temperatures (150, 250, 350 and 450 °C) (Table 2). The absorption bands at 1455 and 1546 cm−1 are attributed to pyridine coordinated to Lewis acid sites and pyridinium ions ring vibration due to pyridine bound to Brønsted acid sites, respectively.55–57 The values of the integrated extinction coefficients (ε) are intrinsic to the adsorbed molecule and are not affected by the catalyst structure or acid strength.57 In order to estimate the ratio between Brønsted and Lewis acid sites at different temperatures, the εB/εL ratio is considered constant between 150 and 450 °C. Table 2 shows that the ratio between Brønsted and Lewis acid sites diminishes at higher desorption temperatures. For a given temperature, both supports present similar ratios. The total acidity ratio between S70 and S80 was estimated previously at 1.01 using pyridine TPD54 while the actual results of pyridine FTIR study give a value of 1.06 at 150 °C (desorption temperature allowing to determine the total acidity of the samples). Selli et al. have already reported that Py-TPD and Py-FTIR are in good agreement when both experiments are conducted at equilibrium conditions at 150 °C.58
| Support | T (°C) | Acid sites (μmol of Py gCat−1) | Brønsted/Lewis ratio | |
|---|---|---|---|---|
| Lewis | Brønsted | |||
| S70 | 150 | 148 | 81 | 0.55 |
| 250 | 100 | 28 | 0.28 | |
| 350 | 65 | 4 | 0.06 | |
| 450 | 33 | 0 | — | |
| S80 | 150 | 144 | 72 | 0.50 |
| 250 | 92 | 26 | 0.28 | |
| 350 | 65 | 5 | 0.08 | |
| 450 | 44 | 2 | 0.05 | |
Fig. 2 shows desorption profiles of pyridine for each Rh(X)/Sy catalyst. It is clear that the largest amount of sites corresponds to moderate acidity sites (with a desorption temperature between 300 and 500 °C). At higher Rh loads, desorption peaks are shifted to lower temperatures on both supports. Catalysts supported on SIRAL 70 have a total acidity slightly higher than those supported on SIRAL 80 for a given metallic content, as it can be seen in Table 3. The model reaction of skeletal isomerization of 33DM1B was used to characterize the Brønsted acid sites. Indeed, as demonstrated by Kemball et al., the Lewis centers are not involved in this reaction and the 33DM1B isomerization is likely to occur through a pure protonic mechanism.59,60 Otherwise, additional 33DM1B isomerization tests performed on the Rh(1.0)/S70 and Rh(1.0)/S80 catalysts (results not shown) indicate a higher activity in the case of the S70 catalyst, resulting from a greater Brønsted acidity (site number and/or strength) on this sample.
 | ||
| Fig. 2 Pyridine TPD profiles of the catalysts: (A) Rh(X)/S80; (B) Rh(X)/S70. | ||
| Catalyst | Total acidity (μmol Py gcat−1) | Acid amount (μmol Py gcat−1) | ||
|---|---|---|---|---|
| Weak | Moderate | Strong | ||
| Rh(1.0)/S70 | 1809 | 222 | 1578 | 9 |
| Rh(1.5)/S70 | 1579 | 235 | 1317 | 27 |
| Rh(2.0)/S70 | 1465 | 243 | 1200 | 22 |
| Rh(1.0)/S80 | 1753 | 129 | 1477 | 147 |
| Rh(1.5)/S80 | 1508 | 252 | 1240 | 16 |
| Rh(2.0)/S80 | 1412 | 293 | 1108 | 11 |
Fig. 3 shows the TPR profiles of the two catalysts series. In both cases, the area of the reduction peaks increases with increasing metallic content and indicates the total reduction of the metallic phase. The Rh(X)/S70 series presents a single reduction peak with a maximum between 100 and 115 °C. It can thus be inferred that the size of the metallic particles and metal–support interactions are of the same order of magnitude on these three samples. On the other hand, the Rh(X)/S80 series presents notable differences between the Rh(1.0)/S80, Rh(1.5)/S80 and Rh(2.0)/S80 catalysts. While Rh(2.0)/S80 exhibits a single reduction peak centered at 103 °C, the other samples lead to an additional peak with a maximum at 173 °C. It is possible to conclude that on this support, a higher heterogeneity is present at low Rh contents, regarding the size distribution and metal interactions with the support, in accordance with the TEM results of Rh(1.5)/S80.
 | ||
| Fig. 3 TPR profiles of the catalysts: (A) Rh(X)/S80; (B) Rh(X)/S70. | ||
Decalin ring opening was performed at 325 and 350 °C. The pristine calcined S70 and S80 supports were evaluated for this reaction in a previous work and an increase in the reaction temperature was found to increase the conversion by more than 25% in both cases, favoring cracking, dehydrogenation, as well as ring opening reactions.54 Conversion rates and yields to dehydrogenated products were practically the same for both supports. However, S70 presents a higher yield to decalin ring contraction products at both temperatures, while the yield to cracking products is higher for S80.30
Fig. 4 gives decalin conversion values at 325 and 350 °C as a function of the cyclopentane hydrogenolysis reaction rates (i.e. the values of the metallic function activity given in Table 1) and as a function of the total acidity (determined from TPD of adsorbed pyridine, Table 3). As expected, a higher conversion is observed at the highest temperature for both Rh(X)/Sy catalysts series. At 350 °C, the decalin conversion evolves between 75% (Rh1/S80) and 82% (Rh1/S70), and is then practically unaffected by changes in the acidity or metallic function activity. On the other hand, at 325 °C, the conversion of decalin goes through a maximum as a function of the metal activity and total acidity. A bifunctional mechanism has been proposed for decalin ring opening. The first step is a ring contraction over the acid sites from a 6 to a 5-membered ring. These rings are then more easily hydrogenated over the metallic sites.61,62 As a consequence, the present results can be explained considering that the metallic activity (hydrogenolysis) and the acid activity follow opposite tendencies regarding Rh content. The metal activity rises at increasing Rh loads (Table 1) but at the expense of the acid sites (Table 3 and Fig. 2). At 325 °C, the ring contraction step can be limited by a too low support acidity, and moreover, this lower temperature also reduces the metallic activity. At 350 °C, the hydrogenolytic activity increases substantially, according to its high activation energy.63 Under these conditions, the reaction is no longer limited by the formation of 5-membered carbon rings.
 | ||
| Fig. 4 Decalin conversion at 325 and 350 °C as a function of (A) total acid sites and (B) cyclopentane reaction rate for the Rh(X)/Sy catalysts series. | ||
Fig. 5 shows the influence of the catalyst acidity on the yield to cracking products. The cracking reactions are known to be promoted by acid sites and to present a high activation energy. As a consequence, the cracking product yield rises at increasing reaction temperatures and catalyst acidities as reported in Fig. 5.
 | ||
| Fig. 5 Yields of cracking products as a function of the total acidity at 325 and 350 °C (decalin reaction temperature). | ||
Fig. 6 shows that the yield to dehydrogenated products increases at higher reaction temperatures since dehydrogenation is an endothermic and reversible reaction.63 At 325 °C, the yield to dehydrogenated products decreases slightly at increasing metallic activities (expressed as CP μmoles converted by gram of catalyst and by second), probably because the competing hydrogenolysis reactions are more favored. At 350 °C, a remarkable increase in the yield to dehydrogenated products is seen, the S80 series being the more dehydrogenating catalysts. Fig. 7 shows that the yield to ring contraction products is smaller at 350 °C, due to a greater conversion of these products into ring opening, dehydrogenated and cracking products at higher temperature. At both reaction temperatures, the yield to RC products increases slightly at increasing acidity and scarcely decreases at increasing metallic activity. Again, this phenomenon can be explained by the fact that the ring contraction reaction is catalyzed by the acid sites while the metallic sites convert this five-membered rings into ring opening products. Although cracking and dehydrogenation reactions are favored at higher temperature, the yield to ring opening products at 350 °C is higher than at 325 °C (Fig. 8). Also, the yield to RO products goes through a maximum expressed as a function of the total acidity or as a function of the metallic activity. According to the bifunctional opening mechanism, a first acid-controlled ring contraction step is followed by ring opening, catalyzed by the metallic sites. As a consequence, an ideal catalyst should possess high acidity and hydrogenolytic activity. The analysis performed on these catalysts shows that the acidity diminishes at increasing Rh contents. An optimum Rh load exists at a 1.5 wt% (theoretic) content, presenting an optimum balance between the two functions. Thus, the highest yield in RO products, 54%, was obtained with Rh(1.5)/S70 at 350 °C, 3 MPa of total pressure and initial decalin/catalyst = 22 w/w, for a decalin conversion of 78%, which corresponds to a selectivity to RO products of 69%. For the same catalyst, the highest selectivity obtained at 325 °C was similar, 68%, although the decalin conversion was lower, 64%. It is difficult to compare these results to those of the literature, since, in addition to the nature of the catalyst, the reaction conditions, as for example the type of reactor, total pressure, reaction temperature, the decalin/catalyst and H2/decalin ratio, vary across studies. For example, Kubicka et al. compared bifunctional Pt and Ir catalysts supported over various zeolites in decalin conversion in a batch reactor for the same decalin/catalyst ratio as in the present study.9 At an extrapolated value of 78% of decalin conversion, the highest selectivity in RO products was ca. 56% at 6 MPa and 250 °C for Ir/H-Beta, while increasing the temperature or decreasing the total pressure led to a decrease in selectivity.9 Also on Ir/H-Beta, Alzaid and Smith obtained a selectivity to RO products of ca. 44% at a pressure of 3 MPa and 350 °C, i.e. in conditions apparently similar to the ones used in the present study, in terms of temperature and pressure but very different for the other operating conditions, especially with a decalin/catalyst ratio = 176.16
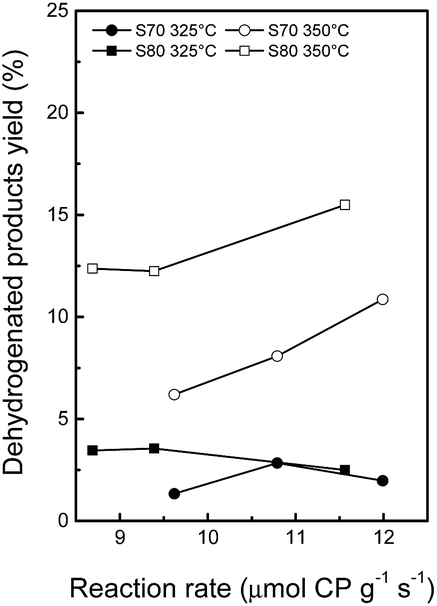 | ||
| Fig. 6 Yields of dehydrogenated products as a function of metal activity (measured in cyclopentane hydrogenolysis) at 325 and 350 °C (decalin reaction temperature). | ||
 | ||
| Fig. 7 Yields of ring contraction products as a function of (A) the total acidity and (B) the metal activity at 325 and 350 °C (decalin reaction temperature). | ||
 | ||
| Fig. 8 Yields of ring opening products as a function of (A) the total acidity and (B) the metal activity at 325 and 350 °C (decalin reaction temperature). | ||
Deactivation by coke deposition is rarely addressed in selective ring opening studies on non-zeolitic catalytic materials. Here, DSC was used to evaluate the coke deposited on the catalysts during decalin ring opening test performed at 325 °C (Fig. 9). The combustion temperature of coke depends on its nature and location (metallic sites or support). All catalysts (except Rh(1.0)/S80) show similar combustion profiles, i.e. a large combustion peak with a maximum around 340–375 °C. Rh(1.0)/S80 presents two peaks at higher temperatures (close to 400 and 500 °C). This particularity for this sample possessing the greatest amount of strong acid sites (Table 3) can be attributed to the presence of a highly polymerized coke, which burns at a higher temperature.64,65 It is known that coke polymerization occurs over sites with the greatest acid strength.63 Nonetheless, these high temperature DSC peaks could also be due to the diffusional limitations induced by pore blocking by polyaromatic compounds.66 When the coke content increases, it becomes more stable and the DSC peak shifts to higher temperatures.67 It is important to notice that all peaks are below 500 °C, and may be attributed to amorphous coke.68 Whatever the catalyst, carbon combustion starts at low temperatures (below 250 °C), and can be assigned to coke deposited over or near metallic particles, which catalyze the combustion reaction.69,70 Coke deposition was also studied by TPO on the same used catalysts. Fig. 10 shows that the coke combustion profiles are similar for catalysts with a similar metallic load, irrespective of the support, except for Rh(1.0)/S80. Table 4 displays the carbon concentrations determined from TPO analysis.
 | ||
| Fig. 9 DSC profiles of the Rh(1.0), Rh(1.5) and Rh(2.0) catalysts supported on S70 and S80, after 6 h of decalin reaction at 325 °C. | ||
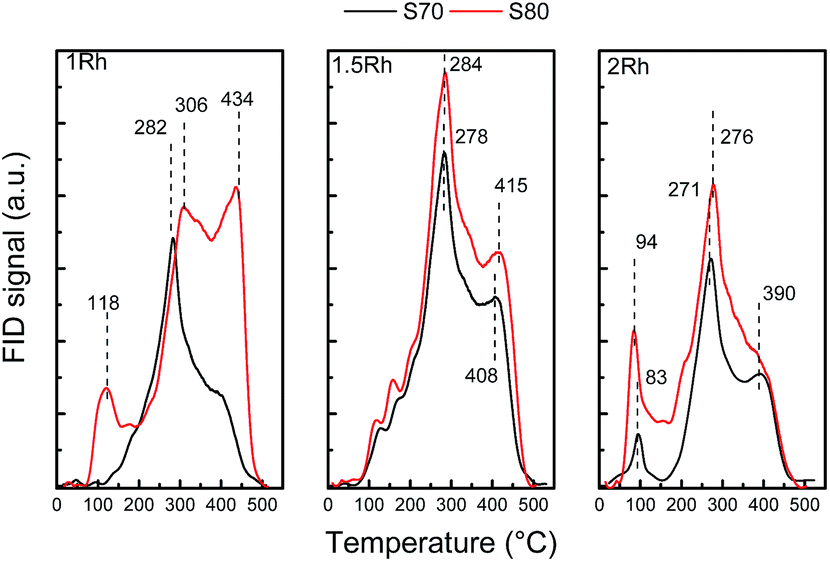 | ||
| Fig. 10 TPO profiles of the Rh(1.0), Rh(1.5) and Rh(2.0) catalysts supported on S70 and S80, after 6 h of decalin reaction at 325 °C. | ||
| Catalyst | C (wt%) | Catalyst | C (wt%) |
|---|---|---|---|
| 1Rh/S70 | 2.05 | 1Rh/S80 | 4.08 |
| 1.5Rh/S70 | 3.62 | 1.5Rh/S80 | 4.65 |
| 2Rh/S70 | 2.03 | 2Rh/S80 | 3.27 |
It is clear that catalysts supported over SIRAL 80 suffer greater coke deposition than those over SIRAL 70 during the reaction of decalin ring opening at 325 °C, and that a deactivation is very likely for both catalysts series, and higher for the S80 series. It is interesting to analyze the relation between coke deposition and the formation of dehydrogenated products during decalin RO reaction, since these products have been proposed as coke formation precursors. They are produced on the metallic sites and then polymerized over the acid sites of the support.70,71 Fig. 11 shows a direct relation between the carbon content deposited on the used samples and their yield to dehydrogenation products.
 | ||
| Fig. 11 Carbon percentage as a function of yield to dehydrogenated products. Decalin reaction temperature: 325 °C. | ||
Conclusion
Selective ring opening of decalin was studied at 325 and 350 °C using rhodium catalysts supported on two different SiO2–Al2O3 supports (SIRAL 70, S70, and SIRAL 80, S80) and with various Rh contents. In term of RO yield, a Rh load of 1.5 wt% was found to be optimum for the two sets of catalysts. The best metal/support combination was finally obtained for the Rh(1.5)/S70 sample. These results show that there is an optimum ratio between the metallic and acid functions, favoring ring opening products. An increase in the reaction temperature favors cracking and dehydrogenation products. A linear correlation was obtained between the amount of coke deposited on the used catalysts and the formation of dehydrogenated compounds.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the ECOS-MINCyT program no. A12E02 (2013–2015) which allowed the collaboration between the two research teams, and the “Bernardo Houssay” program for the post-doc fellowship.References
- W. C. Baird, J. G. Chen and G. B. McVicker, US Pat., 6623626, 2001, assigned to Exxon Mobil.
- G. B. McVicker, M. Daage, M. S. Touvelle, C. W. Hudson, D. P. Klein, W. C. Baird, B. R. Cook, J. G. Chen, S. Hantzer, D. E. W. Vaughan, E. S. Ellis and O. C. Feeley, J. Catal., 2002, 210, 137 CrossRef CAS.
- R. C. Santana, P. T. Do, M. Santikunaporn, W. E. Alvarez, J. D. Taylor, E. L. Sughrue and D. E. Resasco, Fuel, 2006, 85, 643 CrossRef CAS.
- P. T. Do, W. E. Alvarez and D. E. Resasco, J. Catal., 2006, 238, 477 CrossRef CAS.
- B. H. Cooper and B. B. L. Donnis, Appl. Catal., A, 1996, 137, 203 CrossRef CAS.
- V. Calemma, M. Ferrari, S. Rabl and J. Weitkamp, Fuel, 2013, 111, 763 CrossRef CAS.
- L. J. Yuan, S. Q. Guo, Z. R. Li, H. T. Cui, H. Y. Dong, L. F. Zhao and J. W. Wang, RSC Adv., 2017, 7, 9446 RSC.
- M. Santikunaporn, J. Herrera, S. Jongpatiwut, D. Resasco, W. Alvarez and E. Sughrue, J. Catal., 2004, 228, 100 CrossRef CAS.
- D. Kubicka, M. Kangas, N. Kumar, M. Tiita, M. Lindblad and D. Y. Murzin, Top. Catal., 2010, 53, 1438 CrossRef CAS.
- S. Rabl, A. Haas, D. Santi, C. Flego, M. Ferrari, V. Calemma and J. Weitkamp, Appl. Catal., A, 2011, 400, 131 CrossRef CAS.
- L. Di Felice, N. Catherin, L. Piccolo, D. Laurenti, E. Blanco, E. Leclerc, C. Geantet and V. Calemna, Appl. Catal., A, 2016, 512, 43 CrossRef CAS.
- D. Kubicka, T. Salmi, M. Tiita and D. Y. Murzin, Fuel, 2009, 52, 380 Search PubMed.
- A. Martinez, M. A. Arribas and S. B. C. Perher, Catal. Sci. Technol., 2016, 6, 2528 CAS.
- S. Rabl, A. Haas, D. Santi, C. Flego, M. Ferrari, V. Calemna and J. Weitkamp, Appl. Catal., A, 2011, 400, 131 CrossRef CAS.
- D. Santi, T. Holl, V. Calemna and J. Weitkamp, Appl. Catal., A, 2013, 455, 46 CrossRef CAS.
- A. H. Alzaid and K. J. Smith, Appl. Catal., A, 2013, 450, 243 CrossRef CAS.
- H. Du, C. Fairbridge, H. Yang and Z. Ring, Appl. Catal., A, 2005, 294, 1 CrossRef CAS.
- G. Maire, G. Plouidy, J. C. Prudhomme and F. G. Gault, J. Catal., 1965, 4, 556 CrossRef CAS.
- G. Maire, C. Corolleur, D. Juttard and F. G. Gault, J. Catal., 1971, 21, 250 CrossRef CAS.
- F. G. Gault, Adv. Catal., 1981, 30, 1 CAS.
- J. L. Carter, J. A. Cusumano and J. H. Sinfelt, J. Catal., 1971, 20, 223 CrossRef CAS.
- C. G. Walter, B. Coq, F. Figueras and M. Boulet, Appl. Catal., A, 1995, 133, 95 CrossRef CAS.
- J. R. Anderson and Y. Shimoyama, in Proc. 5th ICC, ed. J. W. Hightower, North-Holland, Amsterdam, 1972, vol. 1, p. 695 Search PubMed.
- G. Del Angel, B. Coq, R. Dutartre and F. Figueras, J. Catal., 1984, 87, 27 CrossRef CAS.
- H. Ziaei-Azad and A. Sayari, J. Catal., 2016, 344, 729 CrossRef CAS.
- N. Kumar, P. Mäki-Arvela, N. Musakka, D. Kubicka, M. Kangas, M. Tiita, H. Österholm, A.-R. Leino, K. Kordas, T. Heikkilä, T. Salmi and D. Y. Murzin, Catal. Ind., 2013, 5, 11 Search PubMed.
- H. Zimmer and Z. Paal, J. Mol. Catal., 1989, 51, 261 CrossRef CAS.
- Y. J. Lee, E. S. Kim, J. R. Kim, J. W. Kim, T. W. Kim, H. J. Chae, C. U. Kim, C. H. Lee and S. Y. Jeong, J. Nanosci. Nanotechnol., 2016, 16, 4335 CrossRef CAS PubMed.
- A. Galadima and O. Muraza, Fuel, 2016, 181, 618 CrossRef CAS.
- H. S. Cerqueira, P. C. Mihindou-Koumba, P. Magnoux and M. Guisnet, Ind. Eng. Chem. Res., 2001, 40, 1032 CrossRef CAS.
- A. Corma, V. Gonzalez-Alfaro and A. V. Orchillés, J. Catal., 2001, 200, 34 CrossRef CAS.
- D. Kubicka, N. Kumar, P. Mäki-Arvela, M. Tiitta, V. Niemi, H. Karhu, T. Salmi and D. Y. Murzin, J. Catal., 2004, 227, 313 CrossRef CAS.
- D. Kubicka, N. Kumar, P. Mäki-Arvela, M. Tiitta, V. Niemi, T. Salmi and D. Y. Murzin, J. Catal., 2004, 222, 65 CrossRef CAS.
- L. Piccolo, S. Nassredine, G. Toussaint and C. Geantet, ChemSusChem, 2012, 5, 1717 CrossRef CAS PubMed.
- S. Nassredine, L. Massin, M. Aouine, C. Geantet and L. Piccolo, J. Catal., 2011, 278, 253 CrossRef.
- S. Albonetti, G. Baldi, A. Barzanti, E. Rodríguez-Castellon, A. J. Lopez, D. E. Quesada and A. Vaccari, Catal. Lett., 2006, 108, 197 CrossRef CAS.
- L. S. Carvalho, K. C. S. Conceição, V. A. Mazzieri, P. Reyes, C. L. Pieck and M. C. Rangel, Appl. Catal., A, 2012, 419–420, 156 CrossRef.
- M. Guisnet, P. Ayrault, C. Coutanceau, M. F. Alvarez and J. Datka, J. Chem. Soc., Faraday Trans., 1997, 93, 1661 RSC.
- M. Guisnet, P. Ayrault and J. Datka, Pol. J. Chem., 1997, 71, 1455 CAS.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH, Weinheim, 2nd edn, 2008, pp. 738–765 Search PubMed.
- Y.-F. Y. Yao, J. Catal., 1984, 87, 152 CrossRef CAS.
- Y. Cao, R. Ran, X. Wu, B. Zhao and D. Weng, J. Environ. Sci., 2017, 52, 197 CrossRef PubMed.
- F. P. Bouxin, X. Zhang, I. N. Kings, A. F. Lee, M. J. H. Simmons, K. Wilson and S. D. Jackson, Appl. Catal., A, 2017, 539, 29 CrossRef CAS.
- S. A. D'Ippolito, C. Especel, L. Vivier, F. Epron and C. L. Pieck, Appl. Catal., A, 2014, 469, 532 CrossRef.
- S. A. D'Ippolito, L. B. Gutierrez and C. L. Pieck, Appl. Catal., A, 2012, 445, 195 CrossRef.
- S. A. D'Ippolito, C. Especel, F. Epron, P. Marécot and C. L. Pieck, Appl. Catal., A, 2010, 388, 272 CrossRef.
- F. Solymosi and M. Pasztor, J. Phys. Chem., 1985, 89, 4789 CrossRef CAS.
- P. B. Rasband and W. C. Hecker, J. Catal., 1993, 139, 551 CrossRef CAS.
- H. F. J. Van't Blik, J. B. A. D. Van Zon, T. Hulzinga, J. C. Vis, J. C. Koningsberger and R. Prins, J. Phys. Chem., 1983, 87, 2264 CrossRef.
- M. Efstathiou, T. Chafok, D. Bianchi and C. O. Bennett, J. Catal., 1994, 147, 24 CrossRef.
- J. H. Sinfelt and G. H. Via, J. Catal., 1979, 56, 1 CrossRef CAS.
- A. Roberti, V. Ponec and W. M. H. Sachtler, J. Catal., 1973, 29, 381 CrossRef.
- B. Biloen, J. Helle, H. Verbeek, F. Dautzenberg and W. Sachtler, J. Catal., 1980, 63, 112 CrossRef.
- S. A. D'Ippolito, A. Ballarini and C. L. Pieck, Energy Fuels, 2017, 31, 5461 CrossRef.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401 RSC.
- T. R. Hughes and H. M. White, J. Phys. Chem., 1967, 71, 2192 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- E. Selli and L. Forni, Microporous Mesoporous Mater., 1999, 31, 129 CrossRef CAS.
- C. S. John, C. Kemball and R. A. Rajadhayarsha, J. Catal., 1979, 57, 264 CrossRef CAS.
- D. Martin and D. Duprez, J. Mol. Catal. A: Chem., 1997, 118, 113 CrossRef CAS.
- H. Du, C. Fairbridge, H. Yang and Z. Ring, Appl. Catal., A, 2005, 294, 1 CrossRef CAS.
- R. Moraes, K. Thomas, S. Thomas, S. Van Donk, G. Grasso, J.-P. Gilson and M. J. Houalla, J. Catal., 2013, 299, 30 CrossRef CAS.
- J. M. Parera and N. S. Fígoli, in Catalytic naphtha reforming: science and technology. ed. G. J. Antos, A. M. Aitani and J. M. Parera, Marcel Dekker Inc, New York, 1995 Search PubMed.
- J. Barbier, P. Marecot, N. Martin, L. E. Lassa and R. Maurel, Stud. Surf. Sci. Catal., 1980, 6, 53 CrossRef CAS.
- J. M. Parera, N. S. Figoli, E. M. Traffano, J. Beltramini and E. E. Martinelli, Appl. Catal., 1983, 5, 33 CrossRef CAS.
- P. Magnoux and M. Guisnet, Zeolites, 1989, 9, 329 CrossRef CAS.
- F. J. Maldonado-Hódar, M. F. Ribeiro, J. M. Silva, A. P. Antunes and F. R. Ribeiro, J. Catal., 1998, 178, 1 CrossRef.
- C. Wang, N. Sun, N. Zhao, W. Wei, Y. Sun, C. Sun, H. Liu and C. E. Snape, Fuel, 2015, 43, 527 CrossRef.
- J. M. Parera, N. S. Figoli and E. M. Traffano, J. Catal., 1983, 79, 481 CrossRef CAS.
- J. Barbier, in Catalyst Deactivation 1987, ed. B. Delmon and G. Froment, Elsevier, Amsterdam, 1987, p. 1 Search PubMed.
- J. Barbier, Appl. Catal., 1986, 23, 225 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |